
Genome-Wide Association Analysis Revealed Candidate Genes Related to Early Growth Traits in Inner Mongolia Cashmere Goats

 , , , , ,
, , , , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
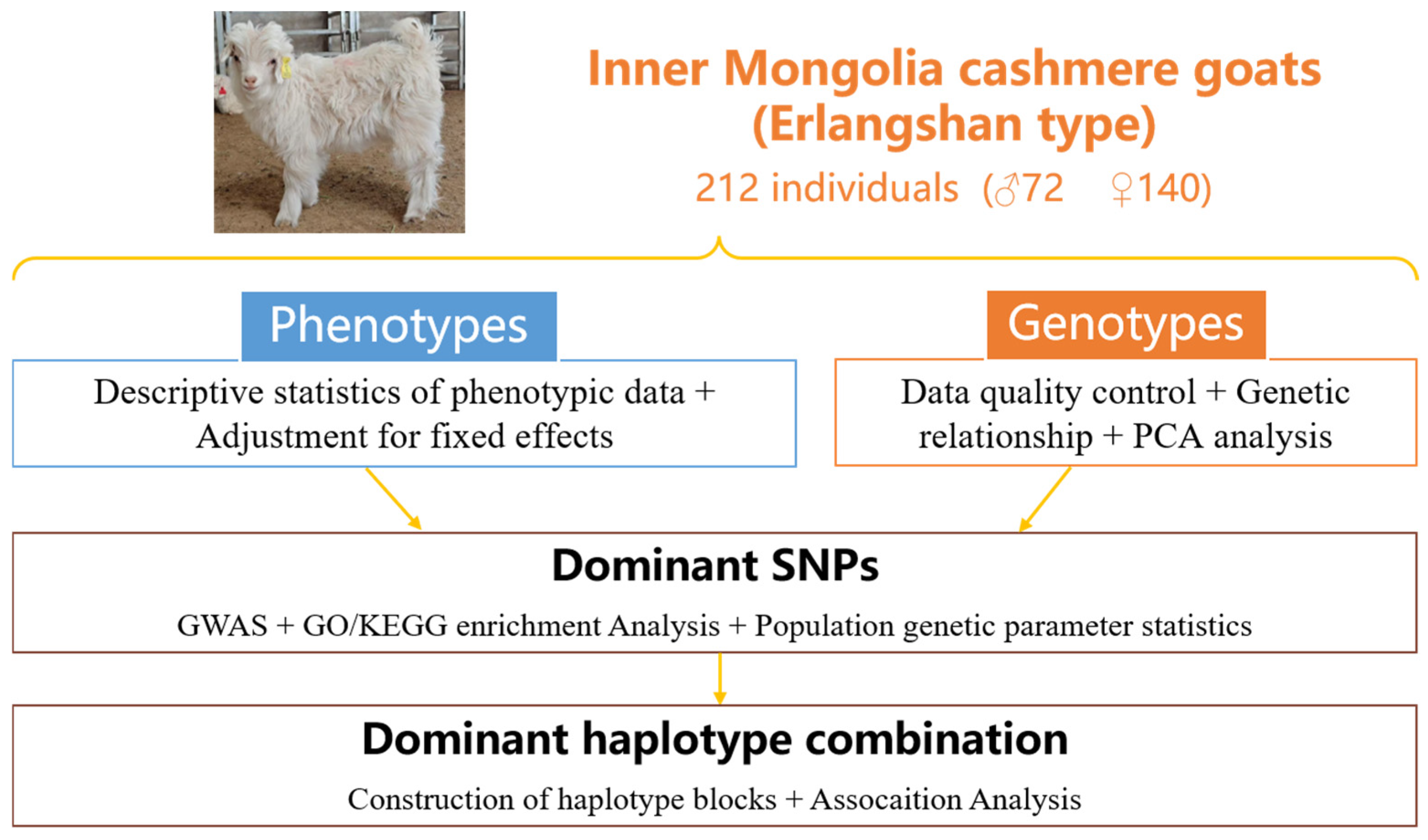
2.1. Source of Experimental Animal and Phenotype Data
2.2. Genomic DNA Extraction, Library Construction, and Online Sequencing
2.3. Single-Nucleotide Polymorphism Calling
2.4. Data Quality Control, Genetic Relationship Analysis Based on the IBS Distance Matrix and G Matrix, and PCA Analysis
2.5. Genome-Wide Association Study
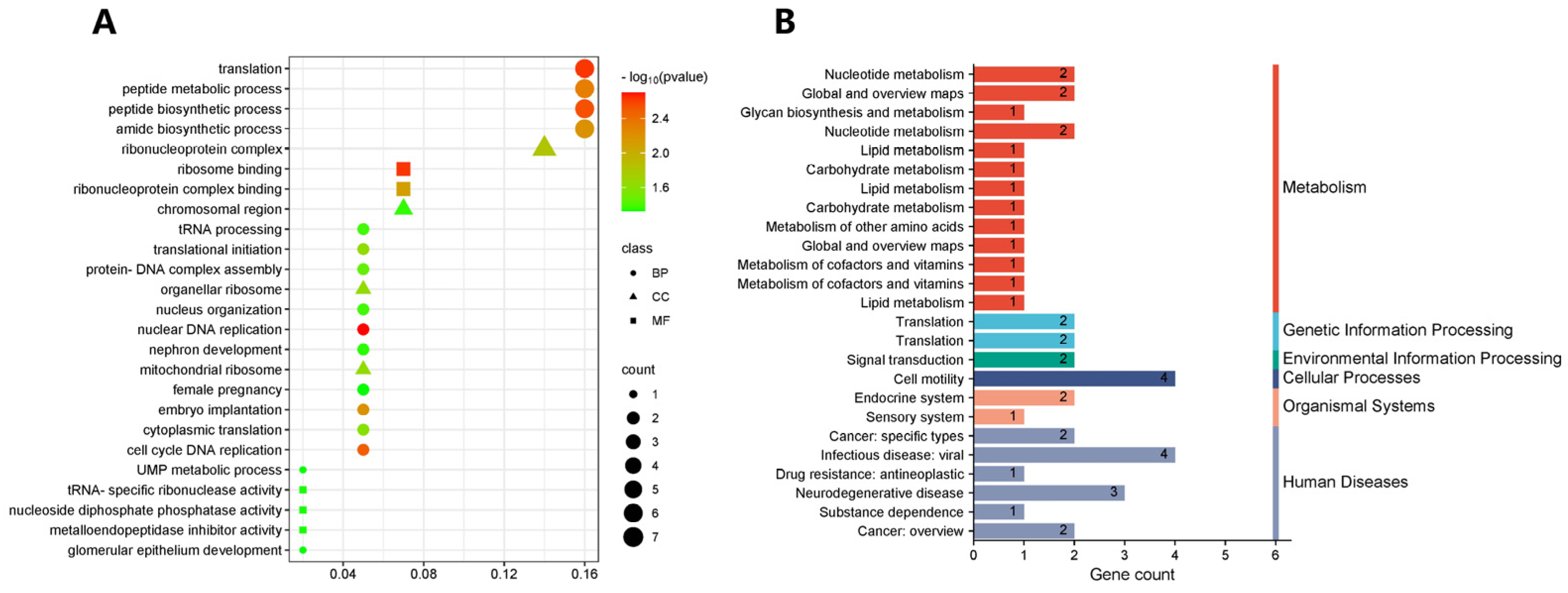
2.6. GO Functional Annotation and KEGG Enrichment Analysis of Candidate Genes
2.7. Statistical Analysis of Population Genetic Parameters for Significant SNPs
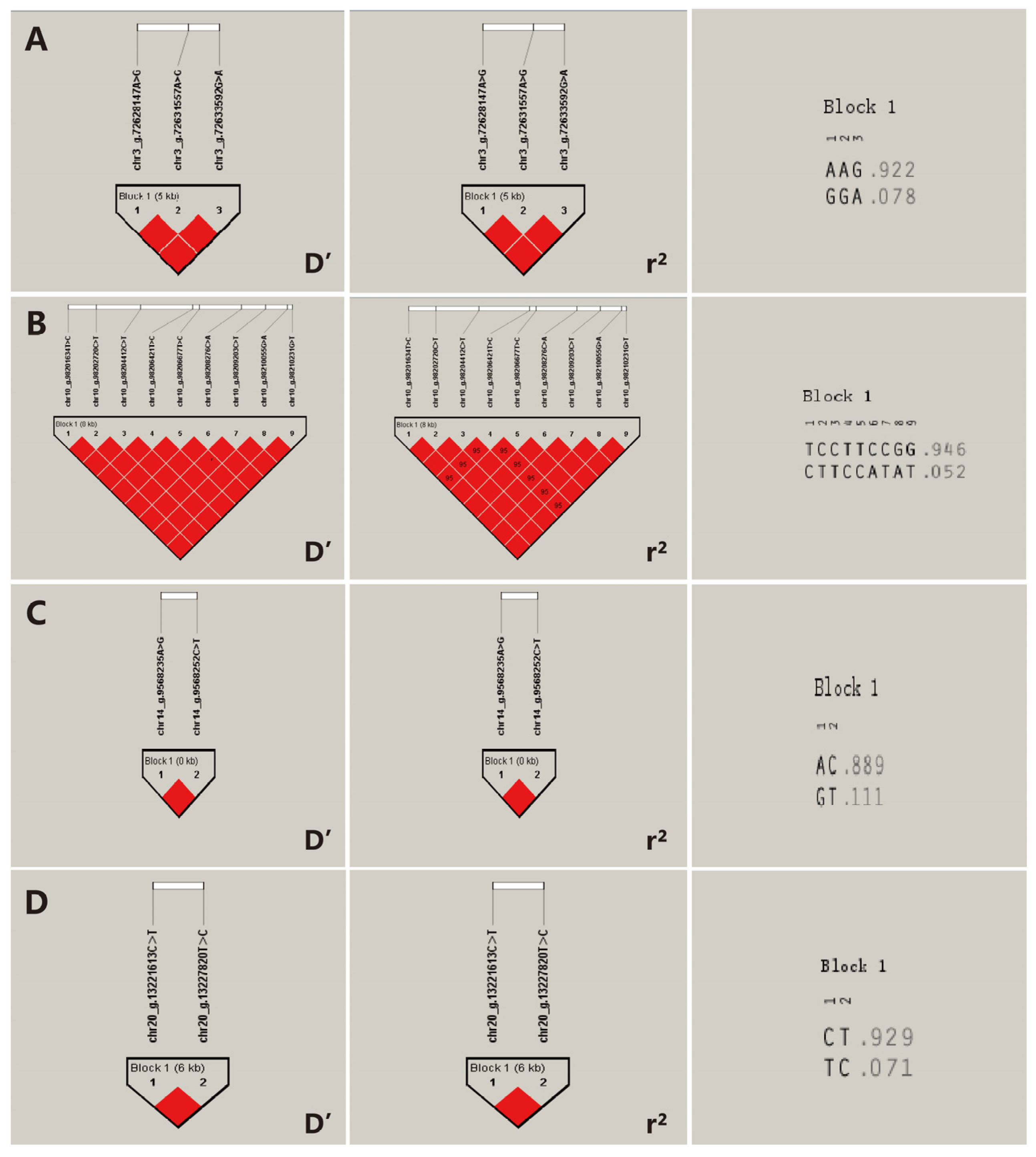
2.8. Construction of Haplotype Block and Analysis of Linkage Disequilibrium
2.9. Association Analysis of Haplotype Combination with Birth Weight and Weaning Weight Traits
3. Results
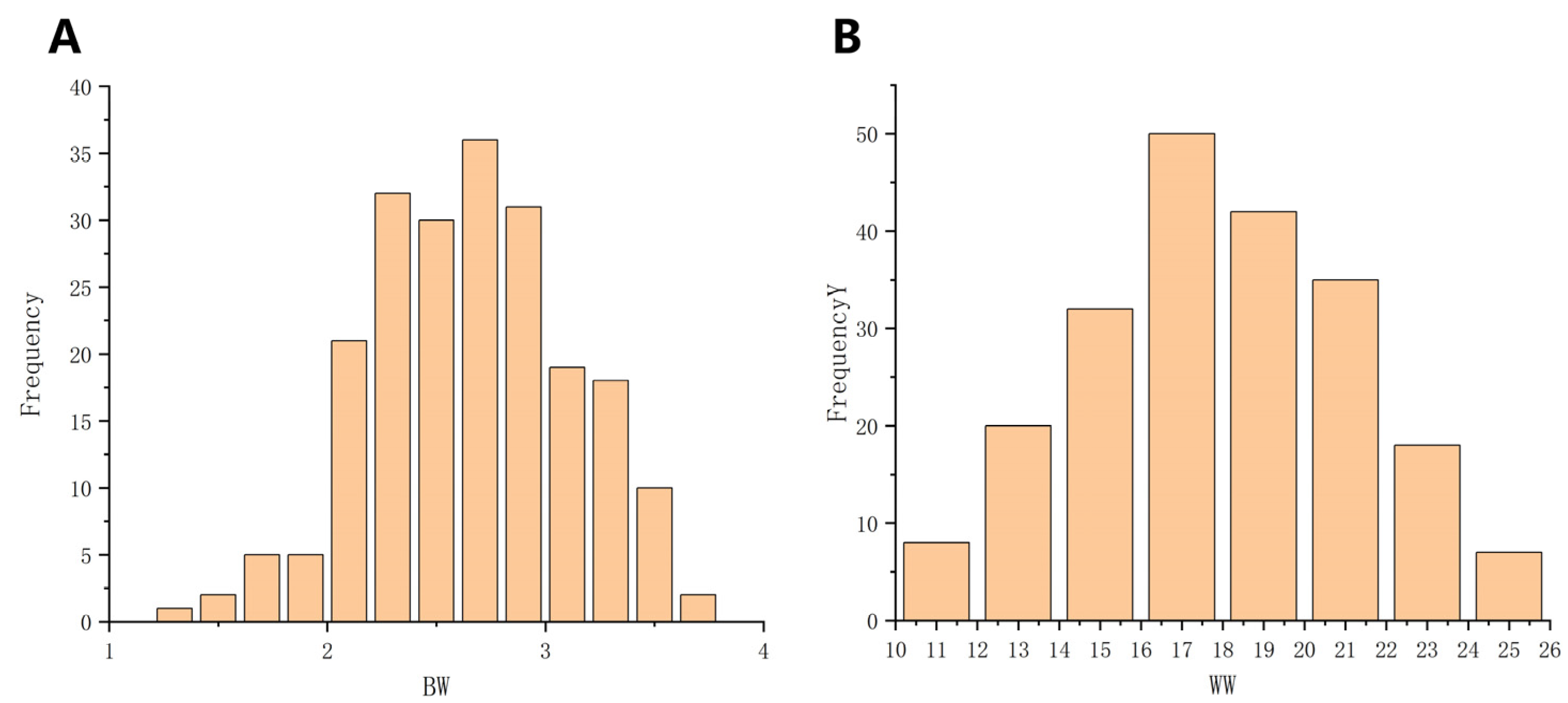
3.1. Descriptive Statistics of Phenotypic Data
3.2. Adjustment for Fixed Effects
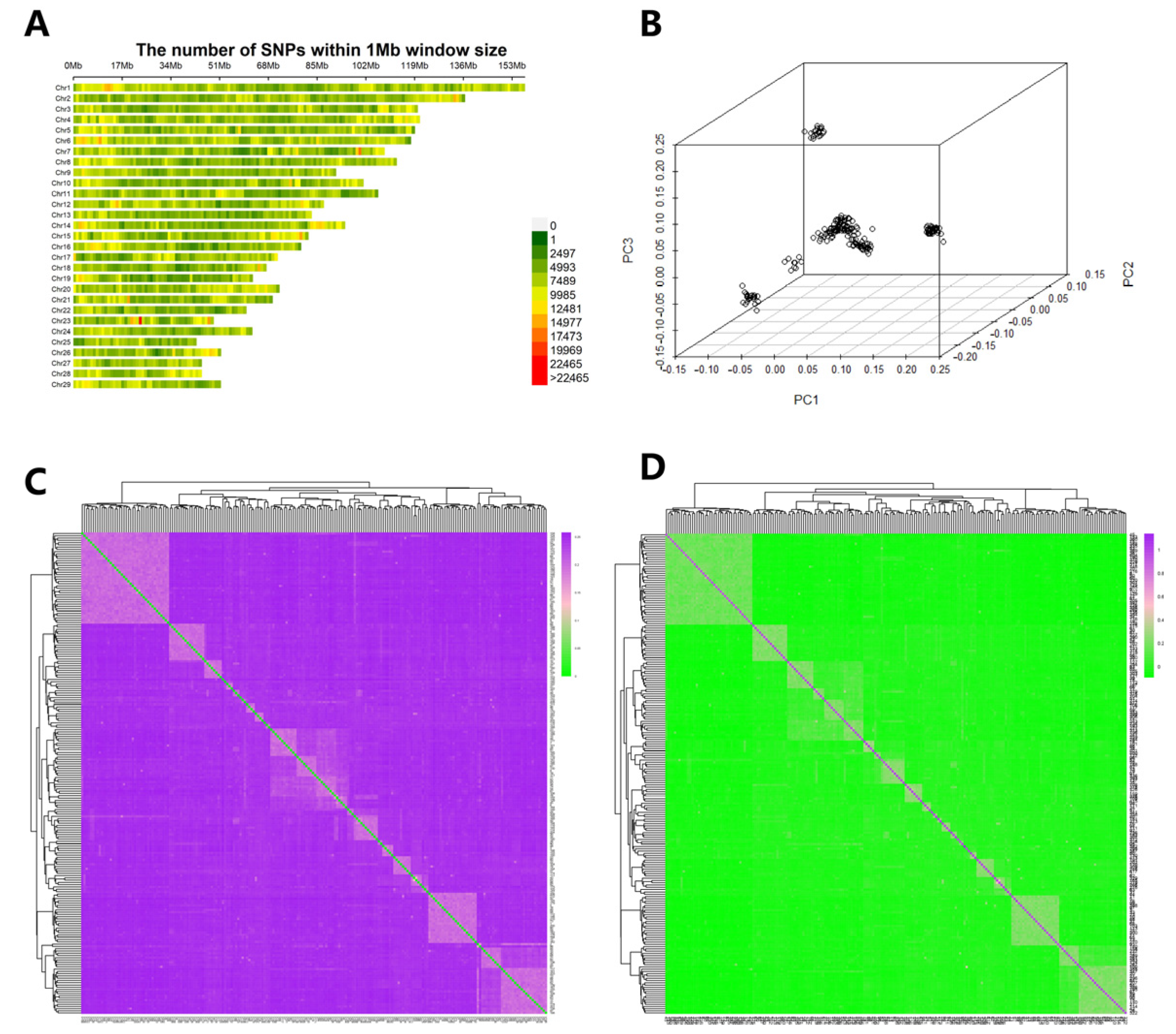
3.3. Data Quality Control, Genetic Relationship, and PCA Analysis
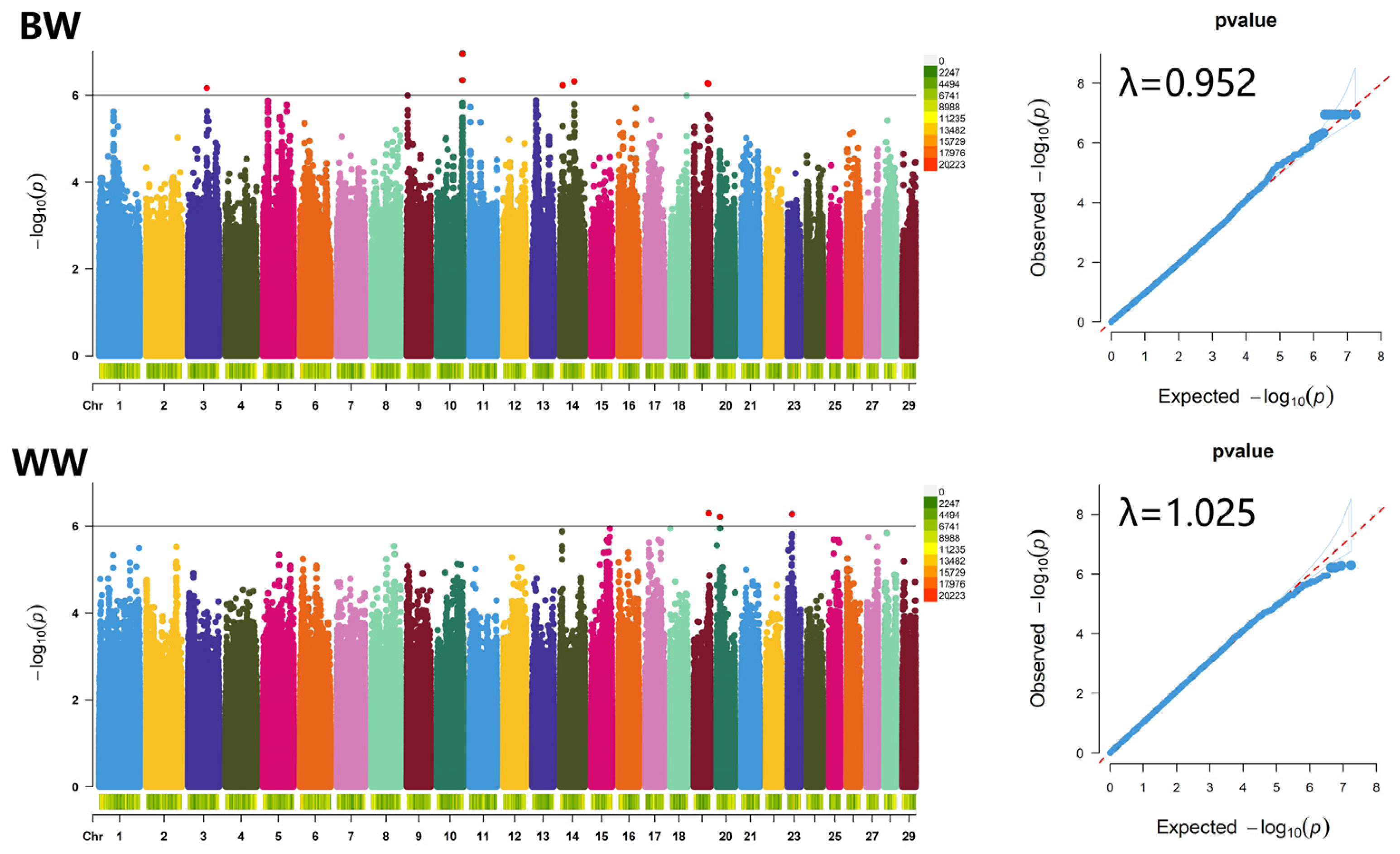
3.4. GWAS for Birth Weight and Weaning Weight Traits
3.4.1. Birth Weight Trait
3.4.2. Weaning Weight Trait
3.5. Annotation and Enrichment Analysis of Candidate Genes
3.6. Population Genetic Parameter Statistics
3.7. Haplotype Analysis
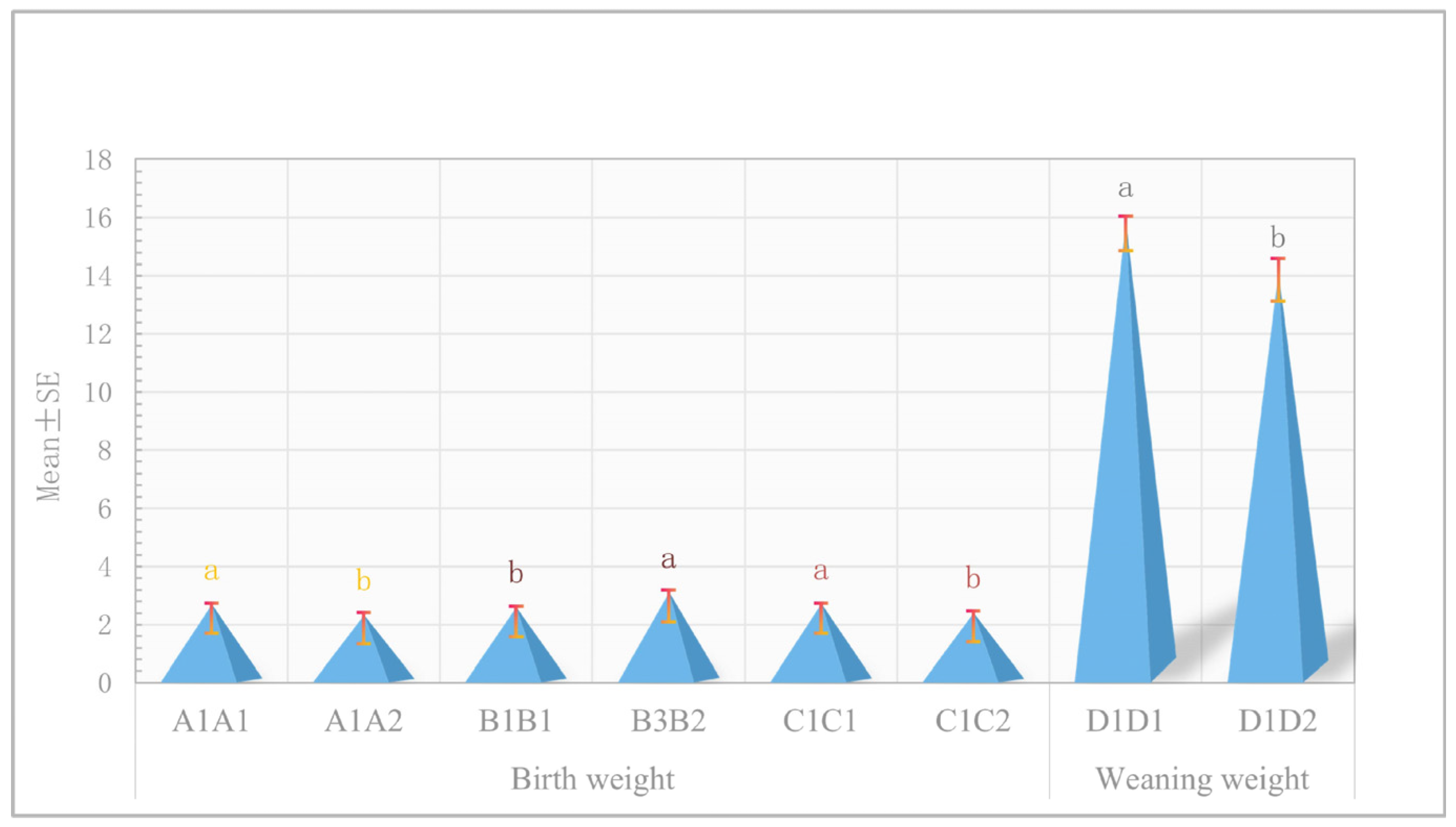
3.8. Association Analysis of Haplotype Combinations with Birth Weight and Weaning Weight Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| WW | weaning weight |
| BW | birth weight |
| GWAS | genome-wide associated study |
| SNPs | single-nucleotide polymorphisms |
| LD | linkage disequilibrium |
| IMCGs | Inner Mongolian cashmere goats |
| HWE | Hardy–Weinberg equilibrium test |
| Ho | homozygosity |
| He | heterozygosity |
| PIC | polymorphism information content |
| Ne | effective allele number |
| MAF | minor allele frequency |
| IBS | identity by state |
References
- Zhao, Q.; Huang, C.; Chen, Q.; Su, Y.; Zhang, Y.; Wang, R.; Su, R.; Xu, H.; Liu, S.; Ma, Y.; et al. Genomic Inbreeding and Runs of Homozygosity Analysis of Cashmere Goat. Animals 2024, 14, 1246. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hai, E.; Di, Z.; Ma, R.; Shang, F.; Wang, M.; Liang, L.; Rong, Y.; Pan, J.; Su, R.; et al. Chi-miR-130b-3p regulates Inner Mongolia cashmere goat skin hair follicles in fetuses by targeting Wnt family member 10A. G3 2021, 11, jkaa023. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Yang, F.; Wu, Z.; Guo, F.; Zhang, J.; Hai, E.; Shang, F.; Su, R.; Wang, R.; Wang, Z.; et al. Inner Mongolian Cashmere Goat Secondary Follicle Development Regulation Research Based on mRNA-miRNA Co-analysis. Sci. Rep. 2020, 10, 4519. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tan, H.; Zhang, M.; Zhao, R.; Wang, S.; Qin, Q.; Wang, J.; Zhang, C.; Tao, M.; Ma, M.; et al. The Hybrid Genome of a New Goldfish-Like Fish Lineage Provides Insights Into the Origin of the Goldfish. Front. Genet. 2020, 11, 122. [Google Scholar] [CrossRef]
- Zhuang, Z.; Xu, L.; Yang, J.; Gao, H.; Zhang, L.; Gao, X.; Li, J.; Zhu, B. Weighted Single-Step Genome-Wide Association Study for Growth Traits in Chinese Simmental Beef Cattle. Genes 2020, 11, 189. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, H.; Lei, C.; Pan, C.; Chen, H.; Lin, Q.; Lan, X. Effects of genetic variations within goat PITX2 gene on growth traits and mRNA expression. Anim. Biotechnol. 2020, 31, 107–114. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, C.; Guo, Y.; She, S.; Wang, B.; Jiang, Y.; Bai, Y.; Song, X.; Li, L.; Shi, L.; et al. Screening of Deletion Variants within the Goat PRDM6 Gene and Its Effects on Growth Traits. Animals 2020, 10, 208. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, F.; Gao, G.; Yan, X.; Liu, H.; Liu, Z.; Wang, Z.; He, L.; Lv, Q.; Wang, Z.; et al. Genome-Wide Association Study of Body Weight Traits in Inner Mongolia Cashmere Goats. Front. Vet. Sci. 2021, 8, 752746. [Google Scholar] [CrossRef]
- Gu, B.; Sun, R.; Fang, X.; Zhang, J.; Zhao, Z.; Huang, D.; Zhao, Y.; Zhao, Y. Genome-Wide Association Study of Body Conformation Traits by Whole Genome Sequencing in Dazu Black Goats. Animals 2022, 12, 548. [Google Scholar] [CrossRef]
- Easa, A.A.; Selionova, M.; Aibazov, M.; Mamontova, T.; Sermyagin, A.; Belous, A.; Abdelmanova, A.; Deniskova, T.; Zinovieva, N. Identification of Genomic Regions and Candidate Genes Associated with Body Weight and Body Conformation Traits in Karachai Goats. Genes 2022, 13, 1773. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.J.; Hawkins, J.R. DNA variation and the future of human genetics. Nat. Biotechnol. 1998, 16, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.M.; Terwilliger, J.D. How many diseases does it take to map a gene with SNPs? Nat. Genet. 2000, 26, 151–157. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, K.; Zhao, H. Haplotype-association analysis. Adv. Genet. 2008, 60, 335–405. [Google Scholar] [PubMed]
- Almasi, M.; Zamani, P.; Mirhoseini, S.Z.; Moradi, M.H. Genome-wide association study for postweaning weight traits in Lori-Bakhtiari sheep. Trop. Anim. Health Prod. 2021, 53, 163. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Gondro, C.; Porto-Neto, L.R.; Lee, S.H. R for genome-wide association studies. Methods Mol. Biol. 2013, 1019, 1–17. [Google Scholar] [PubMed]
- Jiang, L.; Zheng, Z.; Qi, T.; Kemper, K.E.; Wray, N.R.; Visscher, P.M.; Yang, J. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet. 2019, 51, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Barrett, J.C. Haploview: Visualization and analysis of SNP genotype data. Cold Spring Harb. Protoc. 2009, 2009, pdb.ip71. [Google Scholar] [CrossRef]
- Moser, E.B.; Saxton, A.M.; Geaghan, J.P. Biological applications of the SAS system: An overview. Comput. Appl. Biosci. CABIOS 1988, 4, 233–238. [Google Scholar] [CrossRef]
- Craddock, N.; Sklar, P. Genetics of bipolar disorder. Lancet 2013, 381, 1654–1662. [Google Scholar] [CrossRef]
- Jiang, N.; Wang, Z.; Guo, X.; Peng, Z.; He, Y.; Wang, Q.; Wu, H.; Cui, Y. Hepatic Runx1t1 improves body fat index after endurance exercise in obese mice. Sci. Rep. 2023, 13, 19427. [Google Scholar] [CrossRef]
- Jiao, Y.; Huang, B.; Chen, Y.; Hong, G.; Xu, J.; Hu, C.; Wang, C. Integrated Analyses Reveal Overexpressed Notch1 Promoting Porcine Satellite Cells’ Proliferation through Regulating the Cell Cycle. Int. J. Mol. Sci. 2018, 19, 271. [Google Scholar] [CrossRef]
- Abe, A.; Yamamoto, Y.; Katsumi, A.; Okamoto, A.; Tokuda, M.; Inaguma, Y.; Yamamoto, K.; Yanada, M.; Kanie, T.; Tomita, A.; et al. Rearrangement of VPS13B, a causative gene of Cohen syndrome, in a case of RUNX1-RUNX1T1 leukemia with t(8;12;21). Int. J. Hematol. 2018, 108, 208–212. [Google Scholar] [CrossRef]
- Wu, L.; Gao, L.; Zhang, D.; Yao, R.; Huang, Z.; Du, B.; Wang, Z.; Xiao, L.; Li, P.; Li, Y.; et al. C1QTNF1 attenuates angiotensin II-induced cardiac hypertrophy via activation of the AMPKa pathway. Free Radic. Biol. Med. 2018, 121, 215–230. [Google Scholar] [CrossRef]
- Kim, D.; Park, S.Y. C1q and TNF related protein 1 regulates expression of inflammatory genes in vascular smooth muscle cells. Genes Genom. 2019, 41, 397–406. [Google Scholar] [CrossRef]
- Fukushima, K.; Nakamura, A.; Ueda, H.; Yuasa, K.; Yoshida, K.; Takeda, S.; Ikeda, S. Activation and localization of matrix metalloproteinase-2 and -9 in the skeletal muscle of the muscular dystrophy dog (CXMDJ). BMC Musculoskelet. Disord. 2007, 8, 54. [Google Scholar] [CrossRef]
- So, K.K.H.; Huang, Y.; Zhang, S.; Qiao, Y.; He, L.; Li, Y.; Chen, X.; Sham, M.H.; Sun, H.; Wang, H. seRNA PAM controls skeletal muscle satellite cell proliferation and aging through trans regulation of Timp2 expression synergistically with Ddx5. Aging Cell 2022, 21, e13673. [Google Scholar] [CrossRef]
- Yu, M.; Han, S.; Wang, M.; Han, L.; Huang, Y.; Bo, P.; Fang, P.; Zhang, Z. Baicalin protects against insulin resistance and metabolic dysfunction through activation of GALR2/GLUT4 signaling. Phytomedicine 2022, 95, 153869. [Google Scholar] [CrossRef]
- Leciejewska, N.; Pruszyńska-Oszmałek, E.; Mielnik, K.; Głowacki, M.; Lehmann, T.P.; Sassek, M.; Gawęda, B.; Szczepankiewicz, D.; Nowak, K.W.; Kołodziejski, P.A. Spexin Promotes the Proliferation and Differentiation of C2C12 Cells In Vitro-The Effect of Exercise on SPX and SPX Receptor Expression in Skeletal Muscle In Vivo. Genes 2021, 13, 81. [Google Scholar] [CrossRef]
- Fang, P.; Zhang, L.; Yu, M.; Sheng, Z.; Shi, M.; Zhu, Y.; Zhang, Z.; Bo, P. Activiated galanin receptor 2 attenuates insulin resistance in skeletal muscle of obese mice. Peptides 2018, 99, 92–98. [Google Scholar] [CrossRef]
- Lv, X.; Chen, W.; Wang, S.; Cao, X.; Yuan, Z.; Getachew, T.; Mwacharo, J.M.; Haile, A.; Sun, W. Whole-genome resequencing of Dorper and Hu sheep to reveal selection signatures associated with important traits. Anim. Biotechnol. 2023, 34, 3016–3026. [Google Scholar] [CrossRef]
- Zhang, Q.; Lee, H.G.; Han, J.A.; Kim, E.B.; Kang, S.K.; Yin, J.; Baik, M.; Shen, Y.; Kim, S.H.; Seo, K.S.; et al. Differentially expressed proteins during fat accumulation in bovine skeletal muscle. Meat Sci. 2010, 86, 814–820. [Google Scholar] [CrossRef]
- Liu, R.; Sun, Y.; Zhao, G.; Wang, F.; Wu, D.; Zheng, M.; Chen, J.; Zhang, L.; Hu, Y.; Wen, J. Genome-wide association study identifies Loci and candidate genes for body composition and meat quality traits in Beijing-You chickens. PLoS ONE 2013, 8, e61172. [Google Scholar] [CrossRef]
- Lu, Y.; Zhou, J.; Li, F.; Cao, H.; Zhang, X.; Yu, D.; He, Z.; Ji, H.; Lv, K.; Wu, G.; et al. The Integration of Genome-Wide DNA Methylation and Transcriptomics Identifies the Potential Genes That Regulate the Development of Skeletal Muscles in Ducks. Int. J. Mol. Sci. 2023, 24, 15476. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shin, J.; McGee, M.; Unger, S.; Bando, N.; Sato, J.; Vandewouw, M.; Patel, Y.; Branson, H.M.; Paus, T.; et al. Intake of mother’s milk by very-low-birth-weight infants and variation in DNA methylation of genes involved in neurodevelopment at 5.5 years of age. Am. J. Clin. Nutr. 2022, 116, 1038–1048. [Google Scholar] [CrossRef]
- Hunsucker, S.A.; Spychala, J.; Mitchell, B.S. Human cytosolic 5′-nucleotidase I: Characterization and role in nucleoside analog resistance. J. Biol. Chem. 2001, 276, 10498–10504. [Google Scholar] [CrossRef]
- D’Ambrosio, C.; Hongo, A.; Li, S.; Baserga, R. The role of Grb2 in the growth and transformation of mouse embryo cells. Oncogene 1996, 12, 371–378. [Google Scholar]
- Maina, F.; Casagranda, F.; Audero, E.; Simeone, A.; Comoglio, P.M.; Klein, R.; Ponzetto, C. Uncoupling of Grb2 from the Met receptor in vivo reveals complex roles in muscle development. Cell 1996, 87, 531–542. [Google Scholar] [CrossRef]
- Howey, R.; Cordell, H.J. Imputation without doing imputation: A new method for the detection of non-genotyped causal variants. Genet. Epidemiol. 2014, 38, 173–190. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, Q.; Liu, Y.; Gilbert, E.R.; Li, D.; Yin, H.; Wang, Y.; Yang, Z.; Wang, Z.; Yuan, Y.; et al. Polymorphisms in the Perilipin Gene May Affect Carcass Traits of Chinese Meat-type Chickens. Asian-Australas. J. Anim. Sci. 2015, 28, 763–770. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Birth Type | Dam Age | Number | Mean | Max | Min | SD | CV (%) |
|---|---|---|---|---|---|---|---|---|
| Birth weight | Singletons | 3 | 183 | 2.71 | 3.73 | 1.38 | 0.44 | 16.18 |
| Twins | 3 | 27 | 2.23 | 2.87 | 1.7 | 0.32 | 14.33 | |
| Triplets | 3 | 2 | 2.00 | 2.04 | 1.95 | 0.06 | 3.12 | |
| Weaning weight | Singletons | 3 | 183 | 15.98 | 24.53 | 8.69 | 2.59 | 16.22 |
| Twins | 3 | 27 | 13.43 | 19.23 | 9.86 | 2.30 | 17.16 | |
| Triplets | 3 | 2 | 12.36 | 14.81 | 9.91 | 3.46 | 28.02 |
| Trait | SNP_Number | SNP | BETA | p-Value | r2 (%) | Distance (bp) | Structure_Type | Gene |
|---|---|---|---|---|---|---|---|---|
| Birth weight | SNP1 | chr3_g.72628147A>G | −0.3449 | 6.81 × 10−7 | 2.66 | −361,498 | intergenic | RWDD3 |
| SNP2 | chr3_g.72631557A>G | −0.3449 | 6.81 × 10−7 | 2.66 | −364,908 | intergenic | RWDD3 | |
| SNP3 | chr3_g.72633592G>A | −0.3449 | 6.81 × 10−7 | 2.66 | −366,943 | intergenic | RWDD3 | |
| SNP4 | chr10_g.98201634T>C | 0.4349 | 1.11 × 10−7 | 2.65 | within | intronic | KCNN2 | |
| SNP5 | chr10_g.98202720C>T | 0.4349 | 1.11 × 10−7 | 2.65 | within | intronic | KCNN2 | |
| SNP6 | chr10_g.98204412C>T | 0.4349 | 1.11 × 10−7 | 2.65 | within | intronic | KCNN2 | |
| SNP7 | chr10_g.98206421T>C | 0.3890 | 4.58 × 10−7 | 2.39 | within | intronic | KCNN2 | |
| SNP8 | chr10_g.98206677T>C | 0.4349 | 1.11 × 10−7 | 2.65 | within | intronic | KCNN2 | |
| SNP9 | chr10_g.98208276C>A | 0.4349 | 1.11 × 10−7 | 2.65 | within | intronic | KCNN2 | |
| SNP10 | chr10_g.98209203C>T | 0.4349 | 1.11 × 10−7 | 2.65 | within | intronic | KCNN2 | |
| SNP11 | chr10_g.98210055G>A | 0.4349 | 1.11 × 10−7 | 2.65 | within | intronic | KCNN2 | |
| SNP12 | chr10_g.98210231G>T | 0.4349 | 1.11 × 10−7 | 2.65 | within | intronic | KCNN2 | |
| SNP13 | chr14_g.9568235A>G | −0.2871 | 5.96 × 10−7 | 2.73 | 228,432 | intergenic | RUNX1T1 | |
| SNP14 | chr14_g.9568252C>T | −0.2871 | 5.96 × 10−7 | 2.73 | 228,415 | intergenic | RUNX1T1 | |
| SNP15 | chr14_g.54137103A>G | −0.1880 | 4.85 × 10−7 | 2.93 | within | intronic | NKAIN3 | |
| SNP16 | chr19_g.52517379T>C | −0.2512 | 5.31 × 10−7 | 2.79 | within | intronic | RBFOX3 | |
| SNP17 | chr19_g.55261375C>T | −0.2657 | 5.44 × 10−7 | 2.77 | within | intronic | CASKIN2 | |
| Weaning weight | SNP18 | chr19_56428712A>T | −1.1205 | 5.09 × 10−7 | 2.58 | within | intronic | TTYH2 |
| SNP19 | chr20_13221613C>T | −1.8399 | 6.11 × 10−7 | 2.52 | −177,193 | intergenic | LOC102178014 | |
| SNP20 | chr20_13227820T>C | −1.8399 | 6.11 × 10−7 | 2.52 | −183,400 | intergenic | LOC102178014 | |
| SNP21 | chr23_16477721T>G | 1.0185 | 5.33 × 10−7 | 2.45 | within | intronic | FARS2 |
| Trait | SNP_Number | Allelic Frequency | HWE | Ho | He | PIC | Ne | ||
|---|---|---|---|---|---|---|---|---|---|
| Major | Minor | χ2 | p | ||||||
| Birth weight | SNP1 | 0.922 | 0.078 | 1.51 | 0.219 | 0.856 | 0.144 | 0.133 | 1.168 |
| SNP2 | 0.922 | 0.078 | 1.51 | 0.219 | 0.856 | 0.144 | 0.133 | 1.168 | |
| SNP3 | 0.922 | 0.078 | 1.51 | 0.219 | 0.856 | 0.144 | 0.133 | 1.168 | |
| SNP4 | 0.948 | 0.052 | 0.635 | 0.426 | 0.902 | 0.098 | 0.094 | 1.109 | |
| SNP5 | 0.948 | 0.052 | 0.635 | 0.426 | 0.902 | 0.098 | 0.094 | 1.109 | |
| SNP6 | 0.948 | 0.052 | 0.635 | 0.426 | 0.902 | 0.098 | 0.094 | 1.109 | |
| SNP7 | 0.946 | 0.054 | 0.254 | 0.615 | 0.897 | 0.103 | 0.097 | 1.114 | |
| SNP8 | 0.948 | 0.052 | 0.635 | 0.426 | 0.902 | 0.098 | 0.094 | 1.109 | |
| SNP9 | 0.948 | 0.052 | 0.635 | 0.426 | 0.902 | 0.098 | 0.094 | 1.109 | |
| SNP10 | 0.948 | 0.052 | 0.635 | 0.426 | 0.902 | 0.098 | 0.094 | 1.109 | |
| SNP11 | 0.948 | 0.052 | 0.635 | 0.426 | 0.902 | 0.098 | 0.094 | 1.109 | |
| SNP12 | 0.948 | 0.052 | 0.635 | 0.426 | 0.902 | 0.098 | 0.094 | 1.109 | |
| SNP13 | 0.889 | 0.111 | 0.178 | 0.673 | 0.803 | 0.197 | 0.178 | 1.246 | |
| SNP14 | 0.889 | 0.111 | 0.178 | 0.673 | 0.803 | 0.197 | 0.178 | 1.246 | |
| SNP15 | 0.535 | 0.465 | 1.731 | 0.188 | 0.503 | 0.497 | 0.374 | 1.99 | |
| SNP16 | 0.856 | 0.144 | 0.116 | 0.733 | 0.754 | 0.246 | 0.216 | 1.327 | |
| SNP17 | 0.874 | 0.126 | 0.181 | 0.67 | 0.78 | 0.22 | 0.196 | 1.281 | |
| Weaning weight | SNP18 | 0.778 | 0.222 | 0.319 | 0.572 | 0.655 | 0.345 | 0.286 | 1.527 |
| SNP19 | 0.929 | 0.071 | 1.229 | 0.268 | 0.869 | 0.131 | 0.123 | 1.151 | |
| SNP20 | 0.929 | 0.071 | 1.229 | 0.268 | 0.869 | 0.131 | 0.123 | 1.151 | |
| SNP21 | 0.736 | 0.264 | 0.182 | 0.67 | 0.611 | 0.389 | 0.313 | 1.636 | |
| Trait | Gene | Tag | Haplotype | Frequency |
|---|---|---|---|---|
| Birth weight | RWDD3 | A1 | AAG | 0.922 |
| A2 | GGA | 0.078 | ||
| KCNN2 | B1 | TCCTTCCGG | 0.946 | |
| B2 | CTTCCATAT | 0.052 | ||
| B3 | TCCCTCCGG | 0.002 | ||
| RUNX1T1 | C1 | AC | 0.889 | |
| C2 | GT | 0.111 | ||
| Weaning weight | LOC102178014 | D1 | CT | 0.929 |
| D2 | TC | 0.071 |
| Trait | Gene | Haplotype Combination | Number | Mean ± SE |
|---|---|---|---|---|
| Birth weight | RWDD3 | AAAAGG (A1A1) | 179 | 2.6989 ± 0.0328 a |
| AGAGGA (A1A2) | 33 | 2.3321 ± 0.0733 b | ||
| KCNN2 | TTCCCCTTTTCCCCGGGG (B1B1) | 190 | 2.5937 ± 0.0316 b | |
| TCCTCTTCTCCACTGAGT (B3B2) | 21 | 3.1 ± 0.0821 a | ||
| RUNX1T1 | AACC (C1C1) | 167 | 2.7072 ± 0.0350 a | |
| AGCT (C1C2) | 43 | 2.4119 ± 0.0583 b | ||
| Weaning weight | LOC102178014 | CCTT (D1D1) | 182 | 15.8704 ± 0.1973 a |
| CTTC (D1D2) | 30 | 14.1227 ± 0.4646 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rong, Y.; Ao, X.; Guo, F.; Wang, X.; Han, M.; Zhang, L.; Xia, Q.; Shang, F.; Lv, Q.; Wang, Z.; et al. Genome-Wide Association Analysis Revealed Candidate Genes Related to Early Growth Traits in Inner Mongolia Cashmere Goats. Vet. Sci. 2025, 12, 192. https://doi.org/10.3390/vetsci12030192
Rong Y, Ao X, Guo F, Wang X, Han M, Zhang L, Xia Q, Shang F, Lv Q, Wang Z, et al. Genome-Wide Association Analysis Revealed Candidate Genes Related to Early Growth Traits in Inner Mongolia Cashmere Goats. Veterinary Sciences. 2025; 12(3):192. https://doi.org/10.3390/vetsci12030192
Chicago/Turabian StyleRong, Youjun, Xiaofang Ao, Furong Guo, Xinle Wang, Mingxuan Han, Lu Zhang, Qincheng Xia, Fangzheng Shang, Qi Lv, Zhiying Wang, and et al. 2025. "Genome-Wide Association Analysis Revealed Candidate Genes Related to Early Growth Traits in Inner Mongolia Cashmere Goats" Veterinary Sciences 12, no. 3: 192. https://doi.org/10.3390/vetsci12030192
APA StyleRong, Y., Ao, X., Guo, F., Wang, X., Han, M., Zhang, L., Xia, Q., Shang, F., Lv, Q., Wang, Z., Su, R., Zhao, Y., Zhang, Y., & Wang, R. (2025). Genome-Wide Association Analysis Revealed Candidate Genes Related to Early Growth Traits in Inner Mongolia Cashmere Goats. Veterinary Sciences, 12(3), 192. https://doi.org/10.3390/vetsci12030192