Abstract
Dutch elm disease (DED), caused by Ophiostoma novo-ulmi (Onu), is a destructive disease of American elm (Ulmus americana L.). The molecular mechanisms of resistance and susceptibility against DED in American elm are still largely uncharacterized. In the present study, we performed a de novo transcriptome (RNA-sequencing; RNA-Seq) assembly of U. americana and compared the gene expression in a resistant genotype, ’Valley Forge’, and a susceptible (S) elm genotype at 0 and 96 h post-inoculation of Onu. A total of 85,863 non-redundant unigenes were identified. Compared to the previously characterized U. minor transcriptome, U. americana has 35,290 similar and 55,499 unique genes. The transcriptomic variations between ‘Valley Forge’ and ‘S’ were found primarily in the photosynthesis and primary metabolism, which were highly upregulated in the susceptible genotype irrespective of the Onu inoculation. The resistance to DED was associated with the activation of RPM1-mediated effector-triggered immunity that was demonstrated by the upregulation of genes involved in the phenylpropanoids biosynthesis and PR genes. The most significantly enriched gene ontology (GO) terms in response to Onu were response to stimulus (GO:0006950), response to stress (GO:0050896), and secondary metabolic process (GO:0008152) in both genotypes. However, only in the resistant genotype, the defense response (GO:0006952) was among the topmost significantly enriched GO terms. Our findings revealed the molecular regulations of DED resistance and susceptibility and provide a platform for marker-assisted breeding of resistant American elm genotypes.
1. Introduction
Ophiostoma species (i.e., Ophiostoma novo-ulmi and Ophiostoma ulmi) are the fungal pathogens causing Dutch elm disease (DED), which is a lethal vascular wilt disease of elms worldwide including American elm (Ulmus americana) [1,2]. O. ulmi caused the original DED epidemic in Europe and North America in the mid-1900s. During the second half of the 20th century, the more aggressive O. novo-ulmi (Onu) largely replaced O. ulmi and is responsible for the ongoing DED pandemic [3]. In North America, overland spread of DED from infected to healthy elms is facilitated by the native elm bark beetle (Hylurgopinus rufipes) and its European counterpart (Scolytus multistriatus), and the symptoms first appear on upper crown branches as wilting and yellowing leaves [4,5,6]. Generally, soon after one branch becomes symptomatic, adjacent branches also show symptoms, which is followed by significant crown dieback. When the bark is peeled off infected branches, longitudinal, brown-colored streaks in the outer rings of the sapwood are visible, and it is the most distinctive symptom of DED observed in the field. These fungi spread within stems and roots of living elms both by passive transport of spores and by the mycelial growth of colonies initiated by spores that germinate in the xylem. Dutch elm disease fungi can also infect healthy elms adjacent to a diseased elm through root grafts [7,8].
Ophiostoma species causing DED grow and reproduce within elms and are generally considered hemibiotrophic fungal pathogens [9]. However, these pathogens can be biotrophic sometimes, feeding on living tissues of the elm tree, and at other times, necrotrophic, getting nutrition from dead elm tissue [9]. Long-term control and management of DED primarily rely on the identification and development of resistant elm genotypes [10,11]. Since the disease appeared, several breeding programs have employed Asian elms such as U. pumila or U. wallichiana as the source of resistance, as they have been shown to be less susceptible to DED. The genetic heritability of resistance has been shown by crossing native and Asian elms [12,13] and crossing susceptible and resistant Iberian U. minor genotypes [14]. The transcriptomic changes of Iberian U. minor genotypes with contrasting resistance to DED showed that the phenylpropanoids biosynthesis pathway played a central role in the tolerance mechanism against DED [15]. Similarly, the transcriptomic analysis of U. americana calli inoculated with Onu showed that the elm transcripts encoding the enzymes involved in the phenylpropanoids metabolism and pathogenesis-related (PR) proteins were enhanced and hence signify the role of phenolics and PR proteins in the DED resistance mechanism [16]. The induction of defense-related genes and plant hormone jasmonic acid (JA) during the necrotrophic phase of DED has also been characterized as a resistance mechanism by comparing resistant and susceptible U. americana genotypes. Furthermore, at the early stage of DED, the application of salicylic acid (SA) enhanced elm resistance in the field and indicated that the coordinated action of SA and JA might be crucial in DED, as reported for resistance to diseases caused by other hemibiotrophic pathogens [9,17,18]. Despite these efforts toward dissecting the defense mechanisms against DED, the molecular responses of resistant and susceptible American elm genotypes to DED at the whole-transcriptmic level are still unknown.
Genome-wide transcriptome analysis has widely been used in several plant pathosystems as an efficient method for elucidating the molecular and genetic mechanisms involved in disease resistance [19,20]. Advances in understanding plant–microbe interactions have been enabled by the availability of plant genome sequences and the development of associated bioinformatics tools and resources [15,19,20]. Comparative transcriptional analysis, using RNA-sequencing (RNA-Seq), is a prominent approach for identifying genes that are differentially expressed between two contrasting treatments/genotypes. However, a comprehensive study of the genes involved in molecular mechanisms of resistance in U. americana has not been performed thus far. The existing variation in U. americana genotypes for resistance and susceptibility traits can be explored by RNA sequencing. This knowledge is indispensable for developing resistant genotypes through breeding to preserve the threatened American elm.
The cultivar ‘Valley Forge’ is considered relatively more resistant to DED than other cultivars such as ‘Princeton’, ‘Delaware’, and ‘New Harmony’ [21]. The induction kinetics of genes after infection with O. novo-ulmi (Onu; MH75-4O) were examined in resistant (Valley Forge) and susceptible genotypes, and indicated that the differences among genotypes are due to timing and level of the gene expression rather than the presence or absence of disease-responsive genes [9]. At 96 h post-inoculation (hpi), genes encoding pathogenesis-related proteins showed ≥2-fold higher expression in ‘Valley Forge’ than the susceptible clone along with abundant growth of conidia and hyphae in the susceptible clone [9]. In the present study, we used Illumina HiSeq 2500 paired-end sequencing followed by a de novo transcriptomes assembly to reveal elm transcriptomic changes in response to Onu. Two contrasting genotypes, including the resistant Valley Forge (V) and a susceptible clone (S), were exposed to Onu, and samples were collected at 0 hpi and 96 hpi. The objective of the work reported herein was to identify genes upregulated and downregulated in response to Onu infection and provide a platform for characterizing them further for developing resistance in American elm against DED. Fungal transcriptomes were also recovered and analyzed; results will be presented in a separate contribution.
2. Materials and Methods
2.1. Experimental Materials, Fungal Isolate, and Inoculation Conditions
All plant material, inoculum (O. novo-ulmi; MH75-4O), inoculation conditions, and tissue collection were described previously [9]. Briefly, four-year-old American elm ‘Valley Forge’ was used as a line with high degree of resistance to DED. The elm genotype highly susceptible to DED was selected from in vitro elm germplasm collection at the Gosling Research Institute for Plant Preservation (GRIPP), University of Guelph, Ontario, Canada. The clones were kept under 16 h light at 24 °C and 8 h darkness at 20 °C, and light intensity was set at 110 μmolm−2 s−1 (LI-250 A, LI-COR; Biosciences, Lincoln, NE, USA) under greenhouse conditions and later moved outside for the experiment. Both clones were inoculated with O. novo-ulmi (Onu) along the main stem, and stem segments 2 cm2 around each inoculation point were collected at 0 and 96 hpi for transcriptome profiling. To prepare the Onu inoculum, yeast spore suspensions were prepared as described elsewhere [9], and the spore density was adjusted to 107 spores/mL based on initial densities determined with a hemocytometer. The highly virulent and sequenced strain O. novo-ulmi ssp novo-ulmi H327 could not be used in this study because of the quarantine regulations in Ontario [22]. For RNA sequencing, three biological replicates were collected for resistant and susceptible genotypes at 0 and 96 hpi, resulting in 12 libraries. The inoculated plants were assessed for the disease incidence and severity at 60 days post-inoculation (dpi).
2.2. RNA Extraction and Sequencing
The detailed protocols for RNA extraction, cDNA synthesis, and quantitative real-time PCR were mentioned in the previous study [9]. Briefly, the CTAB protocol was used for RNA extraction, which is followed by purification with a RNeasy Mini Kit (Qiagen). The cDNA was synthesized using a Superscript VILO cDNA Synthesis kit (Invitrogen, Burlington, ON, Canada). The cDNA libraries were prepared and sequenced using paired-end reads on the Illumina HiSeq 2500 platform.
2.3. Ulmus Americana Transcriptome Analysis
The filtered reads were obtained after removing adapter sequences. The assembled U. americana putative scaffolds were filtered by size (≥300 bp) and annotated using the non-redundant protein database downloaded from NCBI (ftp://ftp.ncbi.nlm.nih.gov/blast/db/, accessed on 1 March 2018) in BLASTx v. 2.8.0 software. Then, filtered reads were mapped to the assembled Ulmus americana transcriptome for quantification purposes using BWA v. 0.7.4-r385 [23]. Afterwards, read counts were assessed in R software [24] for differential gene expression analysis using the package edgeR. Within edgeR, the function plotMDS was implemented to assess for variability among biological replicates in a bi-dimensional scaling plot in which distances correspond to leading log-fold changes between each pair of RNAseq samples. The glmTreat function was employed for a rigorous differential gene expression analysis based on the variability observed among biological replicates. Significantly differentially expressed genes (DEGs) were identified with log2FC > 2 and adjusted p-value < 0.05 as the upregulated DEGs, and log2FC < −2 and adjusted p-value 0.05 as downregulated DEGs. Volcano plots of DEGs were generated using MetaboAnalyst (http://www.metaboanalyst.ca, accessed on 26 November 2021).
2.4. Comparative Analysis between U. americana and U. minor Transcriptomes
The FASTA file corresponding to the U. minor transcriptome, accession number SRR1687227 was downloaded from NCBI Short Read Archive. Thereafter, the FASTA files corresponding to U. minor and U. americana transcriptomes were converted to two local databases using the makeblastdb function of BLAST v.2.8.0 software. The blastn application was used to compare the FASTA files corresponding to each Ulmus transcriptome against their local database counterpart. Shared genes between U. minor and U. americana were filtered from the original FASTA files, and the remaining unique genes were compared against the non-redundant protein database downloaded from NCBI (ftp://ftp.ncbi.nlm.nih.gov/blast/db/, accessed on 1 March 2018) using BLASTx v.2.8.0. The unique genes having E-values ≤ 1 × 10−6 and similarity ≥ 70% were further studied to identify the top 10 species with the highest number of hits produced by BLASTx. Unique contaminant genes unrelated to plant species and potential artifacts were identified and removed using the package taxonomizr in R.
A transcriptome comparison was performed using the FASTA files of U. minor and U. americana transcriptomes in order to determine and annotate unique genes for each species. To this end, the FASTA files for each transcriptome were converted to two local databases (DB), which were used as templates for BLASTX software (U. minor FASTA file vs. U. americana local DB, and vice versa).
2.5. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways Enrichment Analysis
A BLASTx-based approach was run from the command prompt to extract the GenInfo Identifier (GI) numbers from the best BLASTx hits with the Ulmus americana transcriptome, having E-value ≤ 1 × 10−6and similarity ≥ 70%. The Arabidopsis genes corresponding to American elm genes were obtained as the best hit of Arabidopsis. The Significant Differentially Expressed Genes (DEGs) were annotated with gene ontology (GO) by AgriGO v2.0 [25] using Arabidopsis TAIR10 genomes. The KEGG pathway enrichment analysis of DEGs was conducted using the web-based DAVID v6.8 tool (https://david-d.ncifcrf.gov/, accessed on 11 December 2021). The hypergeometric statistical model and p values were adjusted by the Benjamini and Hochberg method. The GO terms and KEGG pathways with FDR < 0.05 were regarded as significantly enriched. The GO enrichment networks were analyzed by BiNGO in Cytoscape [26].
2.6. Validation of Candidate Genes Expression by Quantitative Real-Time PCR
Total RNA isolated as described previously [9] was treated with DNase I, and first-strand cDNA was synthesized from 2.5 μg of DNase treated RNA using the SuperScript® VILO™ cDNA Synthesis Kit (Invitrogen, Burlington, ON, Canada). A 2.5 µL aliquot representing a 20-fold dilution of the cDNA was used as a template for qRT-PCR using gene-specific primers (Table S1). All treatment samples were assayed in duplicate in a 10 µL reaction containing Bio-Rad SYBR Green and 5 pmol of each primer on a CFX Connect Real-Time PCR Detection System (Bio-Rad, Mississauga, ON, Canada). The cycle threshold (Ct) value of each candidate gene was compared to the corresponding reference gene, Splicing factor 3B-F, and 0 h post-inoculation (hpi) in susceptible (S) genotype as treatment control and relative expression values were calculated according to the 2−ΔΔCt method [27]. Results were statistically analyzed using the CFX manager software (Bio-Rad). The RNA used for qRT-PCR investigations was extracted from the same tissues used for transcriptome analysis.
3. Results
3.1. Development of Dutch Elm Disease (DED) Symptoms
In response to Onu, the American elm cultivar ‘Valley Forge’ was found almost symptomless (Figure 1A), whereas the susceptible genotype showed severe DED symptoms at 60 dpi (Figure 1B). Internal DED symptoms appeared as brown and dark brown streaks right under the bark of the susceptible genotype.

Figure 1.
Development of Dutch elm disease symptom in American elm genotypes at 60 days post-inoculation with Ophiostoma novo-ulmi. (A) Resistant ‘Valley Forge’; and (B) susceptible clones.
3.2. Analysis of the American Elm Transcriptomes
A transcriptomic analysis using the RNA-Seq technology was performed to characterize the molecular defense responses in American elm saplings against O. novo-ulmi (Onu). For maximal read depth, all RNA samples were depleted of the highly abundant rRNA, which strongly interferes with the sequencing reactions in Illumina platforms [28,29]. The Illumina HiSeq 2500 platform was used to generate paired-end reads for 12 cDNA libraries representing three biological replicates of two American elm genotypes; DED-resistant ‘Valley Forge’ and DED-susceptible (herein referred to as Susceptible, S) at 0 and 96 h post-inoculation with O. novo-ulmi strain MH75-4 O. For libraries of both American elm genotypes at each post-inoculation time point, the average number of raw sequence reads ranged from 66.4 to 75.8 million (Table 1). Among the total mapped reads, less than 0.22% aligned to more than one location in the reference genome, while the remaining resulted in single mappings. The assembly revealed that approximately 70% of the unmapped reads, representing an average of 50.8 million raw reads, were de novo assembled into U. americana transcriptome.

Table 1.
Overview of raw, mapped, and unmapped reads in Ulmus americana.
3.3. Comparative Transcriptome Analysis
BLASTX analyses aligned approximately 53.5% of the U. minor unigenes to U. americana local DB (n = 85,863; E-value ≤ 1 × 10−6; similarity ≥ 70%) and 41.1% of U. americana unigenes to U. minor local DB (n = 73,917; E-value ≤ 1 × 10−6; similarity ≥ 70%).
Although the similarity cut-off was set at 70%, the majority (91.3%) of common genes between U. minor and U. americana transcriptomes showed similarities higher than 90%. In addition, using the BLASTX software, the unique genes of each transcriptome were compared to the non-redundant protein database downloaded from NCBI, annotating approximately 61% genes from U. minor and 42% genes from U. americana as plant-related transcripts (Figure 2A). The number of hits per plant species in the non-redundant protein database for each Ulmus sp. transcriptome was generated using the BLASTX-based approach. As shown in (Figure 2B,C) for each Ulmus transcriptome, nearly 70% of the best hits produced by BLASTX were concentrated in 10 species. The remaining 30% resulted from 118 and 233 species for U. minor and U. americana transcriptomes, respectively. The top 5 best-hit species for U. minor and U. americana transcriptomes were all classified as Angiosperms within the order Rosales. This suggests that most of the unique genes for U. minor and U. americana, regardless of having similarities lower than 70%, could still have related functions, as these were similar to other transcripts from the same plant species.
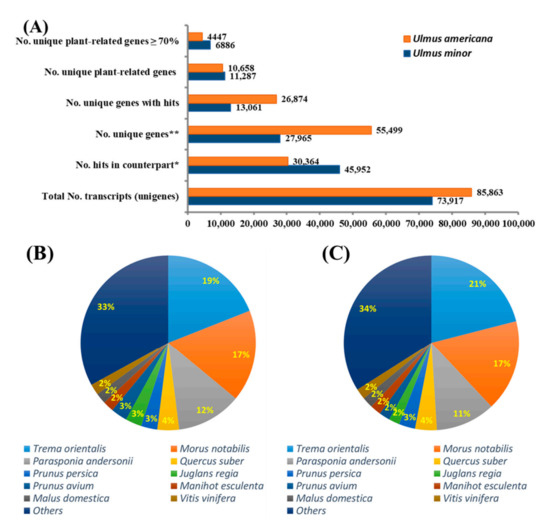
Figure 2.
(A) Comparative transcriptome analysis between Ulmus minor and U. americana. Blastn v.8.0 was used to compare the transcriptome of both Ulmus species. All common genes between the Ulmus spp. showed a percentage of identity higher than 70%. Unique genes that returned no hits between the Ulmus transcriptomes were compared against the non-redundant protein database using a BLASTX approach. (B) The number of hits resulting from a BLASTX analysis of unique plant-related genes from U. minor transcriptome against the non-redundant protein database. (C) The number of hits resulting from a BLASTX analysis of unique plant-related genes from U. americana. * Refers to common genes between U. americana and U. minor. ** Refers to unique genes for each of the two Ulmus sp. transcriptomes that are not similar between them.
3.4. Differential Gene Expression Analysis
A total of 5329 genes were differentially expressed between Valley Forge (V) and Susceptible (S) American elm genotypes in this study. Significant DEGs (upregulated; log2FC > 2 and adjusted p-value < 0.05, and downregulated; log2FC < −2 and adjusted p-value < 0.05) are presented in volcano plots (Figure 3A–D). A total of 165, 130, 64, and 73 significant DEGs were identified from the comparisons between V0h and S0h, V96h and V0h, S96h and S0h, as well as V96h and S96h, respectively (Figure 3).
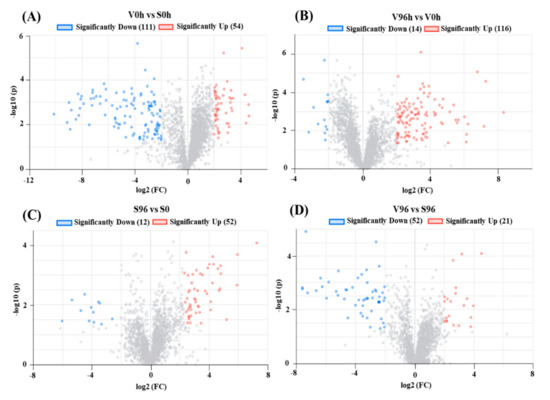
Figure 3.
Volcano plots of differentially expressed genes (DEGs). The X-axis shows the log2 FC in gene expression, (A) between non-inoculated ‘Valley Forge’ (V) and ‘Susceptible’ (S) American elm; (B) between 0 and 96 hpi in ‘Valley Forge’ (V) American elm; (C) between 0 and 96 hpi in ‘Susceptible’ (S) American elm; and (D) between Valley Forge’ (V) and ‘Susceptible’ American elm at 96 hpi. The Y-axis shows the statistical significance of the differences. Splashes represent different genes. FC: Fold change; hpi: h post-inoculation; −log10(p): the corrected p-value.
3.5. GO Enrichment Analysis
Gene ontology (GO) enrichment was performed using AgriGO v2.0 [25] to generate an overview of the functional classifications of the DEGs associated with U. americana responses to Onu. The DEGs between the ‘Valley Forge’ and ‘Susceptible’ American elm genotypes yielded the most significant GO terms regardless of the pathogen inoculations. These GOs were involved in the metabolic process, cellular process, and primary metabolic process (Table 2). On the other hand, in responses to Onu, the most significantly enriched GO terms were the response to stimulus (GO:0050896), response to stress (GO:0006950), and secondary metabolic process (GO:0019748) in both resistant and susceptible genotypes. Interestingly, the defense response (GO:0006952) was the topmost significantly enriched GO term in the resistant genotype exposed to Onu.
Table 2.
Results of gene ontology (GO) enrichment analysis of differentially expressed genes.
3.6. Pathway Enrichment Analysis
Significantly DEGs were analyzed for pathway enrichment using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The photosynthesis (ko00195), carbon metabolism (ko01200), and citrate cycle (ko00020) were among the highly enriched pathway between the resistant and susceptible elm genotypes irrespective of Onu inoculation. The secondary metabolism, mainly the phenylpropanoids biosynthesis (ko00940), was highly annotated in response to Onu infection (Table 3).
Table 3.
Results of Kyoto Encyclopedia of Genes and Genomes pathway (KEGG) enrichment analysis.
3.7. GO Network Analysis
DEGs in resistant (V96h vs. V0h; Figure 4A) and susceptible (S96h vs. S0h; Figure 4B) American elm genotypes in response to Onu were associated with three GO terms: response to stimulus (chemical, abiotic, and biotic), metabolic process (secondary and primary metabolism), and responses to stress (defense response, immune response). The expression pattern of the immune responsive genes is presented as a heatmap (Figure 4C). It showed that American elm genotypic variations in responses to Onu were attributed mainly to genes encoding RPM1 disease resistance protein, phospholipase D beta 1, pathogenesis-related PR4, and thaumatin-like (PR5) proteins. These genes, except the phospholipase D beta 1, were more upregulated in the resistant than the susceptible genotype at 96 hpi (Figure 4C).
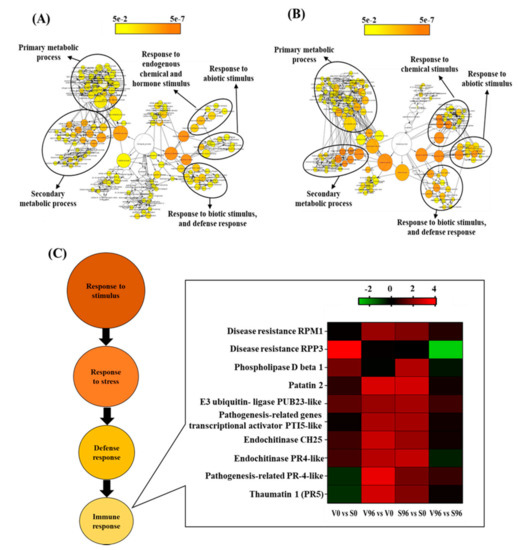
Figure 4.
GO network visualization for the significant DEGs in (A) resistant (V96h vs. V0h) and (B) susceptible (S96h vs. S0h) interactions between Ulmus americana and Ophiostoma novo-ulmi. (C) Expression pattern of the immune-responsive genes in American elm.
3.8. Expression Patterns of the DEGs Involved in Photosynthesis
KEGG pathway enrichment analysis showed that photosynthesis was the most enriched pathway between the resistant and susceptible American elm genotypes at 0 hpi (Figure 5). Among the genes involved in the photosynthesis pathway, 69 genes were significantly differentially expressed, and all of them were downregulated in the resistant compared to the susceptible genotype at 0 hpi with Onu. Both photosystem I and photosystem II genes were significantly downregulated, and the most downregulated gene was photosystem II D1 (log2FC, −7.24). No significant photosynthesis-related DEGs were found in resistant or susceptible genotypes at 96 hpi.
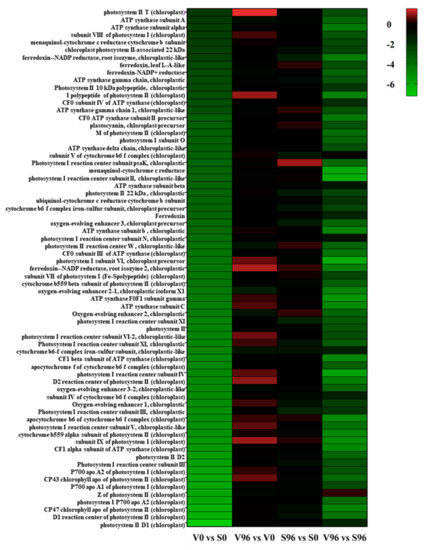
Figure 5.
Heatmap of the significantly differentially expressed genes (DEGs) involved in photosynthesis in resistant (V) and susceptible (S) American elm genotypes at 0 and 96 h post-inoculation (hpi) with Ophiostoma novo-ulmi.
3.9. Expression Profile of the DEGs Involved in the Biosynthesis of Phenylpropanoids
The biosynthesis of secondary metabolites, specifically phenylpropanoids biosynthesis (Figure 6A), was the most Onu-responsive pathway enriched in both resistant and susceptible genotypes. Among the significant DEGs in resistant American elm-Onu pathosystem, ten genes involved in the phenylpropanoids biosynthesis pathway were significantly upregulated, whereas two were downregulated (Figure 6B). Five gene homologs of peroxidase and three of beta-glucosidase were identified among the significant DEGs. Three of the peroxidases showed similar expression profiles in both genotypes, which were upregulated in responses to the pathogen, and the other two were downregulated in the resistant genotype. Transcripts of the aldehyde dehydrogenase, peroxidase P7-like, and peroxidase 4 like genes showed higher log2 fold-change (3.80, 2.70, and 2.67, respectively) in the resistant compared to the susceptible genotype (2.10, 0.50, and 0.47, respectively) in response to Onu.
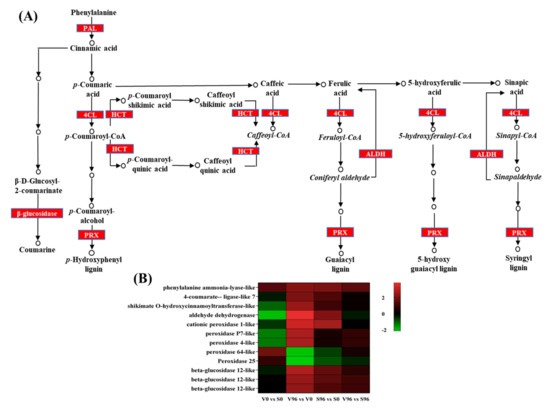
Figure 6.
The expression pattern of genes coding for phenylpropanoid biosynthesis-related enzymes in Ulmus americana - Ophiostoma novo-ulmi interaction. (A) Phenylpropanoid biosynthesis pathway (KEGG database) highlighted significant differentially expressed genes (DEGs). (B) Expression profiles of the phenylpropanoid biosynthesis-related significantly differentially expressed genes in DED-resistant (V) and -susceptible (S) American elm genotypes.
3.10. Validation of RNA-Seq Results by qRT-PCR
From the most significant DEGs between V96 and S96 samples, a representative subset of putative disease-related candidate genes was selected for validation of their expression patterns via qRT-PCR analysis. Following primer efficiency testing, only the primer pairs designed to amplify four upregulated genes (senescence-associated (078107), kinase family (033280), thaumatin (027705), and disease resistance RPP3 (065957)), and three downregulated (photosystem II D2 (045844), rubisco large subunit (014970), and photosystem I P700 apo A2 (000664)) candidate genes were used for the validation. The gene expression analysis was performed using cDNA templates prepared from ‘Valley Forge’ and ‘Susceptible’ American elm mRNAs that were isolated at 0, 48, 96, and 144 h post-inoculation (Figure 7). For comparative purposes, a heatmap was generated with the read count of these genes obtained from the RNA-Seq analysis (Figure 7A). The relative normalized expression patterns of three upregulated genes, thaumatin (027705), kinase family (033280), and disease resistance RPP3 (065957), matched their expression profiles measured via RNA-Seq, although only the kinase family (033280) and disease resistance RPP3 (065957) genes were significantly upregulated in Valley Forge relative to susceptible at each time point (Figure 7B). The highest relative normalized expression among the upregulated candidate genes was detected for disease resistance RPP3 at V0 and V96 relative to S0 and S96 counterparts, which coincided with the expression of this gene in the RNA-Seq data (Figure 7A). Thaumatin and a kinase family gene also displayed comparable gene expression levels between the RNA-Seq data and the qPCR validation approach. In contrast, the expression patterns of a senescence-associated gene behaved differently between the RNA-Seq and the qRT-PCR analysis. While the senescence-associated gene was classified as upregulated in V0 and V96 relative to S0 and S96 samples in the RNA-Seq-based differential gene expression analysis, it appears to be downregulated in V0 relative to S0 samples, when its expression was measured via qRT-PCR (Figure 7B). Furthermore, the downregulated genes photosystem II D2, Rubisco large subunit, and photosystem I P700 apo A2 were also found to be downregulated in the qRT-PCR gene expression analysis, although there was no statistical evidence that they were differentially expressed.
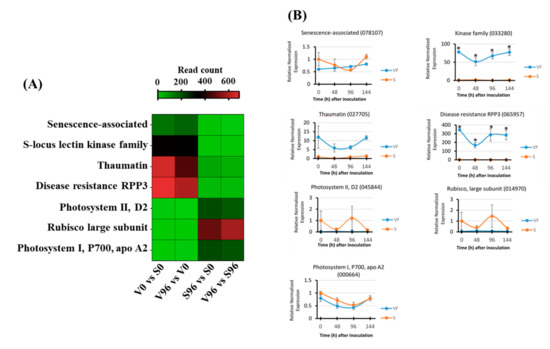
Figure 7.
Validation by qRT-PCR of candidate genes identified by RNA-Seq analysis. (A) Expression of candidate genes via RNA-Seq in Valley Forge (V) and Susceptible (S) American elm genotypes at 0 and 96 hpi with Ophiostoma novo-ulmi. (B) Relative normalized expression of candidate genes for disease resistance and susceptibility in Valley Forge (VF) and Susceptible (S) genotypes quantified via qRT-PCR at 0, 48, 96, and 144 h post-inoculation (hpi). Asterisks (*) represent significant differences between VF and S within post-inoculation time points at p < 0.05, according to Tukey’s HSD test.
4. Discussion
The highly destructive Dutch elm disease (DED) caused by the fungus Optihostoma novo-ulmi poses a significant threat to American elm [1,2,9]. Knowledge of the molecular mechanisms underlying the host–pathogen interactions in this pathosystem is, at best, fragmentary. Previously, transcriptome level changes in American elm calli during compatible interaction with Onu have been reported based on the analysis of a few hundred Expressed Sequence Tags (ESTs) [16]. A subsequent study in which susceptible and resistant lines of U. americana were inoculated with O. novo-ulmi allowed a comparison of molecular responses associated with compatible and incompatible interactions, respectively, but was limited to a small subset of disease-responsive genes that were assayed by qRT-PCR [9]. Therefore, the work reported herein aimed to obtain a comprehensive, genome-wide view of transcriptomic changes in resistant and susceptible genotypes of U. americana in response to DED. The plants of American elm genotypes with contrasting resistance to DED were inoculated with yeast spores of Onu, which germinated within the stem, where hyphae grew and spread through xylem vessels, inducing their cavitational embolism [30,31].
In this transcriptomics study, the BLASTX comparison yielded 55,499 unique genes in U. americana, which was approximately 2-fold higher than the number of unique genes in the U. minor transcriptome (Figure 2A). Previously, only 314 unisequences were isolated from U. americana calli inoculated with Onu [16]. The higher number of U. minor genes aligned to U. americana local DB and the higher number of unique genes in the U. americana transcriptome may be due to the sequencing platform used and the number of reads generated for each transcriptome. The U. minor transcriptome was sequenced in the 454 GS-FLX Titanium System, generating a total of 971,002 raw reads, whereas U. americana was sequenced in the Illumina HiSeq 2500 platform, generating an average of raw reads ranging from 66.4 to 75.8 million. In addition, it is well known that the Illumina HiSeq 2500 platform produces significantly higher sequence coverage than the 454 GS-FLX Titanium System [29,32]. Using the BLASTX software, the unique genes for each transcriptome were compared to the non-redundant protein database downloaded from NCBI, annotating approximately 61% genes from U. minor and 42% genes from U. americana as plant-related transcripts (Figure 2A). The remaining transcripts were annotated with functions from organisms belonging to other kingdoms, indicating the presence of plant endophyte genes that can be characterized in future studies. The lower number of plant-related transcripts in U. americana contrasted with the higher number of unique genes in this species relative to U. minor transcriptome suggests that the use of the Illumina 2500 platform could have generated a more diverse transcriptome of U. americana, which may include related endophytes. The transcriptome reported in this study represents the largest molecular source of information from U. americana obtained so far and would also be very helpful for annotating the U. americana genome that is publicly available in the NCBI database as an unassembled genome.
The transcriptomic analysis yielded a significant number of differentially expressed genes (DEGs) (Figure 3). The functional enrichment analysis of these DEGs indicated that the cellular and primary metabolic processes were the most highly enriched GO terms, wherease photosynthesis, carbon metabolism, and citrate cycle were the most enriched KEGG pathways between resistant ‘Valley Forge’ and susceptible elm genotypes regardless of the Onu inoculation (Table 2 and Table 3). The expression profile of the DEGs involved in the photosynthesis was significantly downregulated in the resistant American elm genotype compared to the susceptible genotype (Figure 5). This suggests that the genotypic variation mainly exists in the primary metabolism, and that enhanced primarily metabolic processes, especially photosynthesis, could be linked to the susceptiblity to DED [11]. In fact, the trade-off between growth and defense has previously been suggested as a defense mechanism against DED in the English elm-Onu pathosystem [33]. In that system, English elm trees compromise primary metabolic function at the expense of activating secondary metabolic processes for a successful plant defense [33,34,35]. GO terms of the Onu-responsive DEGs were enriched in response to stimulus, stress response, secondary metabolic process, and defense response in both the resistant and susceptible elm genotypes (Table 2).
Plants counteract pathogen attacks by activating a complex immune system. The primary immune response is known as pattern-triggered immunity (PTI), which is initiated by the perception of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs) [36,37]. The second layer of defense, known as effector-triggered immunity (ETI), is initiated by plant resistance genes (R-genes) in response to pathogen effector proteins [38]. R-genes are usually characterized by a nucleotide-binding domain and a leucine-rich-repeat domain (NLR). NLRs stimulate rapid yet long-lasting defense responses including hypersensitive response followed by the production of reactive oxygen species (ROS), cell wall rigidification, synthesis of phytoalexins, hormonal signaling, and eventually the induction of PR genes [39,40] for suppressing the growth of invading pathogens. In general, resistance against biotrophic pathogens is regulated by the SA pathway, whereas jasmonic acid/ethylene (JA/ET) pathways provide resistance to necrotrophic pathogens [41]. The coordinated actions of both SA and JA are required to combat the hemibiotrophic pathogens [18], where SA is activated at the early biotrophic phase followed by JA’s induction during the necrotrophic phase [42]. Pathogenesis-related (PR) genes are a diverse group of genes and are considered the signature genes of SA and JA pathways. For instance, increased expression of the PR1, PR2, and PR5 genes represents the SA signaling pathway [43]. In contrast, increased expression of PR3, PR4, and PR12 corresponds to the activation of the JA pathway [44,45]. RPM1 encoding an NLR receptor protein [46] was significantly upregulated in resistant American elm–Onu interaction. On the other hand, phospholipase d beta 1 (PLDβ1), a type of phospholipase d (PLD) previously characterized as a negative regulator of RPM1 [47], was highly expressed in the susceptible American elm genotype in response to Onu (Figure 4). Furthermore, ROS-mediated hypersensitive response (HR), which is an essential mechanism of the plant’s defense against biotrophic and hemibiotrophic pathogens, is also negatively regulated by PLDβ1–PA signaling [48,49,50]. PLDβ1 expression is repressed by salicylic acid (SA), and the mutation of PLDβ1 enhanced SA production and signaling pathways upon pathogen infection [49,50]. On the other hand, PR4 and PR5 (thaumatin 1) genes were significantly upregulated in the resistant genotype, suggesting that resistance to DED in U. americana could be attributed to the simultaneous activation of SA and JA signaling, which agrees with previous findings [9,17]. Hence, it may be inferred that in the American elm-Onu pathosystem, the RPM1-mediated activation of ETI and repression of the PLDβ1 might be crucial for the resistance, and that the Onu pathogen might induce the PLDβ1 in the susceptible, leading to the disease development.
The KEGG enrichment analysis showed that phenylpropanoid biosynthesis was remarkably enriched among the secondary metabolic processes in resistant and susceptible genotypes in response to Onu, which is in agreement with previous studies [15,16]. To estimate the significance of the phenylpropanoids biosynthesis pathway in DED resistance, we further extracted the significant DEGs involved in that pathway (Figure 6B). The biosynthesis of phenylpropanoids starts with the conversion of cinnamic acid from phenylalanine by phenyl ammonia-lyase (PAL), leading to the formation of different forms of phenolics [51,52]. The genes 4-coumarate ligase (4CL), and shikimate O-hydroxycinnamoyl transferase (HCT) encode the enzymes involved in the synthesis of caffeoyl Co-A, feruloyl Co-A, 5-hydroxyferuloyl Co-A, and sinapoyl Co-A that act as precursors in the biosynthesis of different lignins [53], and the expression of these genes was significantly upregulated in the resistant genotype compared to the susceptible one in response to Onu. After a series of deamination, hydroxylation, methylation, and reduction, lignin monomers are produced in the cytoplasm and transported to the apoplast [53,54]. Finally, lignin is generally polymerized with three main types of monolignols (p-coumaryl alcohol, H unit; coniferyl alcohol, G unit; and sinapyl alcohol, S unit) by peroxidase (POD) in the secondary cell wall [55,56]. Interestingly, transcripts for peroxidases showed contrasting accumulation patterns in the resistant genotype challenged with Onu. For instance, peroxidase P7 and peroxidase 4-like transcripts were upregulated, whereas peroxidase 64-like and peroxidase 25 were downregulated (Figure 6B). This indicates that the activation of a specific peroxidase might be crucial for the polymerization of lignin and resistance against DED. Further characterization of these peroxidases and determination of the roles played by particular lignin polymers in resistance to DED warrants further studies.
Phenolic metabolites can be found either in soluble form in the cells or esterified and/or etherified within the cell wall for enhancing the cell-wall cross-linking [52]. The induction of the cell-wall-bound phenolics, lignin, and peroxidase is characterized as a resistance response in Norway spruce trees naturally infected with fungal pathogen Ascocalyx abietina [51]. The plant pathology literature contains several suggestions as to a role for fungal cell-wall-degrading enzymes in pathogenesis [57,58], and DED is no exception. The more aggressive Onu secretes higher amounts of glycosidases and exo-glycanases than the less aggressive O. ulmi under laboratory conditions [59,60]. Therefore, cell-wall reinforcement by inducing the cross-linking of phenolics and polymerization of lignin might be an inducible resistance mechanism of the American elm against DED. The different forms of phenolics and peroxidases for lignin biosynthesis and their contributions to resistance against Dutch elm disease warrant further study.
5. Conclusions
In conclusion, the present study is the first to assess genome-wide transcriptomic changes in American elm during the in vivo colonization of Onu fungus. Our data revealed that prior to infection (at 0 hpi), DEGs associated with photosynthesis and primary cellular metabolism were highly enriched and upregulated in the susceptible genotype, supporting the previously suggested notion of trade-offs between growth and defense in the DED pathosystem. After infection (at 96 hpi), transcriptomic changes in the resistant genotype could be explained in the light of effector-triggered immunity (ETI) as manifested by the enhanced expression of RPM1, PR genes, and genes in the phenylpropanoids biosynthesis pathway and lignin polymerization. In addition to elucidating the significant molecular changes in compatible and incompatible interactions of American elm and Onu, the present study provides transcriptomic data that would assist in the assembly, annotation, and characterization of the American elm genome, which itself is a significant milestone toward developing DED-resistant elm germplasm through breeding and biotechnology.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/jof8020120/s1, Table S1: qRT-PCR primers used in this study; Table S2: Complete list of genes expressed in resistant and susceptible (V0 vs. S0) American elm genotypes at 0 hpi of Ophiostoma novo-ulmi; Table S3: Complete list of genes expressed in comparison between 0 hpi and 96 hpi of Ophiostoma novo-ulmi in resistant (Valley Forge; V) American elm genotype; Table S4: Complete list of genes expressed in comparison between 0 hpi and 96 hpi of Ophiostoma novo-ulmi in susceptible (S) American elm genotype; Table S5: Complete list of genes expressed in resistant and susceptible (V96 vs. S96) American elm genotypes at 96 hpi of Ophiostoma novo-ulmi.
Author Contributions
Investigation, methodology, software, validation, data curation, writing—original draft preparation, M.T.I. and J.F.C.; resources and data curation, M.S.; methodology, formal analysis, writing—review and editing, A.K.D., M.N. and L.B.; conceptualization, project administration, supervision and writing-review and editing, S.M.S. and P.K.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Gosling Foundation, Guelph, ON, Canada, grant number: 050294. The authors thank Virginia Tech’s Open Access Subvention Fund for their help with publication costs.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
RNA-Seq experimental data corresponding to U. americana and O. novo-ulmi raw reads for each biological replicate were deposited at the NCBI Sequence Read Archive as Binary Alignment Files under the accession number SRP149721. The assembled transcriptome of U. americana is also available at NCBI SRA library as a FASTA file under the same accession number.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Paoletti, M.; Buck, K.W.; Brasier, C.M. Selective acquisition of novel mating type and vegetative incompatibility genes via interspecies gene transfer in the globally invading eukaryote Ophiostoma novo-ulmi. Mol. Ecol. 2006, 15, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Ganley, R.J.; Bulman, L.S. Dutch elm disease in New Zealand: Impacts from eradication and management programmes. Plant Pathol. 2016, 65, 1047–1055. [Google Scholar] [CrossRef]
- Brasier, C.M.; Kirk, S.A. Designation of the EAN and NAN races of Ophiostoma novo-ulmi as subspecies. Mycol. Res. 2001, 105, 547–554. [Google Scholar] [CrossRef]
- Webber, J.F. Relative effectiveness of Scolytus scolytus, S. multistriatus and S. kirschi as vectors of Dutch elm disease. Eur. J. For. Pathol. 1990, 20, 184–192. [Google Scholar] [CrossRef]
- Webber, J.F. Experimental studies on factors influencing the transmission of Dutch elm disease. For. Syst. 2004, 13, 197–205. [Google Scholar]
- Lee, J.C.; Seybold, S.J. Host acceptance and larval competition in the banded and European elm bark beetles, Scolytus schevyrewi and S. multistriatus (Coleoptera: Scolytidae): Potential mechanisms for competitive displacement between invasive species. J. Insect Behav. 2010, 23, 19–34. [Google Scholar] [CrossRef]
- Townsend, A.M.; Bentz, S.E.; Johnson, G.R. Variation in response of selected American elm clones to Ophiostoma ulmi. J. Environ. Hortic. 1995, 13, 126. [Google Scholar] [CrossRef]
- Townsend, A.M.; Douglass, L.W. Variation among American elm clones in long-term dieback, growth, and survival following Ophiostoma inoculation. J. Environ. Hortic. 2001, 19, 100–103. [Google Scholar] [CrossRef]
- Sherif, S.M.; Shukla, M.R.; Murch, S.J.; Bernier, L.; Saxena, P.K. Simultaneous induction of jasmonic acid and disease-responsive genes signifies tolerance of American elm to Dutch elm disease. Sci. Rep. 2016, 6, 21934. [Google Scholar] [CrossRef]
- Martín, J.A.; Domínguez, J.; Solla, A.; Brasier, C.M.; Webber, J.F.; Santini, A.; Martínez-Arias, C.; Bernier, L.; Gil, L. Complexities underlying the breeding and deployment of Dutch elm disease resistant elms. New For. 2021, 1–36. [Google Scholar] [CrossRef]
- Martín, J.A.; Sobrino-Plata, J.; Rodríguez-Calcerrada, J.; Collada, C.; Gil, L. Breeding and scientific advances in the fight against Dutch elm disease: Will they allow the use of elms in forest restoration? New For. 2019, 50, 183–215. [Google Scholar] [CrossRef]
- Smalley, E.B.; Guries, R.P. Asian elms: Sources ofdiseaseand insect resistance. In The Elms; Dunn, C.P., Ed.; Springer: Berlin, Germany, 2000; pp. 215–230. [Google Scholar]
- Santini, A.; Pecori, F.; Pepori, A.; Brookes, A. ‘Morfeo’ Elm: A new variety resistant to Dutch elm disease. For. Pathol. 2011, 42, 171–176. [Google Scholar] [CrossRef]
- Venturas, M.; López, R.; Martín, J.; Gascó, A.; Gil, L. Heritability of Ulmus minor resistance to Dutch elm disease and its relationship to vessel size, but not to xylem vulnerability to drought. Plant Pathol. 2014, 63, 500–509. [Google Scholar] [CrossRef]
- Perdiguero, P.; Venturas, M.; Cervera, M.T.; Gil, L.; Collada, C. Massive sequencing of Ulmus minor’s transcriptome provides new molecular tools for a genus under the constant threat of Dutch elm disease. Front. Plant Sci. 2015, 6, 541. [Google Scholar] [CrossRef]
- Aoun, M.; Jacobi, V.; Boyle, B.; Bernier, L. Identification and monitoring of Ulmus americana transcripts during in vitro interactions with the Dutch elm disease pathogen Ophiostoma novo-ulmi. Physiol. Mol. Plant Pathol. 2010, 74, 254–266. [Google Scholar] [CrossRef]
- Martínez-Arias, C.; Sobrino-Plata, J.; Gil, L.; Rodríguez-Calcerrada, J.; Martín, J.A. Priming of Plant Defenses against Ophiostoma novo-ulmi by Elm (Ulmus minor Mill.) Fungal Endophytes. J. Fungi 2021, 7, 687. [Google Scholar] [CrossRef]
- Ding, L.; Xu, H.; Yi, H.; Yang, L.; Kong, Z.; Zhang, L.; Xue, S.; Jia, H.; Ma, Z. Resistance to hemi-biotrophic F. graminearum infection is associated with coordinated and ordered expression of diverse defense signaling pathways. PLoS ONE 2011, 6, e19008. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, L.; Yan, D.H.; He, W. Genome-Wide Transcriptome Analysis Reveals the Comprehensive Response of Two Susceptible Poplar Sections to Marssonina brunnea Infection. Genes 2018, 9, 154. [Google Scholar] [CrossRef]
- Pedro, H.; Maheswari, U.; Urban, M.; Irvine, A.G.; Cuzick, A.; McDowall, M.D.; Staines, D.M.; Kulesha, E.; Hammond-Kosack, K.E.; Kersey, P.J. PhytoPath: An integrative resource for plant pathogen genomics. Nucleic Acids Res. 2016, 44, 688–693. [Google Scholar] [CrossRef]
- Townsend, A.M.; Bentz, S.E.; Douglass, L.W. Evaluation of 19 American elm clones for tolerance to Dutch elm disease. J. Environ. Hortic. 2005, 23, 21–24. [Google Scholar] [CrossRef]
- Comeau, A.M.; Dufour, J.; Bouvet, G.F.; Nigg, M.; Jacobi, V.; Henrissat, B.; Laroche, J.; Levesque, R.C.; Bernier, L. Functional annotation of the Ophiostoma novo-ulmi genome: Insights into the phytopathogenicity of the fungal agent of Dutch elm disease. Genome Biol. Evol. 2015, 7, 410–430. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; Available online: http://www.Rproject.org/ (accessed on 1 March 2018).
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community 2017 update. Nucleic Acids Res. 2017, 45, 122–129. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- O’Neil, D.; Glowatz, H.; Schlumpberger, M. Ribosomal RNA depletion for efficient use of RNA-seq capacity. Curr. Protoc. Mol. Biol. 2013, 103, 4–19. [Google Scholar]
- Li, S.; Tighe, S.W.; Nicolet, C.M.; Grove, D.; Levy, S.; Farmerie, W.; Viale, A.; Wright, C.; Schweitzer, P.A.; Gao, Y.; et al. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nat. Biotechnol. 2014, 32, 915. [Google Scholar] [CrossRef]
- Scheffer, R.J.; Elgersma, D.M. A scanning electron microscope study of cell wall degradation in elm wood by aggressive and non-aggressive isolates of Ophiostoma ulmi. Eur. J. Plant Pathol. 1982, 12, 25–28. [Google Scholar] [CrossRef]
- Newbanks, D.; Bosch, A.; Zimmermann, M.H. Evidence for xylem dysfunction by embolization in Dutch elm disease. Phytopathology 1983, 73, 1060–1063. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [PubMed]
- Perdiguero, P.; Sobrino-Plata, J.; Venturas, M.; Martín, J.A.; Gil, L.; Collada, C. Gene expression trade-offs between defence and growth in English elm induced by Ophiostoma novo-ulmi. Plant Cell Environ. 2018, 41, 198–214. [Google Scholar] [CrossRef]
- Karasov, T.L.; Chae, E.; Herman, J.J.; Bergelson, J. Mechanisms to Mitigate the Trade-Off between Growth and Defense. Plant Cell 2017, 29, 666–680. [Google Scholar] [CrossRef]
- Huot, B.; Yao, J.; Montgomery, B.L.; He, S.Y. Growth-defense tradeoffs in plants: A balancing act to optimize fitness. Mol. Plant. 2014, 7, 1267–1287. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Bigeard, J.; Colcombet, J.; Hirt, H. Signaling mechanisms in pattern-triggered immunity (PTI). Mol. Plant 2015, 8, 521–539. [Google Scholar] [CrossRef]
- Cui, H.; Tsuda, K.; Parker, J.E. Effector-triggered immunity: From pathogen perception to robust defense. Annu. Rev. Plant Biol. 2015, 66, 487–511. [Google Scholar] [CrossRef]
- Van Loon, L.C.; Rep, M.; Pieterse, C.M. Significance of inducible defense-related proteins in infected plants. Annu. Rev. Phytopathol. 2006, 44, 135–162. [Google Scholar] [CrossRef]
- Baggs, E.; Dagdas, G.; Krasileva, K.V. NLR diversity, helpers and integrated domains: Making sense of the NLR ID entity. Curr. Opin. Plant Biol. 2017, 38, 59–67. [Google Scholar] [CrossRef]
- Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–222. [Google Scholar] [CrossRef]
- Islam, M.T.; Mamun, M.A.; Lee, B.R.; La, H.V.; Kim, T.H. Role of salicylic acid signaling in the biotrophy-necrotrophy transition of Xanthomonas campestris pv. campestris infection in Brassica napus. Physiol. Mol. Plant Pathol. 2021, 113, 101578. [Google Scholar] [CrossRef]
- Delaure, S.L.; Hemelrijck, W.V.; Bolle, M.F.C.D.; Cammue, B.P.A.; De Coninck, B.M.A. Building up plant defenses by breaking down proteins. Plant Sci. 2008, 174, 375–385. [Google Scholar] [CrossRef]
- Norman-Setterblad, C.; Vidal, S.; Palva, E.T. Interacting signal pathways control defense gene expression in Arabidopsis in response to cell wall-degrading enzymes from Erwinia carotovora. Mol. Plant Microbe Interact. 2000, 13, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Seo, P.J.; Lee, A.K.; Xiang, F.; Park, C.M. Molecular and functional profiling of Arabidopsis Pathogenesis-related genes: Insights into their roles in salt response of seed germination. Plant Cell Physiol. 2008, 49, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.R.; Godiard, L.; Straube, E.; Ashfield, T.; Lewald, J.; Sattler, A.; Innes, R.W.; Dangl, J.L. Structure of the Arabidopsis RPM1 gene enabling dual specificity disease resistance. Science 1995, 269, 843–846. [Google Scholar] [CrossRef]
- Yuan, X.; Wang, Z.; Huang, J.; Xuan, H.; Gao, Z. Phospholipidase Dδ Negatively Regulates the Function of Resistance to Pseudomonas syringae pv. maculicola 1 (RPM1). Front. Plant Sci. 2019, 9, 1991. [Google Scholar] [CrossRef]
- Bargmann, B.O.R.; Laxalt, A.M.; Ter Riet, B.; Schouten, E.; Van Leeuwen, W.; Dekker, H.L.; De Koster, C.G.; Haring, M.A.; Munnik, T. LePLDβ1 activation and relocalization in suspension-cultured tomato cells treated with xylanase. Plant J. 2006, 45, 358–368. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kuroda, M.; Yamakawa, H.; Ashizawa, T.; Hirayae, K.; Kurimoto, L.; Shinya, T.; Shibuya, N. Suppression of a phospholipase D gene, OsPLDβ1, activates defense responses and increases disease resistance in rice. Plant Physiol. 2009, 150, 308–319. [Google Scholar] [CrossRef]
- Zhao, J.; Devaiah, S.P.; Wang, C.; Li, M.; Welti, R.; Wang, X. Arabidopsis phospholipase Dβ1 modulates defense responses to bacterial and fungal pathogens. New Phytol. 2013, 199, 228–240. [Google Scholar] [CrossRef]
- Cvikrová, M.; Malá, J.; Hrubcová, M. Eder; J. Soluble and cell wall-bound phenolics and lignin in Ascocalyx abietina infected Norway spruces. Plant Sci. 2006, 170, 563–570. [Google Scholar] [CrossRef]
- Islam, M.T.; Lee, B.R.; Park, S.H.; La, V.H.; Jung, W.J.; Bae, D.W.; Kim, T.H. Hormonal regulations in soluble and cell-wall bound phenolics accumulation in two cultivars of Brassica napus contrasting susceptibility to Xanthomonas campestris pv. campestris. Plant Sci. 2019, 285, 132–140. [Google Scholar] [CrossRef]
- Dixon, R.A.; Barros, J. Lignin biosynthesis: Old roads revisited and new roads explored. Open Biol. 2019, 9, 190215. [Google Scholar] [CrossRef]
- Chen, H.C.; Li, Q.; Shuford, C.M.; Liu, J.; Muddiman, D.C.; Sederoff, R.R.; Chiang, V.L. Membrane protein complexes catalyze both 4- and 3-hydroxylation of cinnamic acid derivatives in monolignol biosynthesis. Proc. Natl Acad. Sci. USA 2011, 108, 253–258. [Google Scholar] [CrossRef]
- Duroux, L.; Welinder, K.G. The peroxidase gene family in plants: A phylogenetic overview. J. Mol. Evol. 2003, 57, 397–407. [Google Scholar] [CrossRef]
- Zhao, Q.; Nakashima, J.; Chen, F.; Yin, Y.; Fu, C.; Yun, J.; Shao, H.; Wang, X.; Wang, Z.; Dixon, R.A. LACCASE is necessary and nonredundant with PEROXIDASE for lignin polymerization during vascular development in Arabidopsis thaliana. Plant Cell 2013, 25, 3976–3987. [Google Scholar] [CrossRef]
- Douaiher, M.N.; Nowak, E.; Durand, R.; Halama, P.; Reignault, P. Correlative analysis of Mycosphaerella graminicola pathogenicity and cell wall-degrading enzymes produced in vitro: The importance of xylanase and polygalacturonase. Plant Pathol. 2007, 56, 79–86. [Google Scholar] [CrossRef]
- Kikot, G.E.; Hours, R.A.; Alconada, T.M. Contribution of cell wall degrading enzymes to pathogenesis of Fusarium graminearum: A review. J. Basic Microbiol. 2009, 49, 231–241. [Google Scholar] [CrossRef]
- Przybyl, K.; Dahm, H.; Ciesielska, A.; Molinski, K. Cellulolytic activity and virulence of Ophiostoma ulmi and O. novo-ulmi isolates. For. Pathol. 2006, 36, 58–67. [Google Scholar] [CrossRef]
- Svaldi, R.; Elgersma, D.M. Further studies on the activity of cell wall degrading enzymes of aggressive and non-aggressive isolates of Ophiostoma ulmi. Eur. J. For. Pathol. 1982, 12, 29–36. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).