
Fungal Plasma Membrane H+-ATPase: Structure, Mechanism, and Drug Discovery


{kind=link}
{kind=link}
{kind=link}
{kind=link}
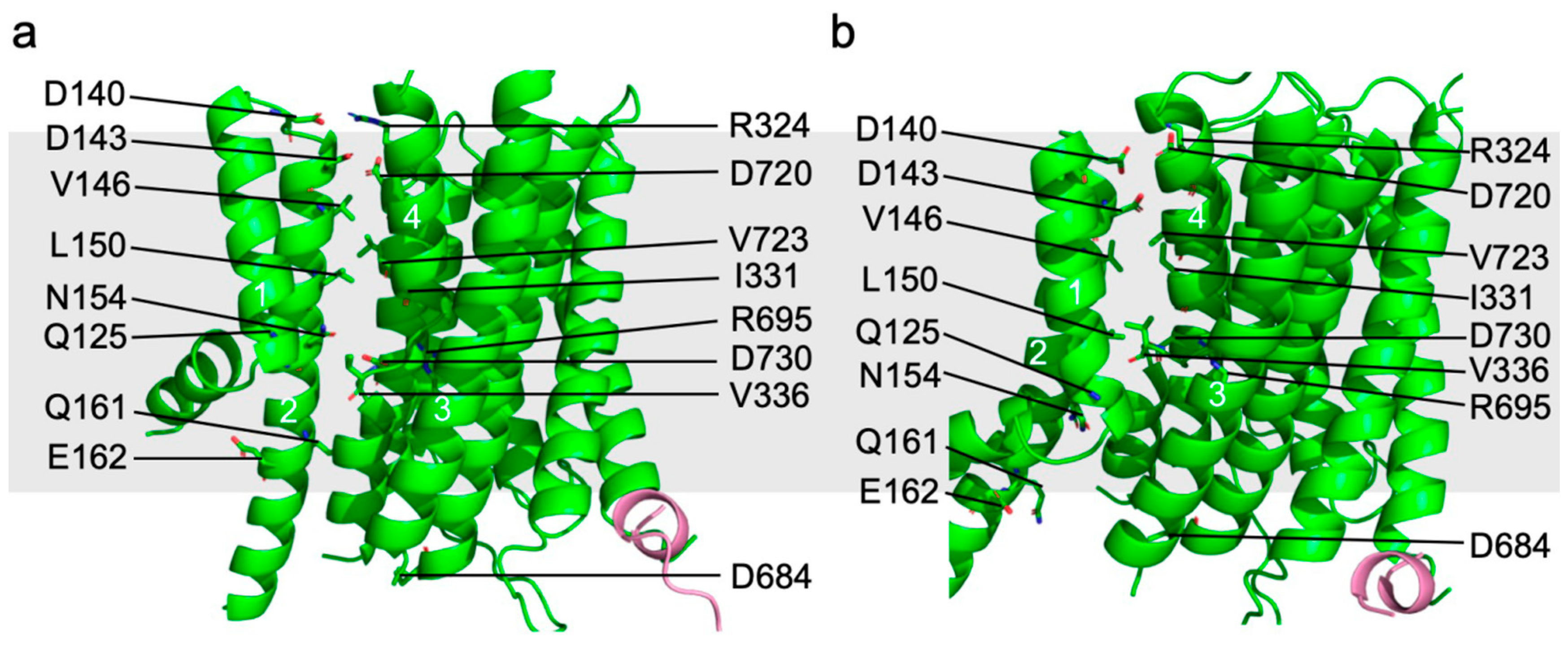{kind=link}
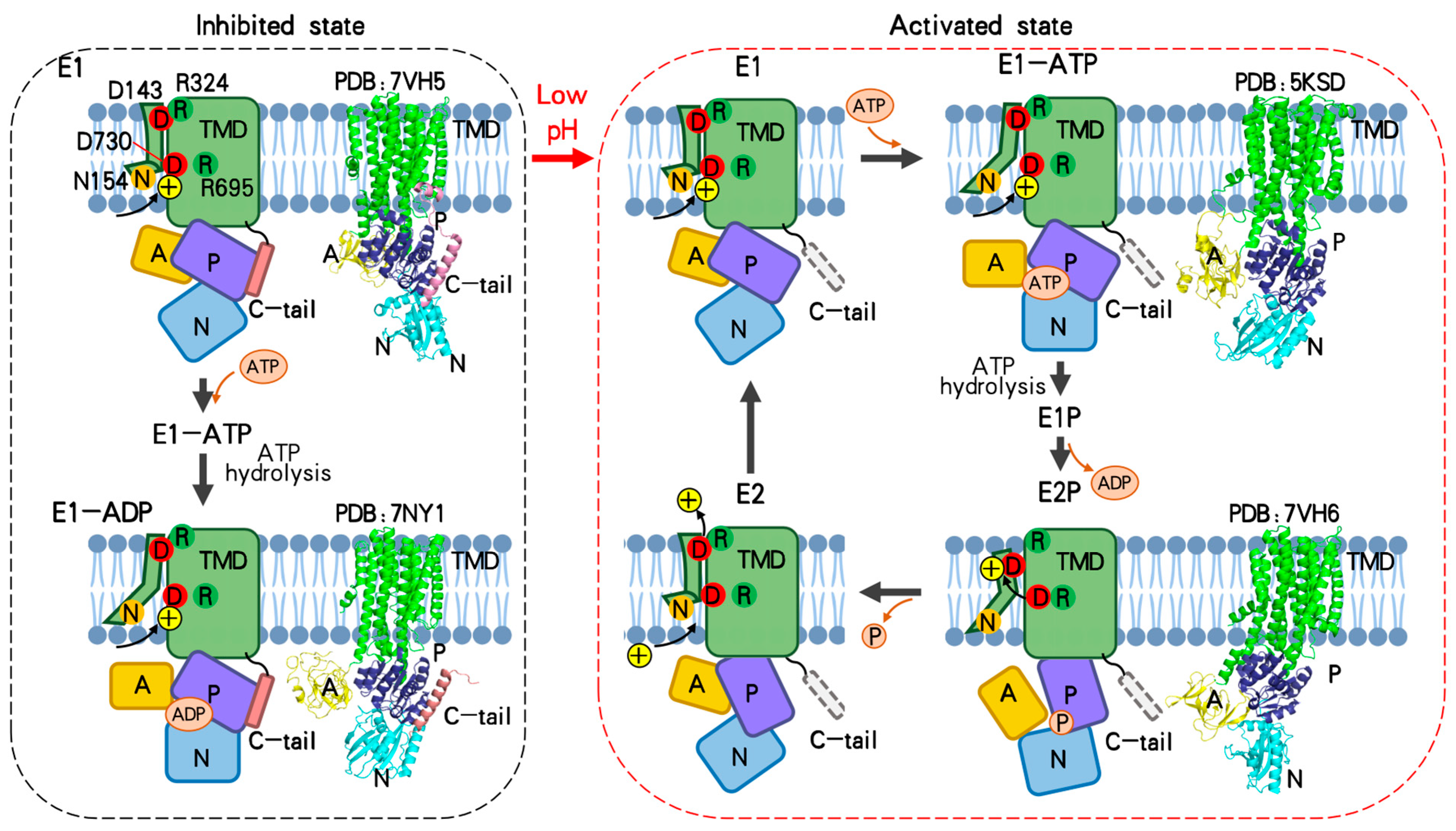{kind=link}
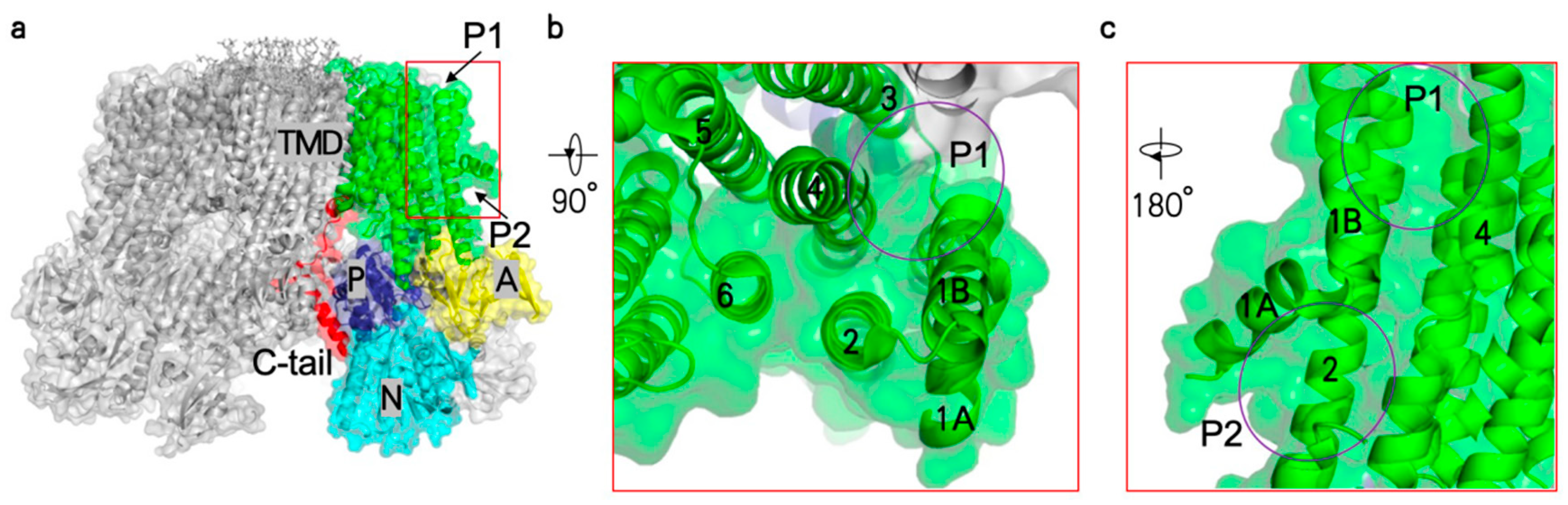{kind=link}
Abstract
1. Introduction
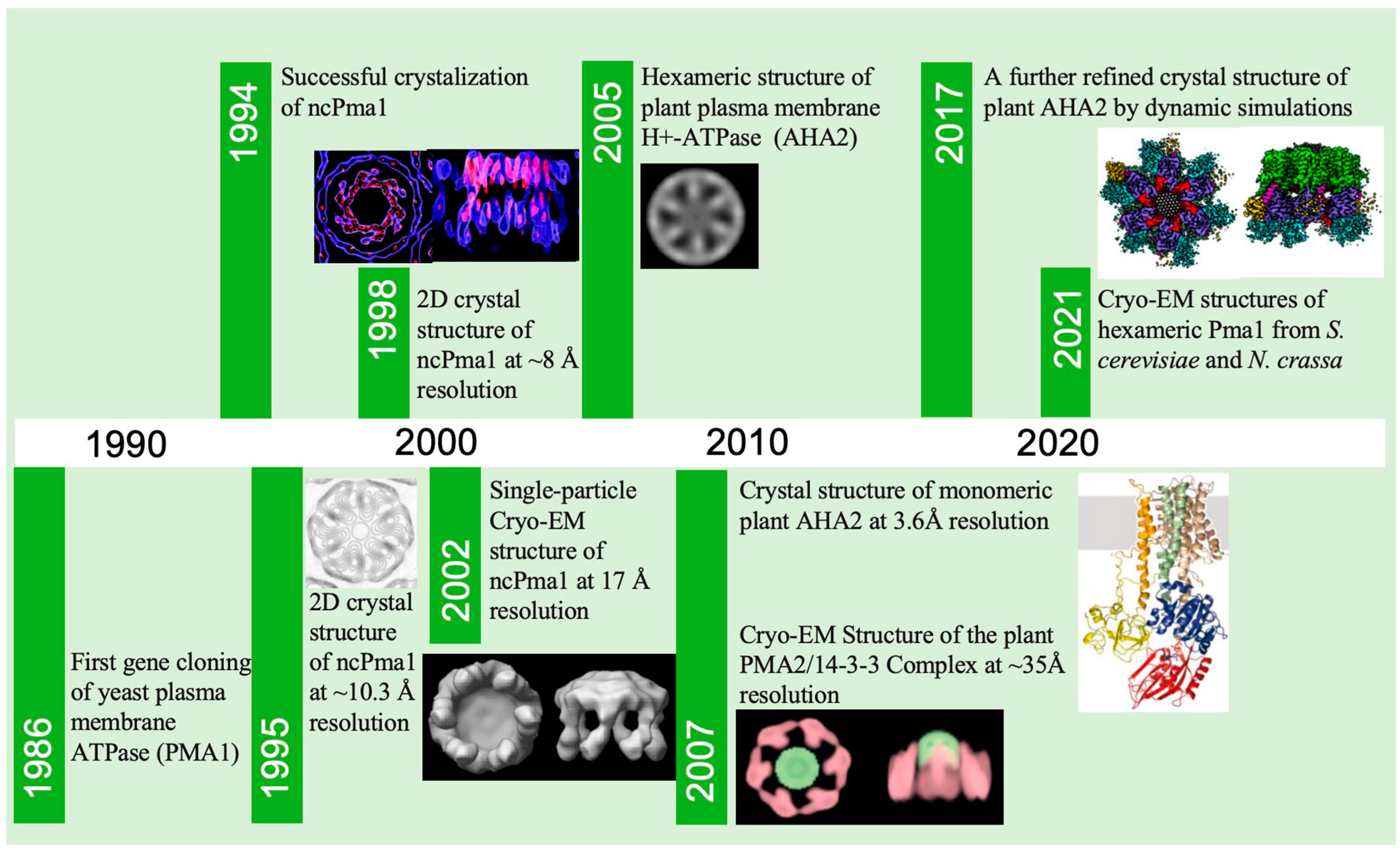
2. A Brief History of Pma1 Structure Determination
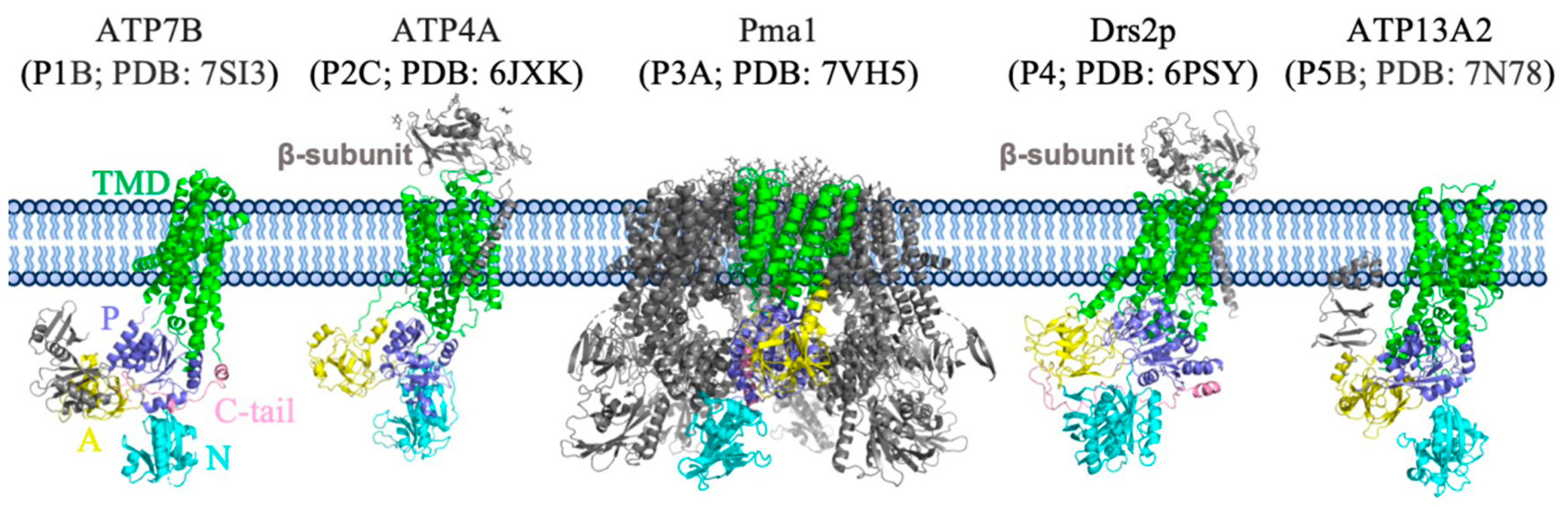
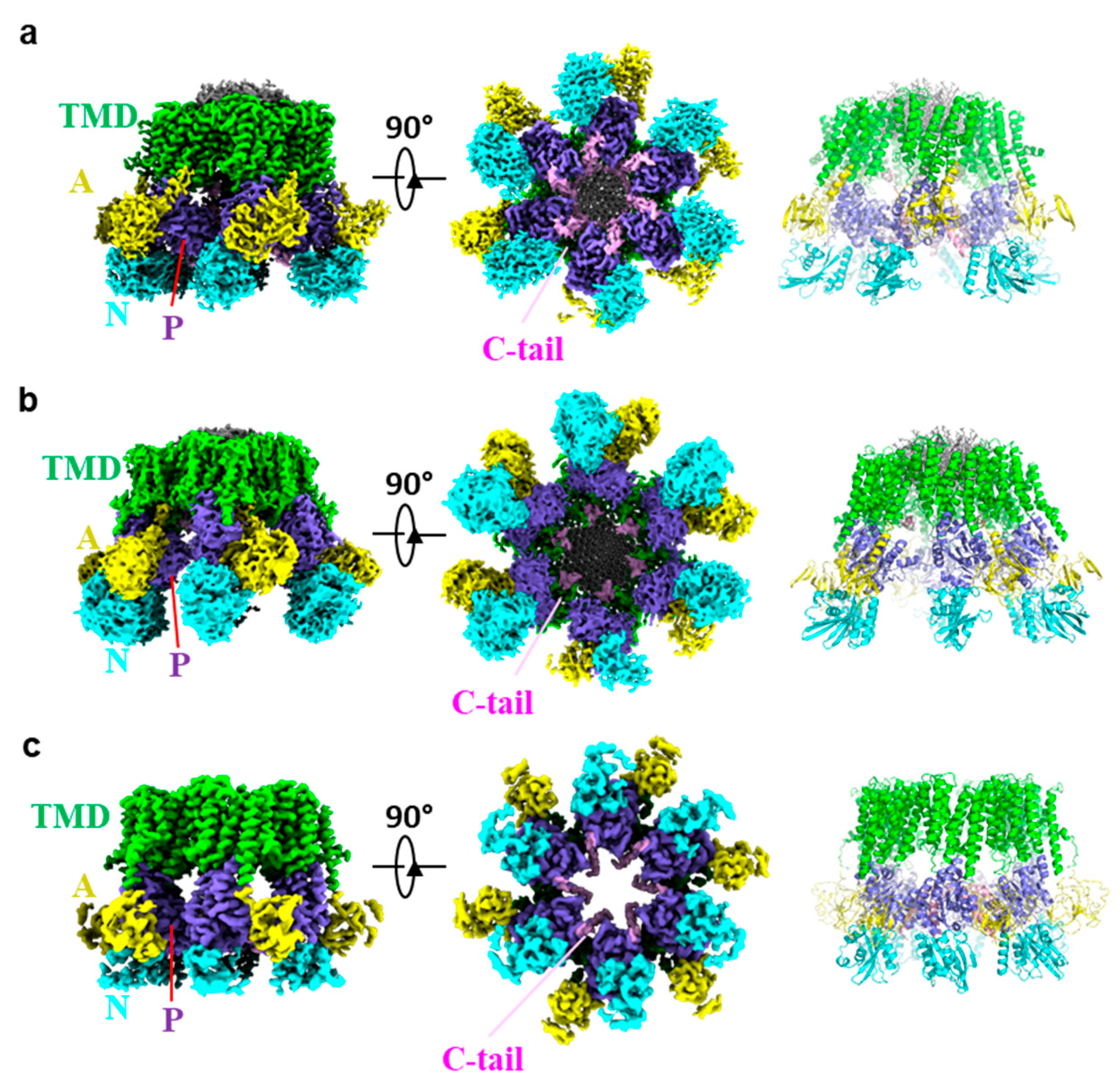
3. Overall Structure of Pma1
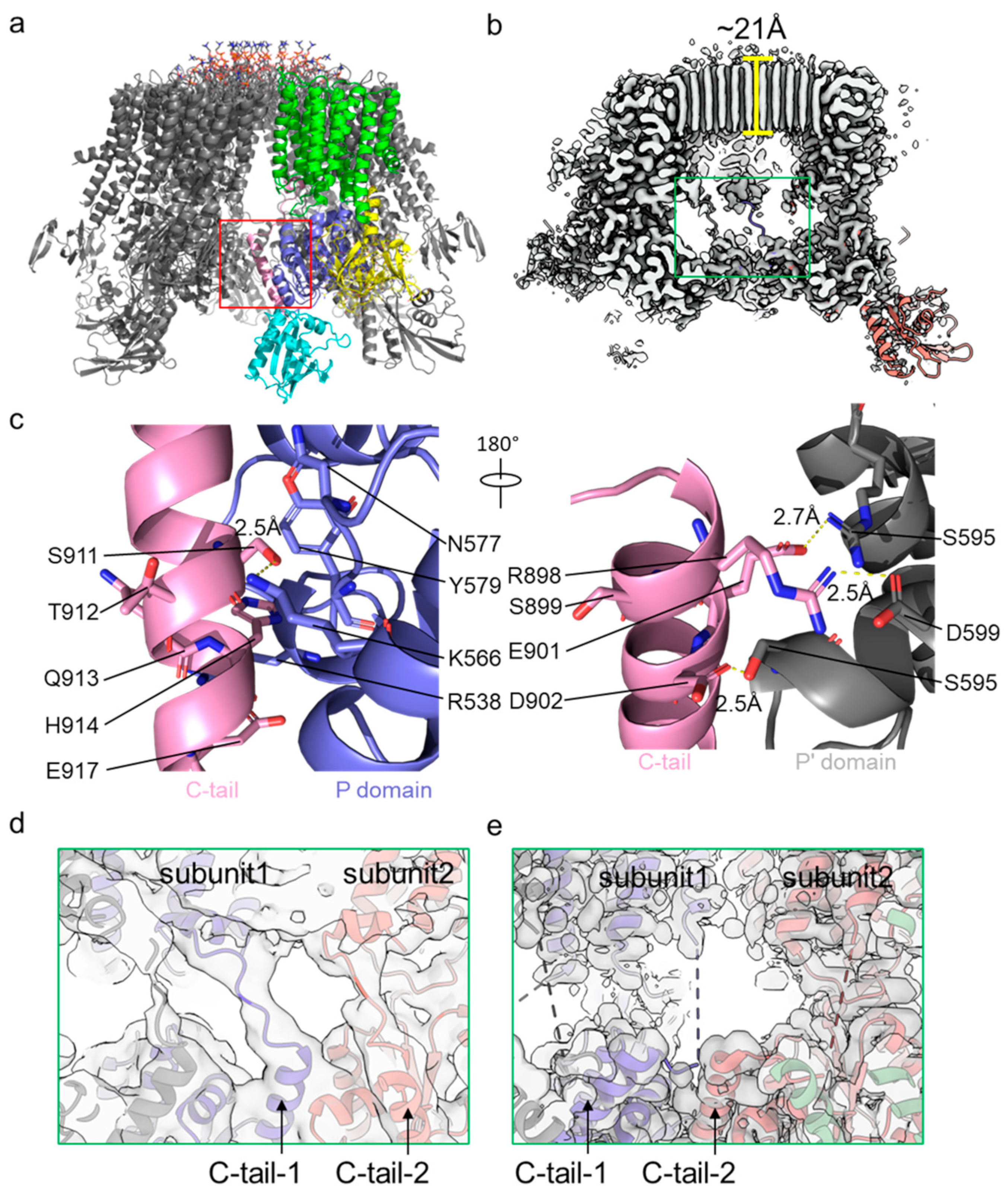
4. Autoinhibition and Activation of Pma1
5. Proton Transport Channel of Pma1
6. Transport Cycle of Pma1
7. Drug Discovery Related to Pma1
8. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [PubMed]
- Almeida, F.; Rodrigues, M.L.; Coelho, C. The Still Underestimated Problem of Fungal Diseases Worldwide. Front. Microbiol. 2019, 10, 214. [Google Scholar] [CrossRef]
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012, 484, 186–194. [Google Scholar] [CrossRef]
- WHO. WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action. 2022. Available online: https://www.who.int/publications/i/item/9789240060241 (accessed on 25 October 2022).
- Young, M.R.; Heit, S.; Bublitz, M. Structure, function and biogenesis of the fungal proton pump Pma1. Biochim. Biophys Acta. Mol. Cell Res. 2024, 1871, 119600. [Google Scholar] [CrossRef] [PubMed]
- Orij, R.; Brul, S.; Smits, G.J. Intracellular pH is a tightly controlled signal in yeast. Biochim. Biophys. Acta 2011, 1810, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Dutra, M.B.; Ambesi, A.; Slayman, C.W. Structure-function relationships in membrane segment 5 of the yeast Pma1 H+-ATPase. J. Biol. Chem. 1998, 273, 17411–17417. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.B.; Allen, K.E.; Slayman, C.W. C-terminal truncations of the Saccharomyces cerevisiae PMA1 H+-ATPase have major impacts on protein conformation, trafficking, quality control, and function. Eukaryot Cell 2014, 13, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Johansson, F.; Sommarin, M.; Larsson, C. Fusicoccin Activates the Plasma Membrane H+-ATPase by a Mechanism Involving the C-Terminal Inhibitory Domain. Plant Cell 1993, 5, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Portillo, F.; Serrano, R. Growth control strength and active site of yeast plasma membrane ATPase studied by site-directed mutagenesis. Eur. J. Biochem. 1989, 186, 501–507. [Google Scholar] [CrossRef]
- Palmgren, M.; Sommarin, M.; Serrano, R.; Larsson, C. Identification of an autoinhibitory domain in the C-terminal region of the plant plasma membrane H+-ATPase. J. Biol. Chem. 1991, 266, 20470–20475. [Google Scholar] [CrossRef]
- Serrano, R.; Kielland-Brandt, M.C.; Fink, G.R. Yeast plasma membrane ATPase is essential for growth and has homology with (Na+ + K+), K+- and Ca2+-ATPases. Nature 1986, 319, 689–693. [Google Scholar] [CrossRef]
- Kuhlbrandt, W. Biology, structure and mechanism of P-type ATPases. Nat. Rev. Mol. Cell Biol. 2004, 5, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, M.G.; Nissen, P. P-type ATPases. Annu. Rev. Biophys. 2011, 40, 243–266. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, M.G.; Axelsen, K.B. Evolution of P-type ATPases. Biochim. Biophys. Acta 1998, 1365, 37–45. [Google Scholar] [CrossRef][Green Version]
- Bublitz, M.; Morth, J.P.; Nissen, P. P-type ATPases at a glance. J. Cell Sci. 2011, 124 Pt 15, 2515–2519. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, M.; Østerberg, J.T.; Nintemann, S.J.; Poulsen, L.R.; López-Marqués, R.L. Evolution and a revised nomenclature of P4 ATPases, a eukaryotic family of lipid flippases. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1135–1151. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhao, Y.; Li, T.; Nie, W.; Yang, X.; Wang, X.; Wu, J.; Liao, J.; Zou, Y. CATP-8/P5A ATPase Regulates ER Processing of the DMA-1 Receptor for Dendritic Branching. Cell Rep. 2020, 32, 108101. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.J.; Sim, S.I.; Ordureau, A.; Wei, L.; Harper, J.W.; Shao, S.; Park, E. The endoplasmic reticulum P5A-ATPase is a transmembrane helix dislocase. Science 2020, 369, 1583. [Google Scholar] [CrossRef] [PubMed]
- Vrijsen, S.; Besora-Casals, L.; van Veen, S.; Zielich, J.; Haute, C.V.D.; Hamouda, N.N.; Fischer, C.; Ghesquière, B.; Tournev, I.; Agostinis, P.; et al. ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc. Natl. Acad. Sci. USA 2020, 117, 31198–31207. [Google Scholar] [CrossRef]
- van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.P.; Gelders, G.; Lambie, E.; et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef]
- Kubler, O.; Gross, H.; Moor, H. Complementary structures of membrane fracture faces obtained by ultrahigh vacuum freeze-fracturing at −196 degrees C and digital image processing. Ultramicroscopy 1978, 3, 161–168. [Google Scholar] [CrossRef]
- Kuhlbrandt, W.; Zeelen, J.; Dietrich, J. Structure, mechanism, and regulation of the Neurospora plasma membrane H+-ATPase. Science 2002, 297, 1692–1696. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Granados, Y.G.; De La Cruz-Torres, V.; Sampedro, J.G. The Oligomeric State of the Plasma Membrane H+-ATPase from Kluyveromyces lactis. Molecules 2019, 24, 958. [Google Scholar] [CrossRef] [PubMed]
- Heit, S.; Geurts, M.M.G.; Murphy, B.J.; Corey, R.A.; Mills, D.J.; Kühlbrandt, W.; Bublitz, M. Structure of the hexameric fungal plasma membrane proton pump in its autoinhibited state. Sci. Adv. 2021, 7, eabj5255. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhao, C.; Chen, D.; Yun, C.; Li, H.; Bai, L. Structure and activation mechanism of the hexameric plasma membrane H+-ATPase. Nat. Commun. 2021, 12, 6439. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, G.A. Large single crystals of the Neurospora crassa plasma membrane H+-ATPase: An approach to the crystallization of integral membrane proteins. Acta Crystallogr. D Biol. Crystallogr. 1994, 50 Pt 4, 643–649. [Google Scholar] [CrossRef]
- Cyrklaff, M.; Auer, M.; Kühlbrandt, W.; Scarborough, G.A. 2-D structure of the Neurospora crassa plasma membrane ATPase as determined by electron cryomicroscopy. EMBO J. 1995, 14, 1854–1857. [Google Scholar] [CrossRef]
- Auer, M.; Scarborough, G.A.; Kuhlbrandt, W. Three-dimensional map of the plasma membrane H+-ATPase in the open conformation. Nature 1998, 392, 840–843. [Google Scholar] [CrossRef] [PubMed]
- Rhee, K.; Scarborough, G.A.; Henderson, R. Domain movements of plasma membrane H+-ATPase: 3D structures of two states by electron cryo-microscopy. EMBO J. 2002, 21, 3582–3589. [Google Scholar] [CrossRef]
- Kanczewska, J.; Marco, S.; Vandermeeren, C.; Maudoux, O.; Rigaud, J.-L.; Boutry, M. Activation of the plant plasma membrane H+-ATPase by phosphorylation and binding of 14-3-3 proteins converts a dimer into a hexamer. Proc. Natl. Acad Sci. USA 2005, 102, 11675–11680. [Google Scholar] [CrossRef]
- Ottmann, C.; Marco, S.; Jaspert, N.; Marcon, C.; Schauer, N.; Weyand, M.; Vandermeeren, C.; Duby, G.; Boutry, M.; Wittinghofer, A.; et al. Structure of a 14-3-3 coordinated hexamer of the plant plasma membrane H+ -ATPase by combining X-ray crystallography and electron cryomicroscopy. Mol. Cell 2007, 25, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.P.; Buch-Pedersen, M.J.; Preben Morth, J.; Palmgren, M.G.; Nissen, P. Crystal structure of the plasma membrane proton pump. Nature 2007, 450, 1111–1114. [Google Scholar] [CrossRef]
- Focht, D.; Croll, T.I.; Pedersen, B.P.; Nissen, P. Improved Model of Proton Pump Crystal Structure Obtained by Interactive Molecular Dynamics Flexible Fitting Expands the Mechanistic Model for Proton Translocation in P-Type ATPases. Front. Physiol. 2017, 8, 202. [Google Scholar] [CrossRef]
- Portillo, F.; de Larrinoa, I.F.; Serrano, R. Deletion analysis of yeast plasma membrane H+-ATPase and identification of a regulatory domain at the carboxyl-terminus. FEBS Lett. 1989, 247, 381–385. [Google Scholar] [CrossRef]
- Palmgren, M.G.; Larsson, C.; Sommarin, M. Proteolytic activation of the plant plasma membrane H+-ATPase by removal of a terminal segment. J. Biol. Chem. 1990, 265, 13423–13426. [Google Scholar] [CrossRef]
- Serrano, R. In vivo glucose activation of the yeast plasma membrane ATPase. FEBS Lett. 1983, 156, 11–14. [Google Scholar] [CrossRef]
- Lecchi, S.; Nelson, C.J.; Allen, K.E.; Swaney, D.L.; Thompson, K.L.; Coon, J.J.; Sussman, M.R.; Slayman, C.W. Tandem phosphorylation of Ser-911 and Thr-912 at the C terminus of yeast plasma membrane H+-ATPase leads to glucose-dependent activation. J. Biol. Chem. 2007, 282, 35471–35481. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Shimazaki, K. Blue light activates the plasma membrane H+-ATPase by phosphorylation of the C-terminus in stomatal guard cells. EMBO J. 1999, 18, 5548–5558. [Google Scholar] [CrossRef]
- Lecchi, S.; Allen, K.E.; Pardo, J.P.; Mason, A.B.; Slayman, C.W. Conformational changes of yeast plasma membrane H+-ATPase during activation by glucose: Role of threonine-912 in the carboxy-terminal tail. Biochemistry 2005, 44, 16624–16632. [Google Scholar] [CrossRef] [PubMed]
- Mazón, M.J.; Eraso, P.; Portillo, F. Specific phosphoantibodies reveal two phosphorylation sites in yeast Pma1 in response to glucose. FEMS Yeast Res. 2015, 15, fov030. [Google Scholar] [CrossRef]
- Eraso, P.; Portillo, F. Molecular mechanism of regulation of yeast plasma membrane H+-ATPase by glucose. Interaction between domains and identification of new regulatory sites. J. Biol. Chem. 1994, 269, 10393–10399. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.A.A.; Tropia, M.J.; Brandao, R.L. New aspects of the glucose activation of the H+-ATPase in the yeast Saccharomyces cerevisiae. Microbiology 2001, 147 Pt 10, 2849–2855. [Google Scholar] [CrossRef] [PubMed]
- Goossens, A.; de la Fuente, N.; Forment, J.; Serrano, R.; Portillo, F. Regulation of yeast H+-ATPase by protein kinases belonging to a family dedicated to activation of plasma membrane transporters. Mol. Cell Biol. 2000, 20, 7654–7661. [Google Scholar] [CrossRef]
- Jahn, T.; Fuglsang, A.T.; Olsson, A.; Bruntrup, I.M.; Collinge, D.B.; Volkmann, D.; Sommarin, M.; Palmgren, M.G.; Larsson, C. The 14-3-3 protein interacts directly with the C-terminal region of the plant plasma membrane H+-ATPase. Plant Cell 1997, 9, 1805–1814. [Google Scholar]
- Oecking, C.; Piotrowski, M.; Hagemeier, J.; Hagemann, K. Topology and target interaction of the fusicoccin-binding 14-3-3 homologs of Commelina communis. Plant J. 2010, 12, 441–453. [Google Scholar] [CrossRef]
- Olivari, C.; Meanti, C.; De Michelis, M.I.; Rasi-Caldogno, F. Fusicoccin binding to its plasma membrane receptor and the activation of the plasma membrane H+-ATPase. IV. Fusicoccin induces the association between the plasma membrane H+-ATPase and the fusicoccin receptor. Plant Physiol. 1998, 116, 529–537. [Google Scholar] [CrossRef][Green Version]
- Fuglsang, A.T.; Visconti, S.; Drumm, K.; Jahn, T.; Stensballe, A.; Mattei, B.; Jensen, O.N.; Aducci, P.; Palmgren, M.G. Binding of 14-3-3 protein to the plasma membrane H+-ATPase AHA2 involves the three C-terminal residues Tyr(946)-Thr-Val and requires phosphorylation of Thr(947). J. Biol. Chem. 1999, 274, 36774–36780. [Google Scholar] [CrossRef]
- Svennelid, F.; Olsson, A.; Piotrowski, M.; Rosenquist, M.; Ottman, C.; Larsson, C.; Oecking, C.; Sommarin, M. Phosphorylation of Thr-948 at the C terminus of the plasma membrane H+-ATPase creates a binding site for the regulatory 14-3-3 protein. Plant Cell 1999, 11, 2379–2391. [Google Scholar] [PubMed]
- Buch-Pedersen, M.J.; Palmgren, M.G. Conserved Asp684 in transmembrane segment M6 of the plant plasma membrane P-type proton pump AHA2 is a molecular determinant of proton translocation. J. Biol. Chem. 2003, 278, 17845–17851. [Google Scholar] [CrossRef]
- Buch-Pedersen, M.J.; Venema, K.; Serrano, R.; Palmgren, M.G. Abolishment of proton pumping and accumulation in the E1P conformational state of a plant plasma membrane H+-ATPase by substitution of a conserved aspartyl residue in transmembrane segment 6. J. Biol. Chem. 2000, 275, 39167–39173. [Google Scholar] [CrossRef]
- Morsomme, P.; Slayman, C.W.; Goffeau, A. Mutagenic study of the structure, function and biogenesis of the yeast plasma membrane H+-ATPase. Biochim. Biophys. Acta 2000, 1469, 133–157. [Google Scholar] [CrossRef] [PubMed]
- Albers, R.W. Biochemical aspects of active transport. Annu. Rev. Biochem. 1967, 36, 727–756. [Google Scholar] [CrossRef]
- Post, R.L.; Hegyvary, C.; Kume, S. Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem. 1972, 247, 6530–6540. [Google Scholar] [CrossRef]
- Toyoshima, C.; Cornelius, F. New crystal structures of PII-type ATPases: Excitement continues. Curr. Opin. Struct. Biol. 2013, 23, 507–514. [Google Scholar] [CrossRef]
- Dyla, M.; Hansen, S.B.; Nissen, P.; Kjaergaard, M. Structural dynamics of P-type ATPase ion pumps. Biochem. Soc. Trans. 2019, 47, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- López-Marqués, R.L.; Gourdon, P.; Pomorski, T.G.; Palmgren, M. The transport mechanism of P4 ATPase lipid flippases. Biochem. J. 2020, 477, 3769–3790. [Google Scholar] [CrossRef] [PubMed]
- Venema, K.; Palmgren, M.G. Metabolic modulation of transport coupling ratio in yeast plasma membrane H+-ATPase. J. Biol. Chem. 1995, 270, 19659–19667. [Google Scholar] [CrossRef]
- Pedersen, J.T.; Kanashova, T.; Dittmar, G.; Palmgren, M. Isolation of native plasma membrane H+-ATPase (Pma1p) in both the active and basal activation states. FEBS Open Bio. 2018, 8, 774–783. [Google Scholar] [CrossRef]
- Kane, P.M. Proton Transport and pH Control in Fungi. Adv. Exp. Med. Biol. 2016, 892, 33–68. [Google Scholar]
- Ferreira, T.; Mason, A.B.; Slayman, C.W. The yeast pma1 proton pump: A model for understanding the biogenesis of plasma membrane proteins. J. Biol. Chem. 2001, 276, 29613–29616. [Google Scholar] [CrossRef]
- Seto-Young, D.; Monk, B.; Mason, A.; Perlin, D.S. Exploring an antifungal target in the plasma membrane H(+)ATPase of fungi. Biochim. Biophys. Acta Biomembr. 1997, 1326, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.C.; Mason, A.B.; Abramochkin, G.; Haber, J.E.; Seto-Young, D.; Perlin, D.S. The Yeast Plasma-Membrane Proton-Pumping Atpase Is a Viable Antifungal Target. 1. Effects of the Cysteine-Modifying Reagent Omeprazole. Biochim. Biophys. Acta Biomembr. 1995, 1239, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.C.; Mason, A.B.; Kardos, T.B.; Perlin, D.S. Targeting the fungal plasma membrane proton pump. Acta Biochim. Pol. 1995, 42, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.C.; Niimi, K.; Lin, S.; Knight, A.; Kardos, T.B.; Cannon, R.D.; Parshot, R.; King, A.; Lun, D.; Harding, D.R.K. Surface-active fungicidal D-peptide inhibitors of the plasma membrane proton pump that block azole resistance. Antimicrob. Agents Chemother. 2005, 49, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Hardej, D.; Santoro, M.; Lau-Cam, C.; Billack, B. Evaluation of the antimicrobial activity of ebselen: Role of the yeast plasma membrane H+-ATPase. J. Biochem. Mol. Toxicol. 2007, 21, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Orie, N.N.; Warren, A.R.; Basaric, J.; Lau-Cam, C.; Piętka-Ottlik, M.; Młochowski, J.; Billack, B. Assessment of the growth and plasma membrane H+-ATPase inhibitory activity of ebselen and structurally related selenium- and sulfur-containing compounds in Candida albicans. J. Biochem. Mol. Toxicol. 2017, 31, e21892. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.A.; Marzouk, M.S.; Moharram, F.A.; El-Sayed, M.M.; Baiuomy, A.R. Phytochemical constituents and hepatoprotective activity of Viburnum tinus. Phytochemistry 2005, 66, 2780–2786. [Google Scholar] [CrossRef] [PubMed]
- Funatogawa, K.; Hayashi, S.; Shimomura, H.; Yoshida, T.; Hatano, T.; Ito, H.; Hirai, Y. Antibacterial activity of hydrolyzable tannins derived from medicinal plants against. Microbiol. Immunol. 2004, 48, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Kongstad, K.T.; Wubshet, S.G.; Kjellerup, L.; Winther, A.M.L.; Staerk, D. Fungal plasma membrane H⁺-ATPase inhibitory activity of o-hydroxybenzylated flavanones and chalcones from Uvaria chamae P. Fitoterapia 2015, 105, 102–106. [Google Scholar] [CrossRef]
- Goldgof, G.M.; Durrant, J.D.; Ottilie, S.; Vigil, E.; Allen, K.E.; Gunawan, F.; Kostylev, M.; Henderson, K.A.; Yang, J.; Schenken, J.; et al. Comparative chemical genomics reveal that the spiroindolone antimalarial KAE609 (Cipargamin) is a P-type ATPase inhibitor. Sci. Rep. 2016, 6, 27806. [Google Scholar] [CrossRef]
- White, N.J.; Pukrittayakamee, S.; Phyo, A.P.; Rueangweerayut, R.; Nosten, F.; Jittamala, P.; Jeeyapant, A.; Jain, J.P.; Lefèvre, G.; Li, R.; et al. Spiroindolone KAE609 for Falciparum and Vivax Malaria. N. Engl. J. Med. 2014, 371, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Ottilie, S.; Goldgof, G.M.; Cheung, A.L.; Walker, J.L.; Vigil, E.; Allen, K.E.; Antonova-Koch, Y.; Slayman, C.W.; Suzuki, Y.; Durrant, J.D. Two inhibitors of yeast plasma membrane ATPase 1 (Pma1p): Toward the development of novel antifungal therapies. J. Cheminformatics 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Kjellerup, L.; Gordon, S.; Cohrt, K.O.; Brown, W.D.; Fuglsang, A.T.; Winther, A.-M.L. Identification of Antifungal H+-ATPase Inhibitors with Effect on Plasma Membrane Potential. Antimicrob. Agents Chemother. 2017, 61, 1128. [Google Scholar] [CrossRef] [PubMed]
- Bublitz, M.; Kjellerup, L.; Cohrt, K.O.; Gordon, S.; Mortensen, A.L.; Clausen, J.D.; Pallin, T.D.; Hansen, J.B.; Fuglsang, A.T.; Dalby-Brown, W.; et al. Tetrahydrocarbazoles are a novel class of potent P-type ATPase inhibitors with antifungal activity. PLoS ONE 2018, 13, e0188620. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Shinoda, T.; Cornelius, F.; Toyoshima, C. Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc. Natl. Acad Sci. USA 2009, 106, 13742–13747. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Irie, K.; Nakanishi, H.; Suzuki, H.; Fujiyoshi, Y. Crystal structures of the gastric proton pump. Nature 2018, 556, 214–218. [Google Scholar] [CrossRef]
- Qiu, D.; Pei, J.V.; Rosling, J.E.O.; Thathy, V.; Li, D.; Xue, Y.; Tanner, J.D.; Penington, J.S.; Aw, Y.T.V.; Aw, J.Y.H.; et al. A G358S mutation in the Plasmodium falciparum Na+ pump PfATP4 confers clinically-relevant resistance to cipargamin. Nat. Commun. 2022, 13, 5746. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, C.-R.; You, Z.-L.; Bai, L. Fungal Plasma Membrane H+-ATPase: Structure, Mechanism, and Drug Discovery. J. Fungi 2024, 10, 273. https://doi.org/10.3390/jof10040273
Zhao C-R, You Z-L, Bai L. Fungal Plasma Membrane H+-ATPase: Structure, Mechanism, and Drug Discovery. Journal of Fungi. 2024; 10(4):273. https://doi.org/10.3390/jof10040273
Chicago/Turabian StyleZhao, Chao-Ran, Zi-Long You, and Lin Bai. 2024. "Fungal Plasma Membrane H+-ATPase: Structure, Mechanism, and Drug Discovery" Journal of Fungi 10, no. 4: 273. https://doi.org/10.3390/jof10040273
APA StyleZhao, C.-R., You, Z.-L., & Bai, L. (2024). Fungal Plasma Membrane H+-ATPase: Structure, Mechanism, and Drug Discovery. Journal of Fungi, 10(4), 273. https://doi.org/10.3390/jof10040273