
Post-Translational Modifications of PCNA: Guiding for the Best DNA Damage Tolerance Choice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction. Proliferating Cell Nuclear Antigen (PCNA)
2. PCNA Structural Features
2.1. Loading and Unloading PCNA onto Duplex DNA
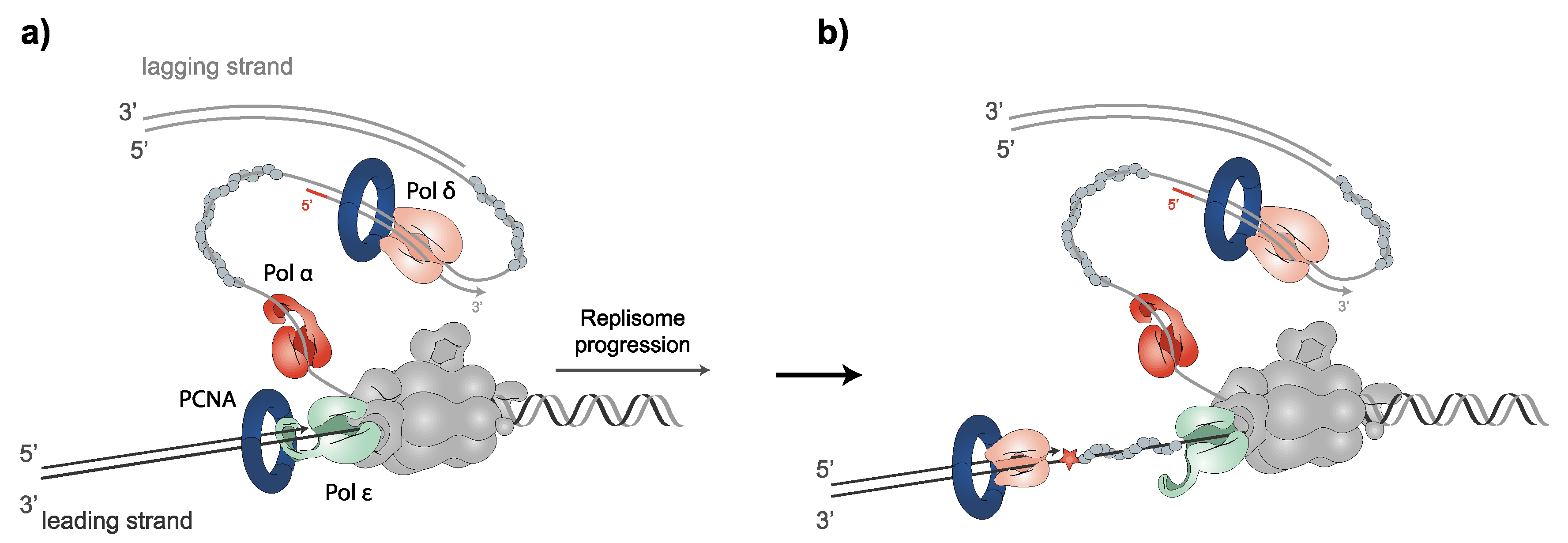
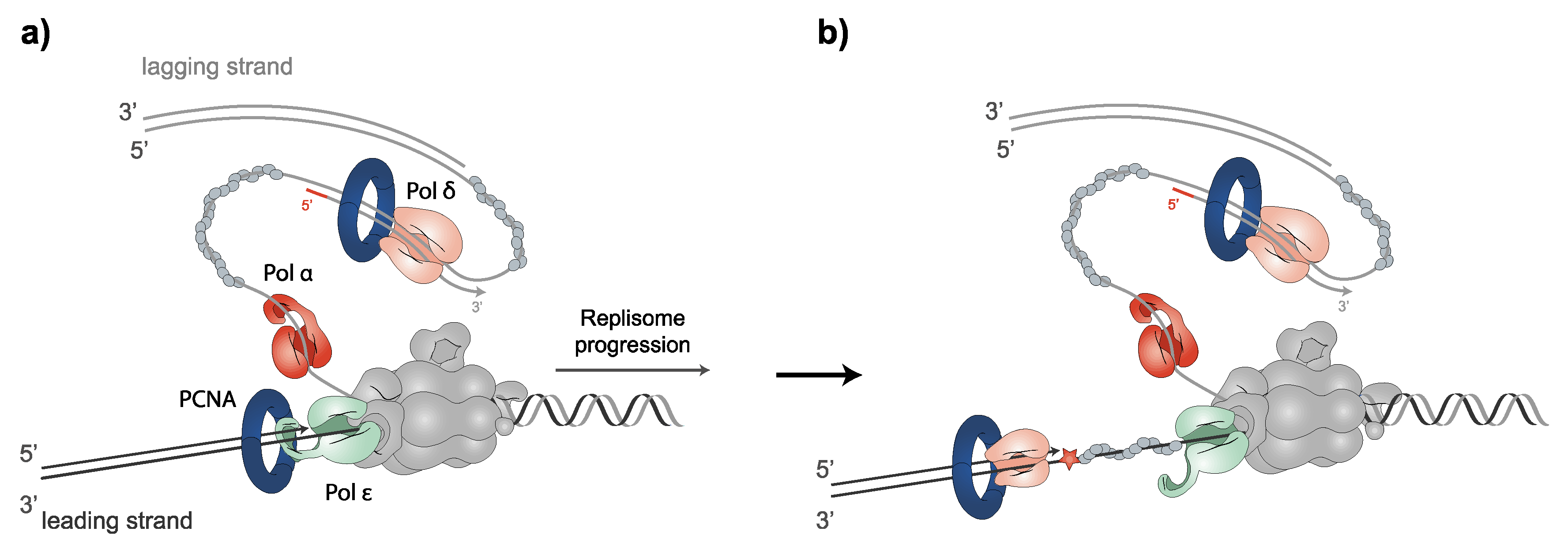
2.2. PCNA and Replicative DNA Polymerases: Leading and Lagging DNA
3. Post-Translational Modifications of PCNA and the DDT Pathways
3.1. Translesion Synthesis (TLS)-Mediated DDT Error-Prone Pathway
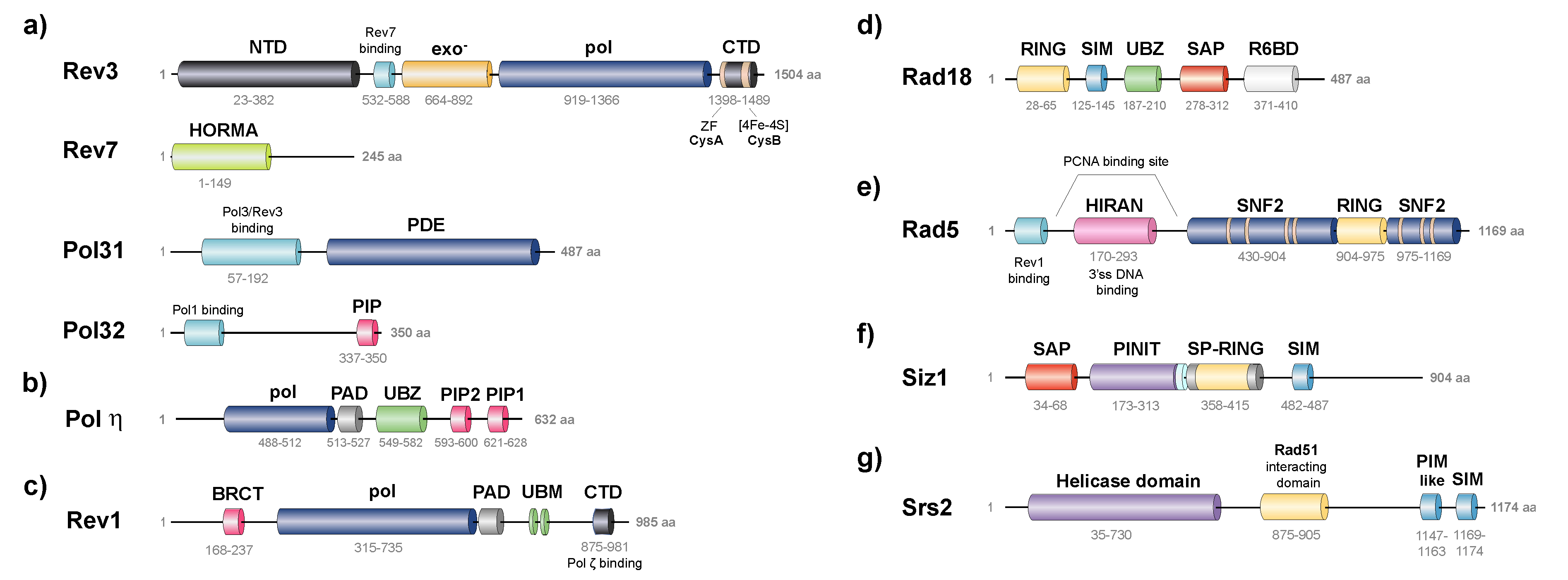
3.1.1. TLS Polymerases Structural Features
3.1.2. Monoubiquitin-PCNA Modification Mediated by Rad6-Rad18
3.1.3. DNA Polymerase Switching during TLS
3.2. Rad5-Mediated Error-Free DDT Bypass Pathway
3.2.1. Polyubiquitinated PCNA by Rad5- Error-Free Pathway
3.2.2. Structural Features of Rad5, Interactions and Associated Activities
3.2.3. Template Switch (TS) Model
3.2.4. Fork Reversal Model
3.3. Alternative Ubiquitination Sites in PCNA
3.4. PCNA Sumoylation: Regulation of Homologous Recombination (HR)
3.4.1. Srs2 Helicase Negatively Regulates the HR Pathway
3.4.2. PCNA Sumoylation by Ubc9-Siz1
3.4.3. Salvage Recombination (SR) Pathway
3.5. PCNA Inner Surface Acetylation in Response to DNA Damage
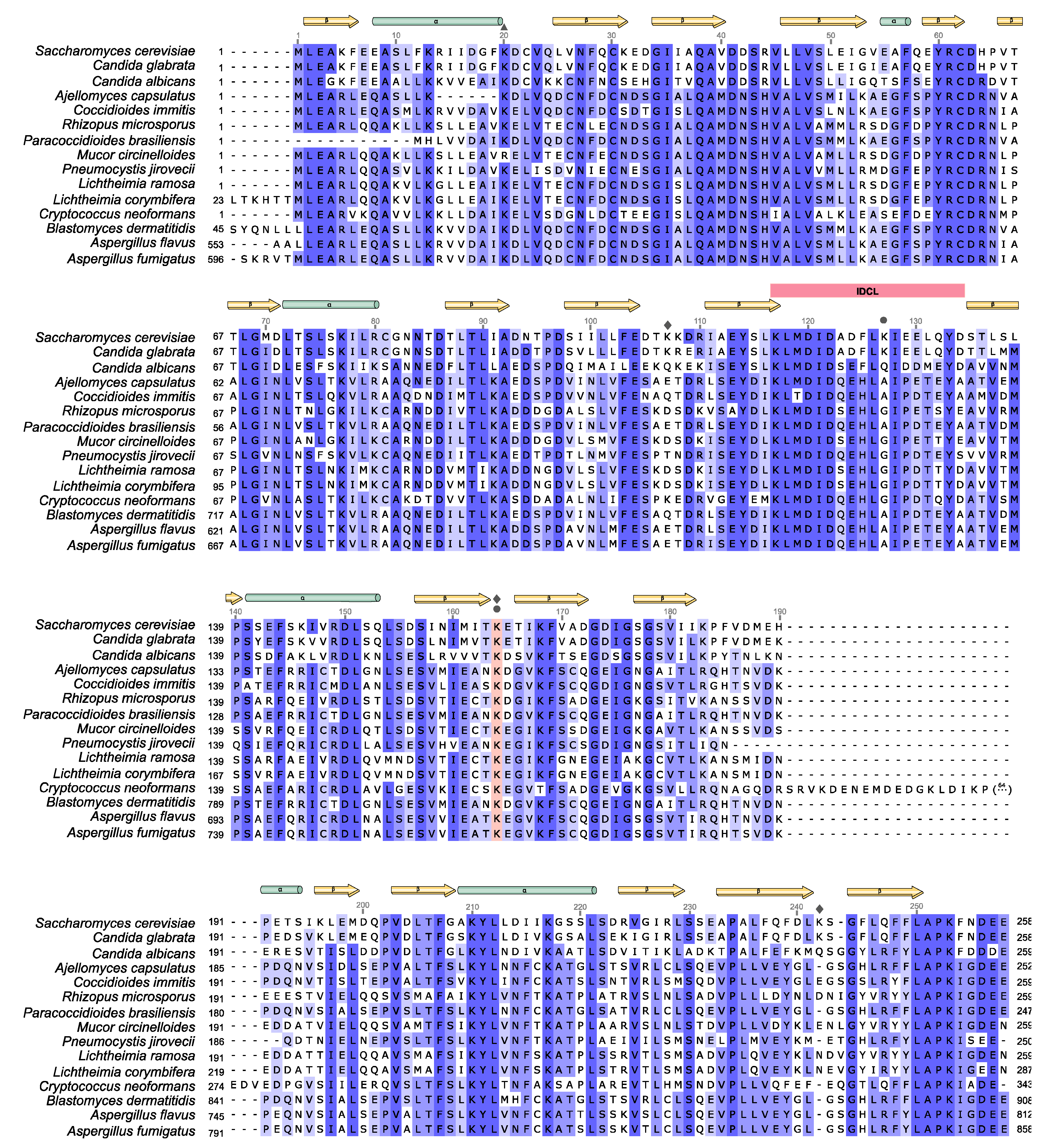
4. Comparative Study of PCNA from Systemic Pathogenic Fungi
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bauer, G.A.; Burgers, M.J. Molecular cloning, structure and expression of the yeast proliferating cell nuclear antigen gene. Nucleic Acids Res. 1990, 18, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Waga, S.; Stillman, B. The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem. 1998, 67, 721–751. [Google Scholar] [CrossRef] [Green Version]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the Maestro of the Replication Fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [Green Version]
- Henderson, D.S.; Banga, S.S.; Grigliatti, T.A.; Boyd, J.B. Mutagen sensitivity and suppression of position-effect variegation result from mutations in mus209, the Drosophila gene encoding PCNA. EMBO J. 1994, 13, 1450–1459. [Google Scholar] [CrossRef]
- Nomura, A. Nuclear distribution of proliferating cell nuclear antigen (PCNA) in fertilized eggs of the starfish Asterina pectinifera. J. Cell Sci. 1994, 107, 3291–3300. [Google Scholar] [CrossRef]
- Poot, R.A.; Bozhenok, L.; van den Berg, D.L.C.; Steffensen, S.; Ferreira, F.; Grimaldi, M.; Gilbert, N.; Ferreira, J.; Varga-Weisz, P.D. The Williams syndrome transcription factor interacts with PCNA to target chromatic remodelling by ISWI to replication foci. Nat. Cell Biol. 2004, 6, 1236–1244. [Google Scholar] [CrossRef]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA Controls Establishment of Sister Chromatid Cohesion during S Phase. Mol. Cell 2006, 23, 723–732. [Google Scholar] [CrossRef]
- Liu, H.W.; Bouchoux, C.; Panarotto, M.; Kakui, Y.; Patel, H.; Uhlmann, F. Division of Labor between PCNA Loaders in DNA Replication and Sister Chromatid Cohesion Establishment. Mol. Cell 2020, 78, 725–738.e4. [Google Scholar] [CrossRef]
- Zuilkoski, C.M.; Skibbens, R.V. PCNA promotes context-specific sister chromatid cohesion establishment separate from that of chromatin condensation. Cell Cycle 2020, 19, 2436–2450. [Google Scholar] [CrossRef]
- Maga, G.; Villani, G.; Tillement, V.; Stucki, M.; Locatelli, G.A.; Frouin, I.; Spadari, S.; Hübscher, U. Okazaki fragment processing: Modulation of the strand displacement activity of DNA polymerase δ by the concerted action of replication protein A, proliferating cell nuclear antigen, and flap endonuclease-1. Proc. Natl. Acad. Sci. USA 2001, 98, 14298–14303. [Google Scholar] [CrossRef] [Green Version]
- Sporbert, A.; Domaing, P.; Leonhardt, H.; Cardoso, M.C. PCNA acts as a stationary loading platform for transiently interacting Okazaki fragment maturation proteins. Nucleic Acids Res. 2005, 33, 3521–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyse, B.; Oshidari, R.; Rowlands, H.; Abbasi, S.; Yankulov, K. RRM3 regulates epigenetic conversions in Saccharomyces cerevisiae in conjunction with Chromatin Assembly Factor I. Nucleus 2016, 7, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenetics 2018, 10, 17. [Google Scholar] [CrossRef]
- Zhang, Z.; Shibahara, K.I.; Stillman, B. PCNA connects DNA replication to epigenetic inheritance in yeast. Nature 2000, 408, 221–225. [Google Scholar] [CrossRef]
- Ehrenhofer-Murray, A.E.; Kamakaka, R.T.; Rine, J. A role for the replication proteins PCNA, RF-C, polymerase ε and Cdc45 in transcriptional silencing in Saccharomyces cerevisiae. Genetics 1999, 153, 1171–1182. [Google Scholar] [CrossRef]
- Iida, T.; Suetake, I.; Tajima, S.; Morioka, H.; Ohta, S.; Obuse, C.; Tsurimoto, T. PCNA clamp facilitates action of DNA cytosine methyltransferase 1 on hemimethylated DNA. Genes Cells 2002, 7, 997–1007. [Google Scholar] [CrossRef]
- Tournier, S.; Leroy, D.; Goubin, F.; Ducommun, B.; Hyams, J.S. Heterologous expression of the human cyclin-dependent kinase inhibitor p21Cip1 in the fission yeast, Schizosaccharomyces pombe reveals a role for PCNA in the chk1+ cell cycle checkpoint pathway. Mol. Biol. Cell 1996, 7, 651–662. [Google Scholar] [CrossRef]
- Miyachi, K.; Fritzler, M.J.; Tan, E.M. Autoantibody to a nuclear antigen in proliferating cells. J. Immunol. 1978, 121, 2228–2234. [Google Scholar]
- Bravo, R.; Fey, S.J.; Bellatin, J.; Larsen, P.M.; Celis, J.E. Identification of a nuclear polypeptide (“cyclin”) whose relative proportion is sensitive to changes in the rate of cell proliferation and to transformation. Prog. Clin. Biol. Res. 1982, 85, 235–248. [Google Scholar]
- Mathews, M.B.; Bernstein, R.M.; Franza, B.R.; Garrels, J.I. Identity of the proliferating cell nuclear antigen and cyclin. Nature 1984, 309, 374–376. [Google Scholar] [CrossRef]
- Tan, C.-K.; Castillo, C.; So, A.G.; Downey, K.M. An auxiliary protein for DNA polymerase-δ from fetal calf thymus. J. Biol. Chem. 1986, 261, 12310–12316. [Google Scholar] [CrossRef]
- Prelich, G.; Tan, C.K.; Kostura, M.; Mathews, M.B.; So, A.G.; Downey, K.M.; Stillman, B. Functional identity of proliferating cell nuclear antigen and a DNA polymerase-δ auxiliary protein. Nature 1987, 326, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Frank, R.; Blundell, P.A.; Macdonald-Bravo, H. Cyclin/PCNA is the auxiliary protein of DNA polymerase-δ. Nature 1987, 326, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Fairman, M.; Prelich, G.; Tsurimoto, T.; Stillman, B. Identification of cellular components required for SV40 DNA replication in vitro. Biochim. Biophys. Acta 1988, 905, 382–387. [Google Scholar] [CrossRef]
- Yoder, B.L.; Burgers, P.M.J. Saccharomyces cerevisiae replication factor C. I. Purification and characterization of its ATPase activity. J. Biol. Chem. 1991, 266, 22689–22697. [Google Scholar] [CrossRef]
- Lee, S.H.; Pan, Z.Q.; Kwong, A.D.; Burgers, P.M.; Hurwitz, J. Synthesis of DNA by DNA polymerase epsilon in vitro. J. Biol. Chem. 1991, 266, 22707–22717. [Google Scholar] [CrossRef]
- Bravo, R.; Macdonald-Bravo, H. Existence of two populations of cyclin/proliferating cell nuclear antigen during the cell cycle: Association with DNA replication sites. J. Cell Biol. 1987, 105, 1549–1554. [Google Scholar] [CrossRef] [Green Version]
- Prelich, G.; Stillman, B. Coordinated leading and lagging strand synthesis during SV40 DNA replication in vitro requires PCNA. Cell 1988, 53, 117–126. [Google Scholar] [CrossRef]
- Celis, J.E.; Madsen, P. Increased nuclear cyclin/PCNA antigen staining of non S-phase transformed human amnion cells engaged in nucleotide excision DNA repair. FEBS Lett. 1986, 209, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Toschi, L.; Bravo, R. Changes in cyclin/proliferating cell nuclear antigen distribution during DNA repair synthesis. J. Cell Biol. 1988, 107, 1623–1628. [Google Scholar] [CrossRef] [Green Version]
- Dresler, S.L.; Kimbro, K.S. 2’, 3’-Dideoxy thymidine 5’-Triphosphate Inhibition of DNA Replication and Ultraviolet-Induced DNA Repair Synthesis in Human Cells: Evidence for Involvement of DNA Polymerase δ. Biochemistry 1987, 26, 2664–2668. [Google Scholar] [CrossRef] [PubMed]
- Nishida, C.; Reinhard, P.; Linn, S. DNA repair synthesis in human fibroblasts requires DNA plymerase δ. J. Biol. Chem. 1988, 263, 501–510. [Google Scholar] [CrossRef]
- Shivji, M.K.K.; Kenny, M.K.; Wood, R.D. Proliferating cell nuclear antigen is required for DNA excision repair. Cell 1992, 69, 367–374. [Google Scholar] [CrossRef]
- Nichols, A.F.; Sancar, A. Purification of PCNA as a nucleotide excision repair protein. Nucleic Acids Res. 1992, 20, 2441–2446. [Google Scholar] [CrossRef]
- Li, R.; Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature 1994, 371, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Buermeyer, A.B.; Simon, J.A.; Thomas, D.C.; Clark, A.B.; Liskay, R.M.; Kunkel, T.A. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell 1996, 87, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.E.; Kovvali, G.K.; Guzder, S.N.; Amin, N.S.; Holm, C.; Habraken, Y.; Sung, P.; Prakash, L.; Prakash, S. Evidence for involvement of yeast proliferating cell nuclear antigen in DNA mismatch repair. J. Biol. Chem. 1996, 271, 27987–27990. [Google Scholar] [CrossRef] [Green Version]
- Lau, P.J.; Kolodner, R.D. Transfer of the MSH2·MSH6 complex from proliferating cell nuclear antigen to mispaired bases in DNA. J. Biol. Chem. 2003, 278, 14–17. [Google Scholar] [CrossRef] [Green Version]
- Klungland, A.; Lindahl, T. Second pathway for completion of human DNA base excision-repair: Reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 1997, 16, 3341–3348. [Google Scholar] [CrossRef] [Green Version]
- Gary, R.; Kim, K.; Cornelius, H.L.; Park, M.S.; Matsumoto, Y. Proliferating cell nuclear antigen facilitates excision in long-patch base excision repair. J. Biol. Chem. 1999, 274, 4354–4363. [Google Scholar] [CrossRef] [Green Version]
- Levin, D.S.; McKenna, A.E.; Motycka, T.A.; Matsumoto, Y.; Tomkinson, A.E. Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Curr. Biol. 2000, 10, 919–922. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y. Molecular mechanism of PCNA-dependent base excision repair. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Lebel, M.; Spillare, E.A.; Harris, C.C.; Leder, P. The Werner syndrome gene product co-purifies with the DNA replication complex and interacts with PCNA and topoisomerase I. J. Biol. Chem. 1999, 274, 37795–37799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath-Loeb, A.S.; Johansson, E.; Burgers, P.M.J.; Loeb, L.A. Functional interaction between the Werner syndrome protein and DNA polymerase δ. Proc. Natl. Acad. Sci. USA 2000, 97, 4603–4608. [Google Scholar] [CrossRef] [Green Version]
- Liberti, S.E.; Andersen, S.D.; Wang, J.; May, A.; Miron, S.; Perderiset, M.; Keijzers, G.; Nielsen, F.C.; Charbonnier, J.B.; Bohr, V.A.; et al. Bi-directional routing of DNA mismatch repair protein human exonuclease 1 to replication foci and DNA double strand breaks. DNA Repair 2011, 10, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Haracska, L.; Johnson, R.E.; Unk, I.; Phillips, B.; Hurwitz, J.; Prakash, L.; Prakash, S. Physical and Functional Interactions of Human DNA Polymerase η with PCNA. Mol. Cell. Biol. 2001, 21, 7199–7206. [Google Scholar] [CrossRef] [Green Version]
- Haracska, L.; Johnson, R.E.; Unk, I.; Phillips, B.B.; Hurwitz, J.; Prakash, L.; Prakash, S. Targeting of human DNA polymerase ι to the replication machinery via interaction with PCNA. Proc. Natl. Acad. Sci. USA 2001, 98, 14256–14261. [Google Scholar] [CrossRef] [Green Version]
- Haracska, L.; Unk, I.; Johnson, R.E.; Phillips, B.B.; Hurwitz, J.; Prakash, L.; Prakash, S. Stimulation of DNA Synthesis Activity of Human DNA Polymerase κ by PCNA. Mol. Cell. Biol. 2002, 22, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Kannouche, P.; Fernández de Henestrosa, A.R.; Coull, B.; Vidal, A.E.; Gray, C.; Zicha, D.; Woodgate, R.; Lehmann, A.R. Localization of DNA polymerases eta and iota to the replication machinery is tightly co-ordinated in human cells. EMBO J. 2003, 22, 1223–1233. [Google Scholar] [CrossRef] [Green Version]
- Maga, G.; Villani, G.; Ramadan, K.; Shevelev, I.; Le Gac, N.T.; Blanco, L.; Blanca, G.; Spadari, S.; Hübscher, U. Human DNA polymerase λ functionally and physically interacts with proliferating cell nuclear antigen in normal and translesion DNA synthesis. J. Biol. Chem. 2002, 277, 48434–48440. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Xu, X.; Chang, C.W.; Liu, Y. TRIM28 functions as the SUMO E3 ligase for PCNA in prevention of transcription induced DNA breaks. Proc. Natl. Acad. Sci. USA 2020, 117, 23588–23596. [Google Scholar] [CrossRef] [PubMed]
- Daigaku, Y.; Davies, A.A.; Ulrich, H.D. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 2010, 465, 951–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, T.S.R.; Kong, X.P.; Gary, S.; Burgers, P.M.; Kuriyan, J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 1994, 79, 1233–1243. [Google Scholar] [CrossRef]
- Kelman, Z.; O’donnell, M. Structural and functional similarities of prokaryotic and eukaryotic DNA polymerase sliding clamps. Nucleic Acids Res. 1995, 23, 3613–3620. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, K.; Morioka, H.; Imajou, S.; Ikeda, S.; Ohtsuka, E.; Tsurimoto, T. Structure-function relationship of the eukaryotic DNA replication factor, proliferating cell nuclear antigen. J. Biol. Chem. 1995, 270, 22527–22534. [Google Scholar] [CrossRef] [Green Version]
- Kondratick, C.M.; Litman, J.M.; Shaffer, K.V.; Washington, M.T.; Dieckman, L.M. Crystal structures of PCNA mutant proteins defective in gene silencing suggest a novel interaction site on the front face of the PCNA ring. PLoS ONE 2018, 13, e0193333. [Google Scholar] [CrossRef]
- Kochaniak, A.B.; Habuchi, S.; Loparo, J.J.; Chang, D.J.; Cimprich, K.A.; Walter, J.C.; van Oijen, A.M. Proliferating cell nuclear antigen uses two distinct modes to move along DNA. J. Biol. Chem. 2009, 284, 17700–17710. [Google Scholar] [CrossRef] [Green Version]
- De March, M.; Merino, N.; Barrera-Vilarmau, S.; Crehuet, R.; Onesti, S.; Blanco, F.J.; De Biasio, A. Structural basis of human PCNA sliding on DNA. Nat. Commun. 2017, 8, 13935. [Google Scholar] [CrossRef] [Green Version]
- Yao, N.Y.; O’Donnell, M. DNA Replication: How Does a Sliding Clamp Slide? Curr. Biol. 2017, 27, R174–R176. [Google Scholar] [CrossRef]
- Hedglin, M.; Pandey, B.; Benkovic, S.J. Stability of the human polymerase δ holoenzyme and its implications in lagging strand DNA synthesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1777–E1786. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Georgescu, R.E.; Li, H.; O’Donnell, M.E. Structure of eukaryotic DNA polymerase δ bound to the PCNA clamp while encircling DNA. Proc. Natl. Acad. Sci. USA 2020, 117, 30344–30353. [Google Scholar] [CrossRef] [PubMed]
- Billon, P.; Côté, J. Novel mechanism of PCNA control through acetylation of its sliding surface. Mol. Cell. Oncol. 2017, 4, e1279724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De March, M.; Barrera-Vilarmau, S.; Crespan, E.; Mentegari, E.; Merino, N.; Gonzalez-Magaña, A.; Romano-Moreno, M.; Maga, G.; Crehuet, R.; Onesti, S.; et al. P15PAF binding to PCNA modulates the DNA sliding surface. Nucleic Acids Res. 2018, 46, 9816–9828. [Google Scholar] [CrossRef] [PubMed]
- Cazzalini, O.; Sommatis, S.; Tillhon, M.; Dutto, I.; Bachi, A.; Rapp, A.; Nardo, T.; Scovassi, A.I.; Necchi, D.; Cardoso, M.C.; et al. CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res. 2014, 42, 8433–8448. [Google Scholar] [CrossRef] [Green Version]
- Warbrick, E. The puzzle of PCNA’s many partners. BioEssays 2000, 22, 997–1006. [Google Scholar] [CrossRef]
- Maga, G.; Hübscher, U. Proliferating cell nuclear antigen (PCNA): A dancer with many partners. J. Cell Sci. 2003, 116, 3051–3060. [Google Scholar] [CrossRef] [Green Version]
- Prestel, A.; Wichmann, N.; Martins, J.M.; Marabini, R.; Kassem, N.; Broendum, S.S.; Otterlei, M.; Nielsen, O.; Willemoës, M.; Ploug, M.; et al. The PCNA interaction motifs revisited: Thinking outside the PIP-box. Cell. Mol. Life Sci. 2019, 76, 4923–4943. [Google Scholar] [CrossRef] [Green Version]
- Warbrick, E. PCNA binding through a conserved motif. BioEssays 1998, 20, 195–199. [Google Scholar] [CrossRef]
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [Google Scholar] [CrossRef]
- Havens, C.G.; Walter, J.C. Docking of a Specialized PIP Box onto Chromatin-Bound PCNA Creates a Degron for the Ubiquitin Ligase CRL4Cdt2. Mol. Cell 2009, 35, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Gilljam, K.M.; Feyzi, E.; Aas, P.A.; Sousa, M.M.L.; Müller, R.; Vågbø, C.B.; Catterall, T.C.; Liabakk, N.B.; Slupphaug, G.; Drabløs, F.; et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 2009, 186, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Hishiki, A.; Hashimoto, H.; Hanafusa, T.; Kamei, K.; Ohashi, E.; Shimizu, T.; Ohmori, H.; Sato, M. Structural basis for novel interactions between human translesion synthesis polymerases and proliferating cell nuclear antigen. J. Biol. Chem. 2009, 284, 10552–10560. [Google Scholar] [CrossRef] [Green Version]
- Lancey, C.; Tehseen, M.; Raducanu, V.S.; Rashid, F.; Merino, N.; Ragan, T.J.; Savva, C.G.; Zaher, M.S.; Shirbini, A.; Blanco, F.J.; et al. Structure of the processive human Pol δ holoenzyme. Nat. Commun. 2020, 11, 1109. [Google Scholar] [CrossRef] [Green Version]
- Sebesta, M.; Cooper, C.D.O.; Ariza, A.; Carnie, C.J.; Ahel, D. Structural insights into the function of ZRANB3 in replication stress response. Nat. Commun. 2017, 16, 15847. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Uchida, M.; Tagata, R.; Yokoyama, H.; Ishikawa, Y.; Hishiki, A.; Hashimoto, H. Structure of proliferating cell nuclear antigen (PCNA) bound to an APIM peptide reveals the universality of PCNA interaction. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74, 214–221. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, P.; Liu, L.; Lee, M.Y.W.T. A novel PCNA-binding motif identified by the panning of a random peptide display library. Biochemistry 2001, 40, 4512–4520. [Google Scholar] [CrossRef]
- Liang, Z.; Diamond, M.; Smith, J.A.; Schnell, M.; Daniel, R. Proliferating cell nuclear antigen is required for loading of the SMCX/KMD5C histone demethylase onto chromatin. Epigenetics Chromatin 2011, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Boehm, E.M.; Washington, M.T. R.I.P. to the PIP: PCNA-binding motif no longer considered specific: PIP motifs and other related sequences are not distinct entities and can bind multiple proteins involved in genome maintenance. BioEssays 2016, 38, 1117–1122. [Google Scholar] [CrossRef]
- Yoon, M.K.; Venkatachalam, V.; Huang, A.; Choi, B.S.; Stultz, C.M.; Chou, J.J. Residual structure within the disordered C-terminal segment of p21 Waf1/Cip1/Sdi1 and its implications for molecular recognition. Protein Sci. 2009, 18, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Cordeiro, T.N.; Chen, P.C.; De Biasio, A.; Sibille, N.; Blanco, F.J.; Hub, J.S.; Crehuet, R.; Bernadó, P. Disentangling polydispersity in the PCNA-p15PAF complex, a disordered, transient and multivalent macromolecular assembly. Nucleic Acids Res. 2017, 45, 1501–1515. [Google Scholar] [CrossRef]
- Click, T.H.; Ganguly, D.; Chen, J. Intrinsically disordered proteins in a physics-based world. Int. J. Mol. Sci. 2010, 11, 5292–5309. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 11, 5292–5309. [Google Scholar] [CrossRef]
- Johnson, A.; O’Donnell, M. Cellular DNA replicases: Components and dynamics at the replication fork. Annu. Rev. Biochem. 2005, 74, 283–315. [Google Scholar] [CrossRef]
- Erzberger, J.P.; Berger, J.M. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 93–114. [Google Scholar] [CrossRef]
- Lee, K.Y.; Fu, H.; Aladjem, M.I.; Myung, K. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J. Cell Biol. 2013, 200, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.; Gali, V.K.; Takahashi, T.S.; Kubota, T. PCNA Retention on DNA into G2/M Phase Causes Genome Instability in Cells Lacking Elg1. Cell Rep. 2016, 16, 684–695. [Google Scholar] [CrossRef] [Green Version]
- Ogura, T.; Wilkinson, A.J. AAA+ superfamily ATPases: Common structure-diverse function. Genes to Cells 2001, 6, 575–597. [Google Scholar] [CrossRef]
- Davey, M.J.; Jeruzalmi, D.; Kuriyan, J.; O’Donnell, M. Motors and switches: AAA+ machines within the replisome. Nat. Rev. Mol. Cell Biol. 2002, 3, 826–835. [Google Scholar] [CrossRef]
- Majka, J.; Burgers, P.M.J. The PCNA-RFC Families of DNA Clamps and Clamp Loaders. Prog Nucleic Acid Res Mol Biol. 2004, 78, 227–260. [Google Scholar] [CrossRef]
- Miyata, T.; Suzuki, H.; Oyama, T.; Mayanagi, K.; Ishino, Y.; Morikawa, K. Open clamp structure in the clamp-loading complex visualized by electron microscopic image analysis. Proc. Natl. Acad. Sci. USA 2005, 102, 13795–13800. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.; Yoder, B.L.; Burgers, P.M.J.; Benkovic, S.J. The structure of a ring-opened proliferating cell nuclear antigen-replication factor C complex revealed by fluorescence energy transfer. Proc. Natl. Acad. Sci. USA 2006, 103, 2546–2551. [Google Scholar] [CrossRef] [Green Version]
- Sakato, M.; Zhou, Y.; Hingorani, M.M. ATP binding and hydrolysis-driven rate-determining events in the RFC-catalyzed PCNA clamp loading reaction. J. Mol. Biol. 2012, 416, 176–191. [Google Scholar] [CrossRef] [Green Version]
- Bylund, G.O.; Burgers, P.M.J. Replication Protein A-Directed Unloading of PCNA by the Ctf18 Cohesion Establishment Complex. Mol. Cell Biol. 2005, 25, 5445–5455. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.S.; Ryu, E.; Lee, S.W.; Park, J.; Ha, N.Y.; Ra, J.S.; Kim, Y.J.; Kim, J.; Abdel-Rahman, M.; Park, S.H.; et al. Regulation of PCNA cycling on replicating DNA by RFC and RFC-like complexes. Nat. Commun. 2019, 10, 2420. [Google Scholar] [CrossRef]
- Kubota, T.; Nishimura, K.; Kanemaki, M.T.; Donaldson, A.D. The Elg1 Replication Factor C-like Complex Functions in PCNA Unloading during DNA Replication. Mol. Cell 2013, 50, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Shiomi, Y.; Shinozaki, A.; Sugimoto, K.; Usukura, J.; Obuse, C.; Tsurimoto, T. The reconstituted human Chl12-RFC complex functions as a second PCNA loader. Genes Cells 2004, 9, 279–290. [Google Scholar] [CrossRef]
- Kubota, T.; Hiraga, S.I.; Yamada, K.; Lamond, A.I.; Donaldson, A.D. Quantitative proteomic analysis of chromatin reveals that Ctf18 acts in the DNA replication checkpoint. Mol. Cell. Proteom. 2011, 10, M110.005561. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; Katou, Y.; Nakato, R.; Shirahige, K.; Donaldson, A.D. Replication-Coupled PCNA Unloading by the Elg1 Complex Occurs Genome-wide and Requires Okazaki Fragment Ligation. Cell Rep. 2015, 12, 774–787. [Google Scholar] [CrossRef] [Green Version]
- Shemesh, K.; Sebesta, M.; Pacesa, M.; Sau, S.; Bronstein, A.; Parnas, O.; Liefshitz, B.; Venclovas, Č.; Krejci, L.; Kupiec, M. A structure-function analysis of the yeast Elg1 protein reveals the importance of PCNA unloading in genome stability maintenance. Nucleic Acids Res. 2017, 45, 3189–3203. [Google Scholar] [CrossRef] [Green Version]
- Kanellis, P.; Agyei, R.; Durocher, D. Elg1 forms an alternative PCNA-interacting RFC complex required to maintain genome stability. Curr. Biol. 2003, 13, 1583–1595. [Google Scholar] [CrossRef] [Green Version]
- Smolikov, S.; Mazor, Y.; Krauskopf, A. ELG1, a regulator of genome stability, has a role in telomere length regulation and in silencing. Proc. Natl. Acad. Sci. USA 2004, 101, 1656–1661. [Google Scholar] [CrossRef] [Green Version]
- Lengronne, A.; McIntyre, J.; Katou, Y.; Kanoh, Y.; Hopfner, K.P.; Shirahige, K.; Uhlmann, F. Establishment of Sister Chromatid Cohesion at the S. cerevisiae Replication Fork. Mol. Cell 2006, 23, 787–799. [Google Scholar] [CrossRef]
- Yao, N.Y.; Johnson, A.; Bowman, G.D.; Kuriyan, J.; O’Donnell, M. Mechanism of proliferating cell nuclear antigen clamp opening by replication factor C. J. Biol. Chem. 2006, 281, 17528–17539. [Google Scholar] [CrossRef] [Green Version]
- Shibahara, K.I.; Stillman, B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 1999, 96, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Hedglin, M.; Perumal, S.K.; Hu, Z.; Benkovic, S. Stepwise assembly of the human replicative polymerase holoenzyme. Elife 2013, 2, e00278. [Google Scholar] [CrossRef]
- Boehm, E.M.; Gildenberg, M.S.; Washington, M.T. The Many Roles of PCNA in Eukaryotic DNA Replication. Enzymes 2016, 39, 231–254. [Google Scholar] [CrossRef] [Green Version]
- Yeeles, J.T.P.; Janska, A.; Early, A.; Diffley, J.F.X. How the Eukaryotic Replisome Achieves Rapid and Efficient DNA Replication. Mol. Cell 2017, 65, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.R.; Pons, C.; Nguyen, H.D.; Costanzo, M.; Boone, C.; Myers, C.L.; Bielinsky, A.K. Genetic Interactions Implicating Postreplicative Repair in Okazaki Fragment Processing. PLoS Genet. 2015, 11, e1005659. [Google Scholar] [CrossRef] [Green Version]
- Thakar, T.; Leung, W.; Nicolae, C.M.; Clements, K.E.; Shen, B.; Bielinsky, A.K.; Moldovan, G.L. Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat. Commun. 2020, 11, 2147. [Google Scholar] [CrossRef]
- Lujan, S.A.; Williams, J.S.; Kunkel, T.A. DNA Polymerases Divide the Labor of Genome Replication. Trends Cell Biol. 2016, 26, 640–654. [Google Scholar] [CrossRef] [Green Version]
- Kunkel, T.A.; Burgers, P.M.J. Arranging eukaryotic nuclear DNA polymerases for replication: Specific interactions with accessory proteins arrange Pols α, δ, and ϵ in the replisome for leading-strand and lagging-strand DNA replication. BioEssays 2017, 38, 1700070. [Google Scholar] [CrossRef]
- Kunkel, T.A. Evolving views of DNA replication (in)fidelity. Cold Spring Harb. Symp. Quant. Biol. 2009, 74, 91–101. [Google Scholar] [CrossRef]
- Flood, C.L.; Rodriguez, G.P.; Bao, G.; Shockley, A.H.; Kow, Y.W.; Crouse, G.F. Replicative DNA Polymerase δ but Not ε Proofreads Errors in Cis and in Trans. PLoS Genet. 2015, 11, e1005049. [Google Scholar] [CrossRef] [Green Version]
- Bulock, C.R.; Xing, X.; Shcherbakova, P.V. DNA polymerase δ proofreads errors made by DNA polymerase e. Proc. Natl. Acad. Sci. USA 2020, 117, 6035–6041. [Google Scholar] [CrossRef] [Green Version]
- Guilliam, T.A.; Yeeles, J.T. The eukaryotic replisome tolerates leading-strand base damage by replicase switching. EMBO J. 2021, 40, e107037. [Google Scholar] [CrossRef]
- Guilliam, T.A. Mechanisms for Maintaining Eukaryotic Replisome Progression in the Presence of DNA Damage. Front. Mol. Biosci. 2021, 8, 712971. [Google Scholar] [CrossRef]
- Mejlvang, J.; Feng, Y.; Alabert, C.; Neelsen, K.J.; Jasencakova, Z.; Zhao, X.; Lees, M.; Sandelin, A.; Pasero, P.; Lopes, M.; et al. New histone supply regulates replication fork speed and PCNA unloading. J. Cell Biol. 2014, 204, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Prakash, S.; Johnson, R.E.; Prakash, L. Eukaryotic translesion synthesis DNA polymerases: Specificity of structure and function. Annu. Rev. Biochem. 2005, 74, 317–353. [Google Scholar] [CrossRef]
- Lehmann, A.R.; Niimi, A.; Ogi, T.; Brown, S.; Sabbioneda, S.; Wing, J.F.; Kannouche, P.L.; Green, C.M. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair 2007, 6, 891–899. [Google Scholar] [CrossRef]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [Green Version]
- Washington, M.T.; Carlson, K.D.; Freudenthal, B.D.; Pryor, J.M. Variations on a theme: Eukaryotic Y-family DNA polymerases. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Boiteux, S.; Jinks-Robertson, S. DNA repair mechanisms and the bypass of DNA damage in Saccharomyces cerevisiae. Genetics 2013, 193, 1025–1064. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, C.W. Cellular roles of DNA polymerase ζ and Rev1 protein. DNA Repair 2002, 1, 425–435. [Google Scholar] [CrossRef]
- Guilliam, T.A.; Yeeles, J.T.P. Reconstitution of translesion synthesis reveals a mechanism of eukaryotic DNA replication restart. Nat. Struct. Mol. Biol. 2020, 27, 450–460. [Google Scholar] [CrossRef]
- Pryor, J.M.; Gakhar, L.; Washington, M.T. Structure and Functional Analysis of the BRCT Domain of Translesion Synthesis DNA Polymerase Rev1. Biochemistry 2013, 52, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Hedglin, M.; Benkovic, S.J. Eukaryotic Translesion DNA Synthesis on the Leading and Lagging Strands: Unique Detours around the Same Obstacle. Chem. Rev. 2017, 117, 7857–7877. [Google Scholar] [CrossRef] [Green Version]
- Prakash, S.; Prakash, L. Translesion DNA synthesis in eukaryotes: A one- or two-polymerase affair. Genes Dev. 2002, 16, 1872–1883. [Google Scholar] [CrossRef] [Green Version]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Woodgate, R. A plethora of lesion-replicating DNA polymerases. Genes Dev. 1999, 13, 2191–2195. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.; Lawrence, C.; Hinkle, D. Thymine-Thymine Dimer Bypass by Yeast DNA Polymerase ζ. Science 1996, 272, 1646–1649. [Google Scholar] [CrossRef]
- Baranovskiy, A.G.; Lada, A.G.; Siebler, H.M.; Zhang, Y.; Pavlov, Y.I.; Tahirov, T.H. DNA polymerase δ and ζ switch by sharing accessory subunits of DNA polymerase δ. J. Biol. Chem. 2012, 287, 17281–17287. [Google Scholar] [CrossRef] [Green Version]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef] [Green Version]
- Makarova, A.V.; Burgers, P.M. Eukaryotic DNA polymerase ζ. DNA Repair 2015, 29, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Llorente, Y.; Malik, R.; Jain, R.; Choudhury, J.R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Ubarretxena-Belandia, I.; Aggarwal, A.K. The Architecture of Yeast DNA Polymerase ζ. Cell Rep. 2013, 5, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Malik, R.; Kopylov, M.; Gomez-Llorente, Y.; Jain, R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Ubarretxena-Belandia, I.; Aggarwal, A.K. Structure and mechanism of B-family DNA polymerase ζ specialized for translesion DNA synthesis. Nat. Struct. Mol. Biol. 2020, 27, 913–924. [Google Scholar] [CrossRef]
- Netz, D.J.A.; Stith, C.M.; Stümpfig, M.; Köpf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.J.; Pierik, A.J. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 2012, 8, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Johansson, E.; Garg, P.; Burgers, P.M.J. The Pol32 Subunit of DNA Polymerase δ Contains Separable Domains for Processive Replication and Proliferating Cell Nuclear Antigen (PCNA) Binding. J. Biol. Chem. 2004, 279, 1907–1915. [Google Scholar] [CrossRef] [Green Version]
- Powers, K.; Washington, M.T. Analyzing the catalytic activities and interactions of eukaryotic translesion synthesis polymerases. Methods Enzym. 2017, 592, 329–356. [Google Scholar] [CrossRef] [Green Version]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [Green Version]
- Kraszewska, J.; Garbacz, M.; Jonczyk, P.; Fijalkowska, I.J.; Jaszczur, M. Defect of Dpb2p, a noncatalytic subunit of DNA polymerase e, promotes error prone replication of undamaged chromosomal DNA in Saccharomyces cerevisiae. Mutat. Res.—Fundam. Mol. Mech. Mutagen. 2012, 737, 34–42. [Google Scholar] [CrossRef]
- Grabowska, E.; Wronska, U.; Denkiewicz, M.; Jaszczur, M.; Respondek, A.; Alabrudzinska, M.; Suski, C.; Makiela-Dzbenska, K.; Jonczyk, P.; Fijalkowska, I.J. Proper functioning of the GINS complex is important for the fidelity of DNA replication in yeast. Mol. Microbiol. 2014, 92, 659–680. [Google Scholar] [CrossRef]
- Garbacz, M.; Araki, H.; Flis, K.; Bebenek, A.; Zawada, A.E.; Jonczyk, P.; Makiela-Dzbenska, K.; Fijalkowska, I.J. Fidelity consequences of the impaired interaction between DNA polymerase epsilon and the GINS complex. DNA Repair 2015, 29, 23–35. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, S.; Kokoska, J.; Masutani, C.; Iwai, S.; Hanaoka, F. Preferential cis–syn thymine dimer bypass by DNA polymerase η occurs with biased fidelity. Nature 2004, 428, 97–100. [Google Scholar] [CrossRef]
- Johnson, R.E.; Prakash, S.; Prakash, L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science 1999, 283, 1001–1004. [Google Scholar] [CrossRef]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 1999, 399, 700–704. [Google Scholar] [CrossRef]
- Ohmori, H.; Hanafusa, T.; Ohashi, E.; Vaziri, C. Separate roles of structured and unstructured regions of Y-family DNA polymerases. Adv. Protein Chem. Struct. Biol. 2009, 78, 99–146. [Google Scholar] [CrossRef] [Green Version]
- Boehm, E.M.; Spies, M.; Washington, M.T. PCNA tool belts and polymerase bridges form during translesion synthesis. Nucleic Acids Res. 2016, 44, 8250–8260. [Google Scholar] [CrossRef] [Green Version]
- Boehm, E.M.; Powers, K.T.; Kondratick, C.M.; Spies, M.; Houtman, J.C.D.; Washington, M.T. The proliferating cell nuclear antigen (PCNA)-interacting Protein (PIP) motif of DNA polymerase η Mediates Its interaction with the C-terminal domain of Rev1. J. Biol. Chem. 2016, 291, 8735–8744. [Google Scholar] [CrossRef] [Green Version]
- Ripley, B.M.; Reusch, D.T.; Washington, M.T. Yeast DNA polymerase η possesses two PIP-like motifs that bind PCNA and Rad6-Rad18 with different specificities. DNA Repair 2020, 95, 102968. [Google Scholar] [CrossRef]
- Haracska, L.; Kondratick, C.M.; Unk, I.; Prakash, S.; Prakash, L. Interaction with PCNA is essential for yeast DNA polymerase η function. Mol. Cell 2001, 8, 407–415. [Google Scholar] [CrossRef]
- Guo, C.; Sonoda, E.; Tang, T.S.; Parker, J.L.; Bielen, A.B.; Takeda, S.; Ulrich, H.D.; Friedberg, E.C. REV1 Protein Interacts with PCNA: Significance of the REV1 BRCT Domain In Vitro and In Vivo. Mol. Cell 2006, 23, 265–271. [Google Scholar] [CrossRef]
- Pustovalova, Y.; MacIejewski, M.W.; Korzhnev, D.M. NMR mapping of PCNA interaction with translesion synthesis DNA polymerase Rev1 mediated by Rev1-BRCT domain. J. Mol. Biol. 2013, 425, 3091–3105. [Google Scholar] [CrossRef]
- Bienko, M.; Green, C.M.; Crosetto, N.; Rudolf, F.; Zapart, G.; Coull, B.; Kannouche, P.; Wider, G.; Peter, M.; Lehmann, A.R.; et al. Biochemistry: Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 2005, 310, 1821–1824. [Google Scholar] [CrossRef]
- Ohashi, E.; Murakumo, Y.; Kanjo, N.; Akagi, J.I.; Masutani, C.; Hanaoka, F.; Ohmori, H. Interaction of hREV1 with three human Y-family DNA polymerases. Genes to Cells 2004, 9, 523–531. [Google Scholar] [CrossRef]
- Ohashi, E.; Hanafusa, T.; Kamei, K.; Song, I.; Tomida, J.; Hashimoto, H.; Vaziri, C.; Ohmori, H. Identification of a novel REV1-interacting motif necessary for DNA polymerase κ function. Genes Cells 2009, 14, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Wojtaszek, J.; Liu, J.; D’Souza, S.; Wang, S.; Xue, Y.; Walker, G.C.; Zhou, P. Multifaceted recognition of vertebrate Rev1 by translesion polymerases ζ and κ. J. Biol. Chem. 2012, 287, 26400–26408. [Google Scholar] [CrossRef] [Green Version]
- Wojtaszek, J.; Lee, C.J.; D’Souza, S.; Minesinger, B.; Kim, H.; D’Andrea, A.D.; Walker, G.C.; Zhou, P. Structural basis of rev1-mediated assembly of a quaternary vertebrate translesion polymerase complex consisting of Rev1, heterodimeric polymerase (Pol) ζ, and Pol κ. J. Biol. Chem. 2012, 287, 33836–33846. [Google Scholar] [CrossRef] [Green Version]
- Fleck, O.; Schär, P. Translesion DNA synthesis: Little fingers teach tolerance. Curr. Biol. 2004, 14, 389–391. [Google Scholar] [CrossRef] [Green Version]
- Sale, J.E. Competition, collaboration and coordination—Determining how cells bypass DNA damage. J. Cell Sci. 2012, 125, 1633–1643. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.C.; Jackson, M.A.; Pata, J.D. Y-family polymerase conformation is a major determinant of fidelity and translesion specificity. Structure 2013, 21, 20–31. [Google Scholar] [CrossRef] [Green Version]
- Boudsocq, F.; Kokoska, R.J.; Plosky, B.B.; Vaisman, A.; Ling, H.; Kunkel, T.A.; Yang, W.; Woodgate, R. Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. J. Biol. Chem. 2004, 279, 32932–32940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCulloch, S.; Kunkel, T.A. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008, 18, 148–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoege, C.; Pfander, B.; Moldovan, G.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Stelter, P.; Ulrich, H.D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 2003, 425, 188–191. [Google Scholar] [CrossRef]
- Plosky, B.S.; Vidal, A.E.; De Henestrosa, A.R.F.; McLenigan, M.P.; McDonald, J.P.; Mead, S.; Woodgate, R. Controlling the subcellular localization of DNA polymerases ι and η via interactions with ubiquitin. EMBO J. 2006, 25, 2847–2855. [Google Scholar] [CrossRef]
- Garg, P.; Burgers, P.M. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases η and REV1. Proc. Natl. Acad. Sci. USA 2005, 102, 18361–18366. [Google Scholar] [CrossRef] [Green Version]
- Cox, B.; Parry, J. The isolation, genetics and survival characteristics of ultraviolet light-sensitive mutants in yeast. Mutat. Res. Mol. Mech. Mutagen. 1968, 6, 37–55. [Google Scholar] [CrossRef]
- Game, J.C. and Mortimer, R.K. A genetic study of x-ray sensitive mutants in yeast. Mutat Res 1974, 24, 281–292. [Google Scholar] [CrossRef]
- Prakash, L. Lack of chemically induced mutation in repair-deficient mutants of yeast. Genetics 1974, 78, 1101–1118. [Google Scholar] [CrossRef]
- Game, J.C.; Zamb, T.; Braun, R.; Resnick, M.; Roth, R. The role of the radiation (rad) genes in meiotic recombination in yeast. Genetics 1980, 94, 51–68. [Google Scholar] [CrossRef]
- Montelone, B.A.; Prakash, S.; Prakash, L. Recombination and mutagenesis in rad6 mutants of Saccharomyces cerevisiae: Evidence for multiple functions of the RAD6 gene. Mol. Gen. Genet. 1981, 184, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Berleth, E.; Pickart, C.; Prakash, S.; Prakash, L. Yeast RAD6 encoded ubiquitin conjugating enzyme mediates protein degradation dependent on the N-end-recognizing E3 enzyme. EMBO J. 1991, 10, 2187–2193. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.; Krogan, N.J.; Dover, J.; Schneider, J.; Heidt, J.; Boateng, M.A.; Dean, K.; Golshani, A.; Zhang, Y.; Greenblatt, J.F.; et al. Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol. Cell 2003, 11, 267–274. [Google Scholar] [CrossRef]
- Bartel, B.; Wunning, I.; Varshavsky, A. The recognition component of the N-end rule pathway. EMBO J. 1990, 9, 3179–3189. [Google Scholar] [CrossRef]
- Broomfield, S.; Hryciw, T.; Xiao, W. DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae. Mutat. Res. Repair 2001, 9, 167–184. [Google Scholar] [CrossRef]
- Tateishi, S.; Sakuraba, O.; Masuyama, S.; Inoue, R.; Yamaizumi, M. Dysfunction of human Rad18 results in defective postreplication repair and hypersensitivity to multiple mutagens. Proc. Natl. Acad. Sci. USA 2000, 97, 7927–7932. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.A.; Salerno, P.E.; Bezsonova, I.; Korzhnev, D.M. NMR Structure of the Human Rad18 Zinc Finger in Complex with Ubiquitin Defines a Class of UBZ Domains in Proteins Linked to the DNA Damage Response. Biochemistry 2014, 53, 5895–5906. [Google Scholar] [CrossRef]
- Zeman, M.K.; Lin, J.R.; Freire, R.; Cimprich, K.A. DNA damage-specific deubiquitination regulates Rad18 functions to suppress mutagenesis. J. Cell Biol. 2014, 206, 183–197. [Google Scholar] [CrossRef]
- Frittmann, O.; Gali, V.K.; Halmai, M.; Toth, R.; Gyorfy, Z.; Balint, E.; Unk, I. The Zn-finger of Saccharomyces cerevisiae Rad18 and its adjacent region mediate interaction with Rad5. G3 Genes Genomes Genet. 2021, 11, jkab041. [Google Scholar] [CrossRef]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [Green Version]
- Bailly, V.; Lauder, S.; Prakash, S.; Prakash, L. Yeast DNA repair proteins Rad6 and Rad18 form a heterodimer that has ubiquitin conjugating, DNA binding, and ATP hydrolytic activities. J. Biol. Chem. 1997, 272, 23360–23365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly, V.; Lamb, J.; Sung, P.; Prakash, S.; Prakash, L. Specific complex formation between yeast RAD6 and RAD18 proteins: A potential mechanism for targeting RAD6 ubiquitin-conjugating activity to DNA damage sites. Genes Dev. 1994, 8, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedglin, M.; Benkovic, S.J. Replication Protein A Prohibits Diffusion of the PCNA Sliding Clamp along Single-Stranded DNA. Biochemistry 2017, 56, 1824–1835. [Google Scholar] [CrossRef]
- Freudenthala, B.; Brogiea, J.; Gakharb, L.; Kondraticka, C.; Washington, T. Crystal structure of SUMO-modified proliferating cell nuclear antigen. Bone 2011, 406, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indiani, C.; McInerney, P.; Georgescu, R.; Goodman, M.F.; O’Donnell, M. A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol. Cell 2005, 19, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Kath, J.E.; Chang, S.; Scotland, M.K.; Wilbertz, J.H.; Jergic, S.; Dixon, N.E.; Sutton, M.D.; Loparo, J.J. Exchange between Escherichia coli polymerases II and III on a processivity clamp. Nucleic Acids Res. 2015, 44, 1681–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cranford, M.T.; Chu, A.M.; Baguley, J.K.; Bauer, R.J.; Trakselis, M.A. Characterization of a coupled DNA replication and translesion synthesis polymerase supraholoenzyme from archaea. Nucleic Acids Res. 2017, 45, 8329–8340. [Google Scholar] [CrossRef]
- Murakumo, Y.; Ogura, Y.; Ishii, H.; Numata, S.I.; Ichihara, M.; Croce, C.M.; Fishel, R.; Takahashi, M. Interactions in the Error-prone Postreplication Repair Proteins hREV1, hREV3, and hREV7. J. Biol. Chem. 2001, 276, 35644–35651. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Todd Washington, M. Translesion synthesis: Insights into the selection and switching of DNA polymerases. Genes 2017, 8, 24. [Google Scholar] [CrossRef]
- Haracska, L.; Washington, M.T.; Prakash, S.; Prakash, L. Inefficient Bypass of an Abasic Site by DNA Polymerase η. J. Biol. Chem. 2001, 276, 6861–6866. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.L.; Simpson, L.J.; Sale, J.E. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res. 2005, 33, 1280–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmunds, C.E.; Simpson, L.J.; Sale, J.E. PCNA Ubiquitination and REV1 Define Temporally Distinct Mechanisms for Controlling Translesion Synthesis in the Avian Cell Line DT40. Mol. Cell 2008, 30, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Prakash, L.; Prakash, S. Pol31 and Pol32 subunits of yeast DNA polymerase δ are also essential subunits of DNA polymerase ζ. Proc. Natl. Acad. Sci. USA 2012, 109, 12455–12460. [Google Scholar] [CrossRef] [Green Version]
- Waisertreiger, I.; Liston, V.; Menezes, M.; Kim, H.; Lobachev, K.; Stepchenknova, E.; Tahirov, T.; Rogozin, I.; Pavlov, Y. Modulation of mutagenesis in eukaryotes by DNA replication fork dynamics and quality of nucleotide pools. Environ. Mol. Mutagen. 2012, 53, 699–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northam, M.R.; Garg, P.; Baitin, D.M.; Burgers, P.M.J.; Shcherbakova, P.V. A novel function of DNA polymerase ζ regulated by PCNA. EMBO J. 2006, 25, 4316–4325. [Google Scholar] [CrossRef] [Green Version]
- Northam, M.R.; Robinson, H.A.; Kochenova, O.V.; Shcherbakova, P.V. Participation of DNA polymerase ζ in replication of undamaged DNA in Saccharomyces cerevisiae. Genetics 2010, 184, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.R.; Nguyen, H.D.; Wang, X.; Bielinsky, A.K. Mcm10 deficiency causes defective-replisome-induced mutagenesis and a dependency on error-free postreplicative repair. Cell Cycle 2014, 13, 1737–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denkiewicz-Kruk, M.; Jedrychowska, M.; Endo, S.; Araki, H.; Jonczyk, P.; Dmowski, M.; Fijalkowska, I.J. Recombination and pol ζ rescue defective dna replication upon impaired cmg helicase—pol ε interaction. Int. J. Mol. Sci. 2020, 21, 9484. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Walker, G.C. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. PNAS 2006, 103, 8971–8976. [Google Scholar] [CrossRef] [Green Version]
- Plachta, M.; Halas, A.; McIntyre, J.; Sledziewska-Gojska, E. The steady-state level and stability of TLS polymerase eta are cell cycle dependent in the yeast S. cerevisiae. DNA Repair 2015, 29, 147–153. [Google Scholar] [CrossRef]
- Wiltrout, M.E.; Walker, G.C. Proteasomal Regulation of the Mutagenic Translesion DNA Polymerase, Saccharomyces cerevisiae Rev1. DNA Repair 2011, 10, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell. 2006, 21, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Belluscio, L.; van Cappellen, W.A.; Kanaar, R.; Essers, J.; Gottifredi, V. DNA damage induced Pol eta recruitment takes place independently of the cell cycle phase. Cell Cycle 2009, 8, 3340–3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation regulates Rad18-mediated template switch. Nature 2008, 456, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, M.; Zwicky, K.; Follonier, C.; Foiani, M.; Lopes, M.; Branzei, D. Visualization of recombination-mediated damage bypass by template switching. Nat. Struct. Mol. Biol. 2014, 21, 884–892. [Google Scholar] [CrossRef]
- Brusky, J.; Zhu, Y.; Xiao, W. UBC13, a DNA-damage-inducible gene, is a member of the error-free postreplication repair pathway in Saccharomyces cerevisiae. Curr. Genet. 2000, 37, 168–174. [Google Scholar] [CrossRef]
- Xiao, W.; Chow, B.L.; Fontanie, T.; Ma, L.; Bacchetti, S.; Hryciw, T.; Broomfield, S. Genetic interactions between error-prone and error-free postreplication repair pathways in Saccharomyces cerevisiae. Mutat. Res.—DNA Repair 1999, 435, 1–11. [Google Scholar] [CrossRef]
- Takahashi, T.S.; Wollscheid, H.P.; Lowther, J.; Ulrich, H.D. Effects of chain length and geometry on the activation of DNA damage bypass by polyubiquitylated PCNA. Nucleic Acids Res. 2020, 48, 3042–3052. [Google Scholar] [CrossRef] [Green Version]
- Ripley, B.M.; Gildenberg, M.S.; Todd Washington, M. Control of DNA damage bypass by ubiquitylation of PCNA. Genes 2020, 11, 138. [Google Scholar] [CrossRef] [Green Version]
- Chavez, D.A.; Greer, B.H.; Eichman, B.F. The HIRAN domain of helicase-like transcription factor positions the DNA translocase motor to drive efficient DNA fork regression. J. Biol. Chem. 2018, 293, 8484–8494. [Google Scholar] [CrossRef] [Green Version]
- Hishiki, A.; Hara, K.; Ikegaya, Y.; Yokoyama, H.; Shimizu, T.; Sato, M.; Hashimoto, H. Structure of a novel DNA-binding domain of Helicase-like Transcription Factor (HLTF) and its functional implication in DNA damage tolerance. J. Biol. Chem. 2015, 290, 13215–13223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achar, Y.J.; Balogh, D.; Neculai, D.; Juhasz, S.; Morocz, M.; Gali, H.; Dhe-Paganon, S.; Venclovas, Č.; Haracska, L. Human HLTF mediates postreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic Acids Res. 2015, 43, 10277–10291. [Google Scholar] [CrossRef] [PubMed]
- Beyer, D.C.; Ghoneim, M.K.; Spies, M. Structure and Mechanisms of SF2 DNA Helicases. Adv. Exp. Med. Biol. 2013, 767, 47–73. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D.; Jentsch, S. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 2000, 19, 3388–3397. [Google Scholar] [CrossRef] [Green Version]
- Unk, I.; Hajdú, I.; Fátyol, K.; Szakál, B.; Blastyák, A.; Bermudez, V.; Hurwitz, J.; Prakash, L.; Prakash, S.; Haracska, L. Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA 2006, 103, 18107–18112. [Google Scholar] [CrossRef] [Green Version]
- Toth, R.; Balogh, D.; Pinter, L.; Jaksa, G.; Szeplaki, B.; Graf, A.; Gyorfy, Z.; Enyedi, M.Z.; Kiss, E.; Haracska, L.; et al. The Rad5 Helicase and RING Domains Contribute to Genome Stability through their Independent Catalytic Activities. J. Mol. Biol. 2022, 434, 167437. [Google Scholar] [CrossRef]
- Xu, X.; Lin, A.; Zhou, C.; Blackwell, S.R.; Zhang, Y.; Wang, Z.; Feng, Q.; Guan, R.; Hanna, M.D.; Chen, Z.; et al. Involvement of budding yeast Rad5 in translesion DNA synthesis through physical interaction with Rev1. Nucleic Acids Res. 2016, 44, 5231–5245. [Google Scholar] [CrossRef] [Green Version]
- Carlile, C.M.; Pickart, C.M.; Matunis, M.J.; Cohen, R.E. Synthesis of free and proliferating cell nuclear antigen-bound polyubiquitin chains by the RING E3 ubiquitin ligase Rad5. J. Biol. Chem. 2009, 284, 29326–29334. [Google Scholar] [CrossRef] [Green Version]
- Pagès, V.; Bresson, A.; Acharya, N.; Prakash, S.; Fuchs, R.P.; Prakash, L. Requirement of Rad5 for DNA polymerase ζ-dependent translesion synthesis in Saccharomyces cerevisiae. Genetics 2008, 180, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Xu, X.; Zhao, X.; Wang, Q.; Xiao, W.; Guo, Y.; Fu, Y.V. Rad5 coordinates translesion DNA synthesis pathway by recognizing specific DNA structures in saccharomyces cerevisiae. Curr. Genet. 2018, 64, 889–899. [Google Scholar] [CrossRef]
- Gallo, D.; Kim, T.H.; Szakal, B.; Saayman, X.; Narula, A.; Park, Y.; Branzei, D.; Zhang, Z.; Brown, G.W. Rad5 Recruits Error-Prone DNA Polymerases for Mutagenic Repair of ssDNA Gaps on Undamaged Templates. Mol. Cell 2019, 73, 900–914.e9. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Bazán, M.Á.; Gallo-Fernández, M.; Saugar, I.; Jiménez-Martín, A.; Vázquez, M.V.; Tercero, J.A. Rad5 plays a major role in the cellular response to DNA damage during chromosome replication. Cell Rep. 2014, 9, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karras, G.I.; Jentsch, S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 2010, 141, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glineburg, M.R.; Chavez, A.; Agrawal, V.; Brill, S.J.; Johnson, F.B. Resolution by unassisted Top3 points to template switch recombination intermediates during DNA replication. J. Biol. Chem. 2013, 288, 33193–33204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberi, G.; Liberi, G.; Maffioletti, G.; Maffioletti, G.; Lucca, C.; Lucca, C.; Chiolo, I.; Chiolo, I.; Baryshnikova, A.; Baryshnikova, A.; et al. Rad51-dependent DNA structures accumulate at damaged replication forks in. Genes Dev. 2005, 19, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Ehmsen, K.T.; Heyer, W.D. Saccharomyces cerevisiae Mus81-Mms4 is a catalytic, DNA structure-selective endonuclease. Nucleic Acids Res. 2008, 36, 2182–2195. [Google Scholar] [CrossRef] [Green Version]
- Fricke, W.M.; Bastin-Shanower, S.A.; Brill, S.J. Substrate specificity of the Saccharomyces cerevisiae Mus81-Mms4 endonuclease. DNA Repair 2005, 4, 243–251. [Google Scholar] [CrossRef]
- Gonzalez-Huici, V.; Szakal, B.; Urulangodi, M.; Psakhye, I.; Castellucci, F.; Menolfi, D.; Rajakumara, E.; Fumasoni, M.; Bermejo, R.; Jentsch, S.; et al. DNA bending facilitates the error-free DNA damage tolerance pathway and upholds genome integrity. EMBO J. 2014, 33, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Kondratick, C.M.; Washington, M.T.; Spies, M. Making choices: DNA replication fork recovery mechanisms. Semin. Cell Dev. Biol. 2021, 113, 27–37. [Google Scholar] [CrossRef]
- Qiu, S.; Jiang, G.; Cao, L.; Huang, J. Replication Fork Reversal and Protection. Front. Cell Dev. Biol. 2021, 9, 670392. [Google Scholar] [CrossRef]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Fumasoni, M.; Zwicky, K.; Vanoli, F.; Lopes, M.; Branzei, D. Error-Free DNA Damage Tolerance and Sister Chromatid Proximity during DNA Replication Rely on the Polα/Primase/Ctf4 Complex. Mol. Cell 2015, 57, 812–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase i poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Menin, L.; Ursich, S.; Trovesi, C.; Zellweger, R.; Lopes, M.; Longhese, M.P.; Clerici, M. Tel1/ ATM prevents degradation of replication forks that reverse after topoisomerase poisoning. EMBO Rep. 2018, 19, e45535. [Google Scholar] [CrossRef]
- Meng, X.; Zhao, X. Replication fork regression and its regulation. FEMS Yeast Res. 2017, 17, fow110. [Google Scholar] [CrossRef] [PubMed]
- Blastyák, A.; Pintér, L.; Unk, I.; Prakash, L.; Prakash, S.; Haracska, L. Yeast Rad5 Protein Required for Postreplication Repair Has a DNA Helicase Activity Specific for Replication Fork Regression. Mol. Cell 2007, 28, 167–175. [Google Scholar] [CrossRef]
- Shin, S.; Hyun, K.; Kim, J.; Hohng, S. ATP Binding to Rad5 Initiates Replication Fork Reversal by Inducing the Unwinding of the Leading Arm and the Formation of the Holliday Junction. Cell Rep. 2018, 23, 1831–1839. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Dhingra, N.; Wang, Q.; Cheng, C.; Zhu, S.; Tian, X.; Yu, J.; Gong, X.; Li, X.; Zhang, H.; et al. Structural basis for the multi-activity factor Rad5 in replication stress tolerance. Nat. Commun. 2021, 12, 321. [Google Scholar] [CrossRef]
- Sun, W.; Nandi, S.; Osman, F.; Ahn, J.S.; Jakovleska, J.; Lorenz, A.; Whitby, M.C. The FANCM Ortholog Fml1 Promotes Recombination at Stalled Replication Forks and Limits Crossing Over during DNA Double-Strand Break Repair. Mol. Cell 2008, 32, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-F.; Prakash, R.; Saro, D.; Longerich, S.; Niu, H.; Sung, P. Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae Mph1 protein. DNA Repair 2011, 10, 1034–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daee, D.L.; Ferrari, E.; Longerich, S.; Zheng, X.F.; Xue, X.; Branzei, D.; Sung, P.; Myung, K. Rad5-dependent DNA repair functions of the Saccharomyces cerevisiae FANCM protein homolog Mph1. J. Biol. Chem. 2012, 287, 26563–26575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Choi, K.; Bonner, J.N.; Szakal, B.; Chen, Y.H.; Papusha, A.; Saro, D.; Niu, H.; Ira, G.; Branzei, D.; et al. Selective modulation of the functions of a conserved DNA motor by a histone fold complex. Genes Dev. 2015, 29, 1000–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Koyi, C.; Bonner, J.N.; Chiba, T.; Kwon, Y.; Xu, Y.; Sanchez, H.; Wyman, C.; Niu, H.; Zhao, X.; et al. Restriction of Replication Fork Regression Activities by a Conserved SMC Complex. Mol Cell. 2014, 56, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Solé-Soler, R.; Torres-Rosell, J. Smc5/6, an atypical SMC complex with two RING-type subunits. Biochem. Soc. Trans. 2020, 48, 2159–2171. [Google Scholar] [CrossRef]
- Rossi, S.E.; Ajazi, A.; Carotenuto, W.; Foiani, M.; Giannattasio, M. Rad53-Mediated Regulation of Rrm3 and Pif1 DNA Helicases Contributes to Prevention of Aberrant Fork Transitions under Replication Stress. Cell Rep. 2015, 13, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Cotta-Ramusino, C.; Fachinetti, D.; Lucca, C.; Doksani, Y.; Lopes, M.; Sogo, J.; Foiani, M. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol. Cell 2005, 17, 153–159. [Google Scholar] [CrossRef]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity after Replication Stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Vujanovic, M.; Krietsch, J.; Raso, M.C.; Terraneo, N.; Zellweger, R.; Schmid, J.A.; Taglialatela, A.; Huang, J.W.; Holland, C.L.; Zwicky, K.; et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell 2017, 67, 882–890.e5. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.A.; Huttner, D.; Daigaku, Y.; Chen, S.; Ulrich, H.D. Activation of Ubiquitin-Dependent DNA Damage Bypass Is Mediated by Replication Protein A. Mol. Cell 2008, 29, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.; Baxley, R.M.; Moldovan, G.L.; Bielinsky, A.K. Mechanisms of DNA damage tolerance: Post-translational regulation of PCNA. Genes 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das-Bradoo, S.; Nguyen, H.D.; Wood, J.L.; Ricke, R.M.; Haworth, J.C.; Bielinsky, A.K. Defects in DNA ligase i trigger PCNA ubiquitylation at Lys 107. Nat. Cell Biol. 2010, 12, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Nguyen, H.; Becker, J.; Thu, Y.M.; Costanzo, M.; Koch, E.N.; Smith, S.; Myung, K.; Myers, C.L.; Boone, C.; Bielinsky, A.K. Unligated Okazaki Fragments Induce PCNA Ubiquitination and a Requirement for Rad59-Dependent Replication Fork Progression. PLoS ONE 2013, 8, e66379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.; Xu, R.; Mongia, P.; Toyofuku, N.; Nakagawa, T. Fission yeast Rad8/HLTF facilitates Rad52-dependent chromosomal rearrangements through PCNA lysine 107 ubiquitination. PLoS Genet. 2021, 17, e1009671. [Google Scholar] [CrossRef]
- Becker, J.R.; Gallo, D.; Leung, W.; Croissant, T.; Thu, Y.M.; Nguyen, H.D.; Starr, T.K.; Brown, G.W.; Bielinsky, A.K. Flap endonuclease overexpression drives genome instability and DNA damage hypersensitivity in a PCNA-dependent manner. Nucleic Acids Res. 2018, 46, 5634–5650. [Google Scholar] [CrossRef]
- Pfander, B.; Moldovan, G.-L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 2005, 436, 428–433. [Google Scholar] [CrossRef]
- Xu, X.; Blackwell, S.; Lin, A.; Li, F.; Qin, Z.; Xiao, W. Error-free DNA-damage tolerance in Saccharomyces cerevisiae. Mutat. Res.—Rev. Mutat. Res. 2015, 764, 43–50. [Google Scholar] [CrossRef]
- Niu, H.; Klein, H.L. Multifunctional roles of Saccharomyces cerevisiae Srs2 protein in replication, recombination and repair. FEMS Yeast Res. 2017, 17, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, C.W.; Christensen, R.B. Metabolic suppressors of trimethoprim and ultraviolet light sensitivities of Saccharomyces cerevisiae rad6 mutants. J. Bacteriol. 1979, 139, 866–876. [Google Scholar] [CrossRef] [Green Version]
- Schiestl, R.H.; Prakash, S.; Prakash, L. The SRS2 Suppressor of rad6 Mutations of Saccharomyces cerevisiae Acts by Channeling DNA Lesions Into the RAD52 DNA Repair Pathway. Genetics 1990, 124, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Chiolo, I.; Carotenuto, W.; Maffioletti, G.; Petrini, J.H.J.; Foiani, M.; Liberi, G. Srs2 and Sgs1 DNA Helicases Associate with Mre11 in Different Subcomplexes following Checkpoint Activation and CDK1-Mediated Srs2 Phosphorylation. Mol. Cell. Biol. 2005, 25, 5738–5751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.A.; Mohideen, F.; Lima, C.D. Recognition of SUMO-modified PCNA requires tandem receptor motifs in Srs2. Nature 2012, 483, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Papouli, E.; Chen, S.; Davies, A.A.; Huttner, D.; Krejci, L.; Sung, P.; Ulrich, H.D. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 2005, 19, 123–133. [Google Scholar] [CrossRef]
- Burkovics, P.; Sebesta, M.; Sisakova, A.; Plault, N.; Szukacsov, V.; Robert, T.; Pinter, L.; Marini, V.; Kolesar, P.; Haracska, L.; et al. Srs2 mediates PCNA-SUMO-dependent inhibition of DNA repair synthesis. EMBO J. 2013, 32, 742–755. [Google Scholar] [CrossRef] [Green Version]
- Richard, G.F.; Kerrest, A.; Lafontaine, I.; Dujon, B. Comparative genomics of hemiascomycete yeasts: Genes involved in DNA replication, repair, and recombination. Mol. Biol. Evol. 2005, 22, 1011–1023. [Google Scholar] [CrossRef] [Green Version]
- Morishita, T.; Furukawa, F.; Sakaguchi, C.; Toda, T.; Carr, A.M.; Iwasaki, H.; Shinagawa, H. Role of the Schizosaccharomyces pombe F-Box DNA Helicase in Processing Recombination Intermediates. Mol. Cell Biol. 2005, 25, 8074–8083. [Google Scholar] [CrossRef] [Green Version]
- Desterro, J.M.P.; Thomson, J.; Hay, R.T. Ubch9 conjugates SUMO but not ubiquitin. FEBS Lett. 1997, 417, 297–300. [Google Scholar] [CrossRef]
- Tsutakawa, S.E.; Yan, C.; Xu, X.; Weinacht, C.P.; Freudenthal, B.D.; Yang, K.; Zhuang, Z.; Washington, M.T.; Tainer, J.A.; Ivanov, I. Structurally distinct ubiquitin- and sumo-modified PCNA: Implications for their distinct roles in the DNA damage response. Structure 2015, 23, 724–733. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.L.; Ulrich, H.D. A SUMO-interacting motif activates budding yeast ubiquitin ligase Rad18 towards SUMO-modified PCNA. Nucleic Acids Res. 2012, 40, 11380–11388. [Google Scholar] [CrossRef]
- Branzei, D.; Szakal, B. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair 2016, 44, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Urulangodi, M.; Sebesta, M.; Menolfi, D.; Szakal, B.; Sollier, J.; Sisakova, A.; Krejci, L.; Branzei, D. Local regulation of the Srs2 helicase by the SUMO-like domain protein Esc2 promotes recombination at sites of stalled replication. Genes Dev. 2015, 29, 2067–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hishida, T.; Iwasaki, H.; Ohno, T.; Morishita, T.; Shinagawa, H. A yeast gene, MGS1, encoding a DNA-dependent AAA+ ATPase is required to maintain genome stability. Proc. Natl. Acad. Sci. USA 2001, 98, 8283–8289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Martín, A.; Saugar, I.; Joseph, C.R.; Mayer, A.; Lehmann, C.P.; Szakal, B.; Branzei, D.; Tercero, J.A. The Mgs1/WRNIP1 ATPase is required to prevent a recombination salvage pathway at damaged replication forks. Sci. Adv. 2020, 6, eaaz3327. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, C.P.; Jiménez-Martín, A.; Branzei, D.; Tercero, J.A. Prevention of unwanted recombination at damaged replication forks. Curr. Genet. 2020, 66, 1045–1051. [Google Scholar] [CrossRef]
- Arbel, M.; Bronstein, A.; Sau, S.; Liefshitz, B.; Kupiec, M. Access to PCNA by Srs2 and Elg1 Controls the Choice between Alternative Repair Pathways in Saccharomyces cerevisiae. MBio 2020, 11, e00705-20. [Google Scholar] [CrossRef]
- Ivanov, I.; Chapados, B.R.; McCammon, J.A.; Tainer, J.A. Proliferating cell nuclear antigen loaded onto double-stranded DNA: Dynamics, minor groove interactions and functional implications. Nucleic Acids Res. 2006, 34, 6023–6033. [Google Scholar] [CrossRef] [Green Version]
- Billon, P.; Li, J.; Lambert, J.P.; Chen, Y.; Tremblay, V.; Brunzelle, J.S.; Gingras, A.C.; Verreault, A.; Sugiyama, T.; Couture, J.F.; et al. Acetylation of PCNA Sliding Surface by Eco1 Promotes Genome Stability through Homologous Recombination. Mol. Cell 2017, 65, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Slade, D. Maneuvers on PCNA rings during DNA replication and repair. Genes 2018, 9, 416. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, R.; Manohar, K.; Patel, S.K.; Acharya, N.; Vasudevan, D. Structural analyses of PCNA from the fungal pathogen Candida albicans identify three regions with species-specific conformations. FEBS Lett. 2021, 595, 1328–1349. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhang, W.; Liu, C.; Lin, Y.; Wu, Q.; Dai, J. Dissecting PCNA function with a systematically designed mutant library in yeast. J. Genet. Genom. 2019, 46, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Nakajima, Y.; Yu, Y.-L.; Xia, W.; Chen, C.-T.; Yang, C.-C.; McIntush, E.W.; Li, L.-Y.; Hawke, D.H.; Kobayashi, R.; et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat. Cell Biol. 2006, 8, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.; Li, J.Y.; Lee, S.; Tong, D.; Gu, L.; Li, G.M. Phosphorylation of PCNA by EGFR inhibits mismatch repair and promotes misincorporation during DNA synthesis. Proc. Natl. Acad. Sci. USA 2015, 112, 5667–5672. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Lo, Y.-H.; Ma, L.; Walz, S.E.; Gray, Y.K.; Hung, M.-C.; Wang, S.-C. Targeting Tyrosine Phosphorylation of PCNA Inhibits Prostate Cancer Growth. Mol. Cancer Ther. 2011, 10, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, A.; Xu, X.; Wang, C.; Yang, J.; Wang, S.; Dai, J.; Ye, L. EZH2 promotes DNA replication by stabilizing interaction of POLδ and PCNA via methylation-mediated PCNA trimerization. Epigenetics Chromatin 2018, 11, 44. [Google Scholar] [CrossRef] [Green Version]
- Dieckman, L. Something’s gotta give: How PCNA alters its structure in response to mutations and the implications on cellular processes. Prog. Biophys. Mol. Biol. 2021, 163, 46–59. [Google Scholar] [CrossRef]
- Zamir, L.; Zaretsky, M.; Fridman, Y.; Ner-Gaon, H.; Rubin, E.; Aharoni, A. Tight coevolution of proliferating cell nuclear antigen (PCNA)-partner interaction networks in fungi leads to interspecies network incompatibility. Proc. Natl. Acad. Sci. USA 2012, 109, E406-14. [Google Scholar] [CrossRef] [Green Version]
- Manohar, K.; Acharya, N. Characterization of proliferating cell nuclear antigen (PCNA) from pathogenic yeast Candida albicans and its functional analyses in S. Cerevisiae Microbial genetics, genomics and proteomics. BMC Microbiol. 2015, 15, 257. [Google Scholar] [CrossRef] [Green Version]
- Marshall, A.C.; Kroker, A.J.; Murray, L.A.M.; Gronthos, K.; Rajapaksha, H.; Wegener, K.L.; Bruning, J.B. Structure of the sliding clamp from the fungal pathogen Aspergillus fumigatus (AfumPCNA) and interactions with Human p21. FEBS J. 2017, 284, 985–1002. [Google Scholar] [CrossRef] [Green Version]
- Kumari, P.; Sundaram, R.; Manohar, K.; Vasudevan, D.; Acharya, N. Interdomain connecting loop and J loop structures determine cross-species compatibility of PCNA. J. Biol. Chem. 2021, 297, 100911. [Google Scholar] [CrossRef]
- Fridman, Y.; Palgi, N.; Dovrat, D.; Ben-Aroya, S.; Hieter, P.; Aharoni, A. Subtle alterations in PCNA-partner interactions severely impair DNA replication and repair. PLoS Biol. 2010, 8, e1000507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, Y.; Gur, E.; Fleishman, S.J.; Aharoni, A. Computational protein design suggests that human PCNA-partner interactions are not optimized for affinity. Proteins 2013, 81, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Boyce, K.J.; Wang, Y.; Verma, S.; Shakya, V.P.S.; Xue, C.; Idnurm, A. Mismatch repair of DNA replication errors contributes to microevolution in the pathogenic fungus Cryptococcus neoformans. MBio 2017, 8, e00595-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milo, S.; Harari-Misgav, R.; Hazkani-Covo, E.; Covo, S.; Stajich, J. Limited DNA Repair Gene Repertoire in Ascomycete Yeast Revealed by Comparative Genomics. Genome Biol. Evol. 2019, 11, 3409–3423. [Google Scholar] [CrossRef] [PubMed]
- Shor, E.; Garcia-rubio, R.; Degregorio, L.; Perlin, D.S. A Noncanonical DNA Damage Checkpoint Response in a Major Fungal Pathogen. MBio 2020, 11, e03044-20. [Google Scholar] [CrossRef]
- Steenwyk, J.L. Evolutionary divergence in dna damage responses among fungi. MBio 2021, 12, e03348-20. [Google Scholar] [CrossRef]
- Hokken, M.W.J.; Zwaan, B.J.; Melchers, W.J.G.; Verweij, P.E. Facilitators of adaptation and antifungal resistance mechanisms in clinically relevant fungi. Fungal Genet. Biol. 2019, 132, 103254. [Google Scholar] [CrossRef]
- Rodrigues, M.L.; Nosanchuk, J.D. Fungal diseases as neglected pathogens: A wake-up call to public health officials. PLoS Negl. Trop. Dis. 2020, 14, e0007964. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellí, G.; Colomina, N.; Castells-Roca, L.; Lorite, N.P. Post-Translational Modifications of PCNA: Guiding for the Best DNA Damage Tolerance Choice. J. Fungi 2022, 8, 621. https://doi.org/10.3390/jof8060621
Bellí G, Colomina N, Castells-Roca L, Lorite NP. Post-Translational Modifications of PCNA: Guiding for the Best DNA Damage Tolerance Choice. Journal of Fungi. 2022; 8(6):621. https://doi.org/10.3390/jof8060621
Chicago/Turabian StyleBellí, Gemma, Neus Colomina, Laia Castells-Roca, and Neus P. Lorite. 2022. "Post-Translational Modifications of PCNA: Guiding for the Best DNA Damage Tolerance Choice" Journal of Fungi 8, no. 6: 621. https://doi.org/10.3390/jof8060621
APA StyleBellí, G., Colomina, N., Castells-Roca, L., & Lorite, N. P. (2022). Post-Translational Modifications of PCNA: Guiding for the Best DNA Damage Tolerance Choice. Journal of Fungi, 8(6), 621. https://doi.org/10.3390/jof8060621