
The Botrytis cinerea Gene Expression Browser
 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA-Seq Datasets Available for Botrytis cinerea
2.2. Data Pre-Processing and RNA-Seq Experiment Mapping
2.3. Gene Expression Metadata Construction
2.4. Gene Expression Analysis and the BEB Transcriptional Profile Database
2.5. BEB Server Implementation
2.6. Additional Bioinformatics Analyses
3. Results
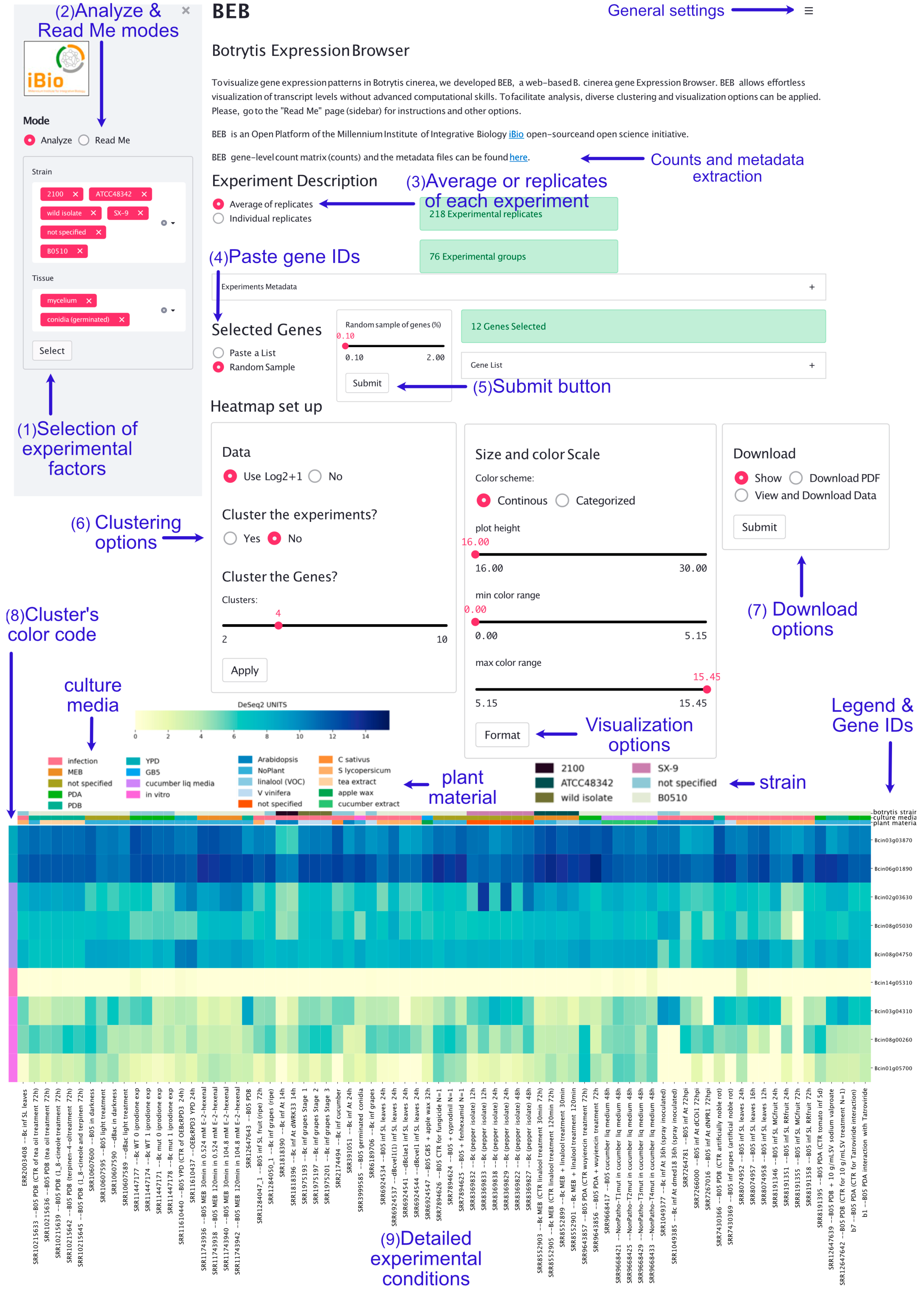
3.1. A Glimpse into the B. cinerea Expression Browser Graphical User Interface
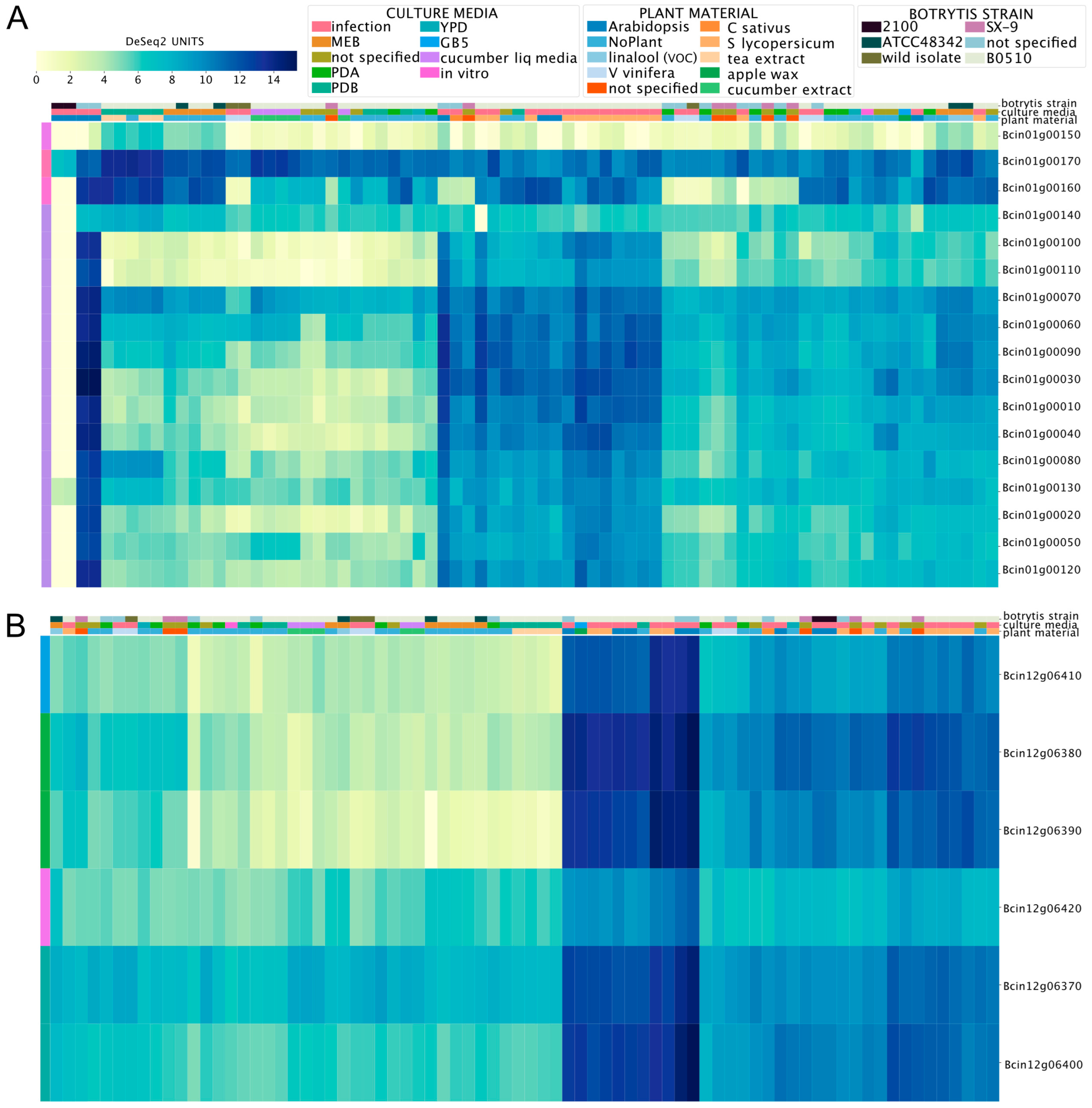
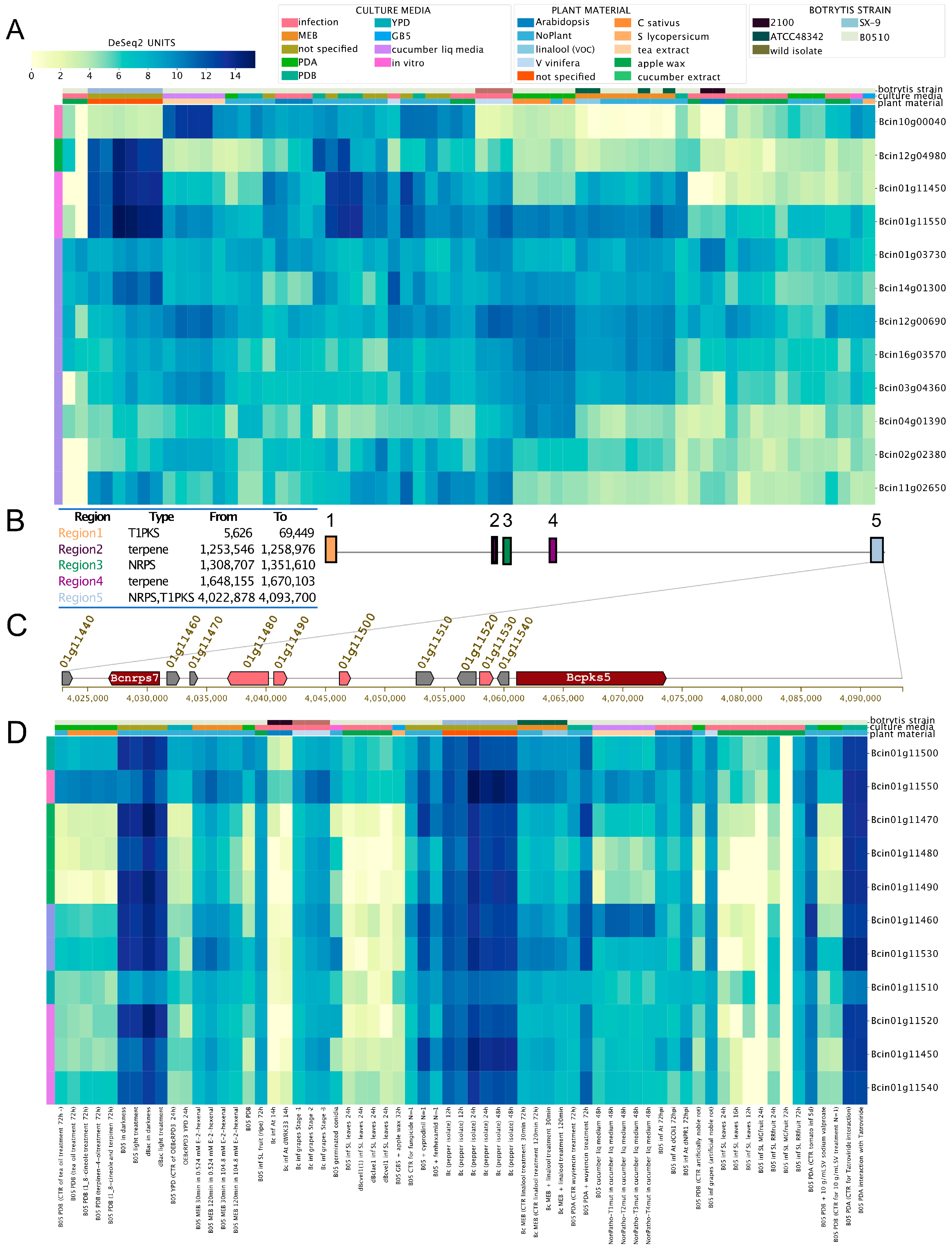
3.2. Global Gene Expression Patterns of Phytotoxic Secondary Metabolite Gene Clusters in B. cinerea
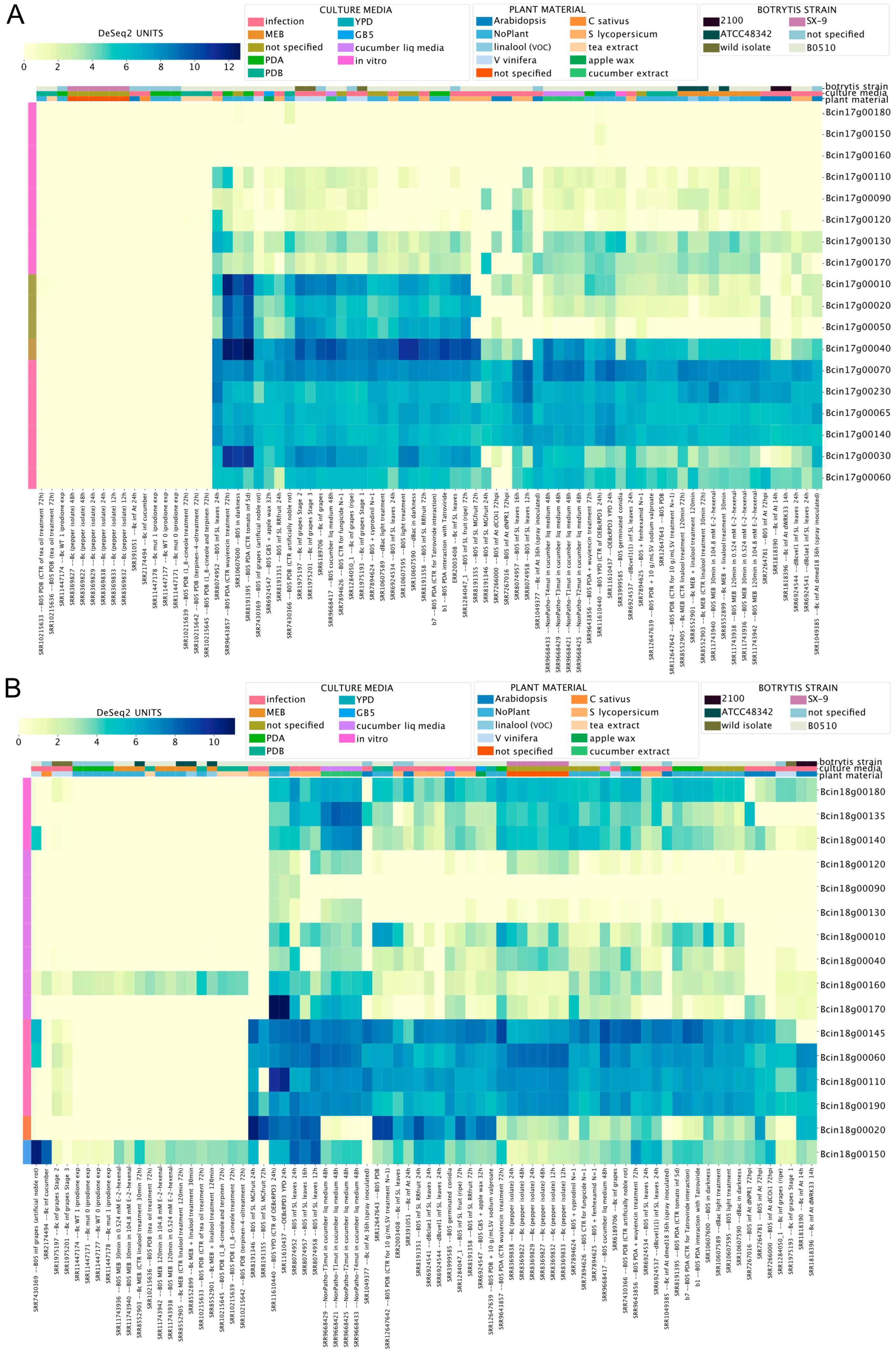
3.3. Gene Expression of Orphan Secondary Metabolite Gene Clusters
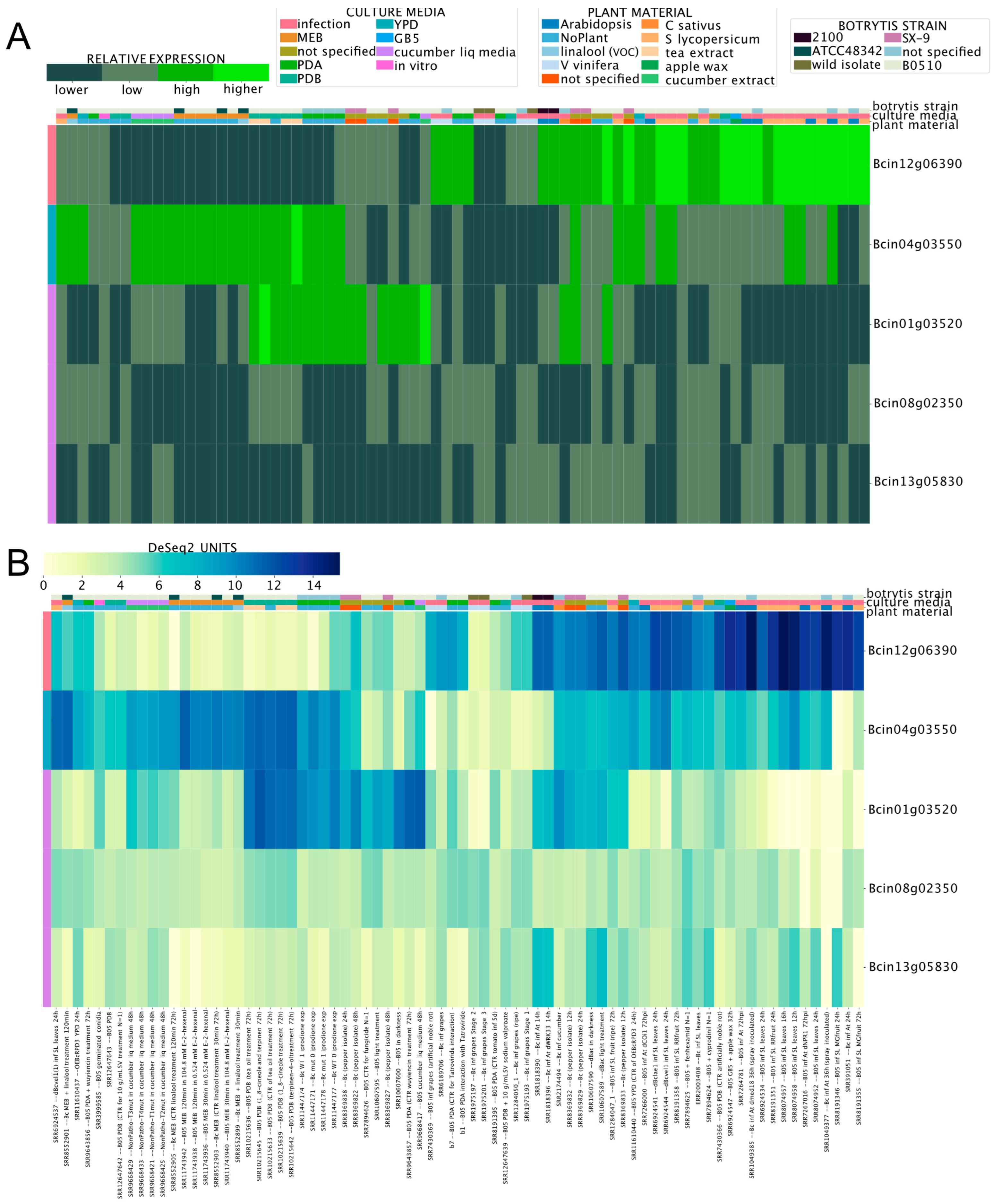
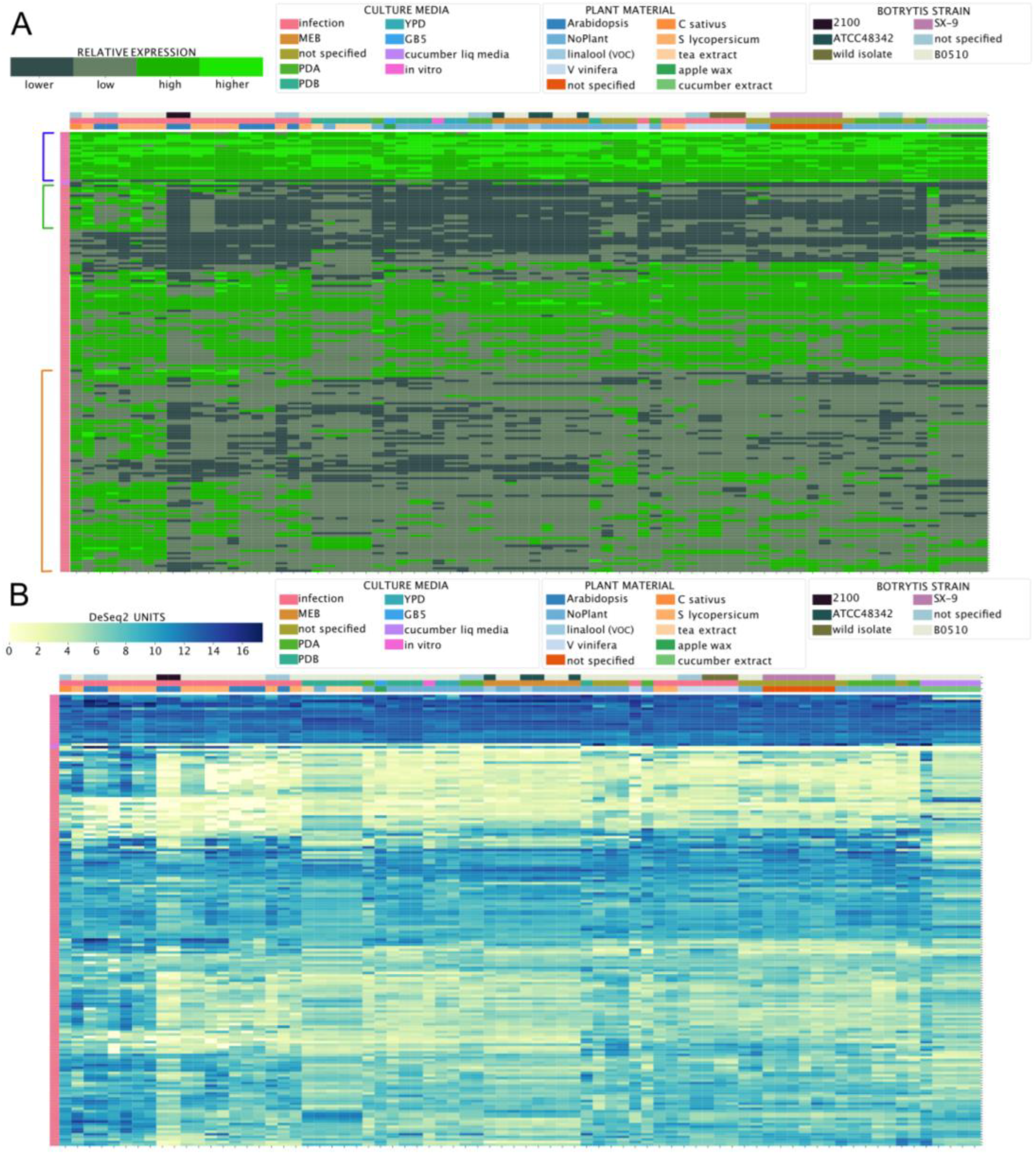
3.4. Chromosome-Wide Gene Expression Analysis
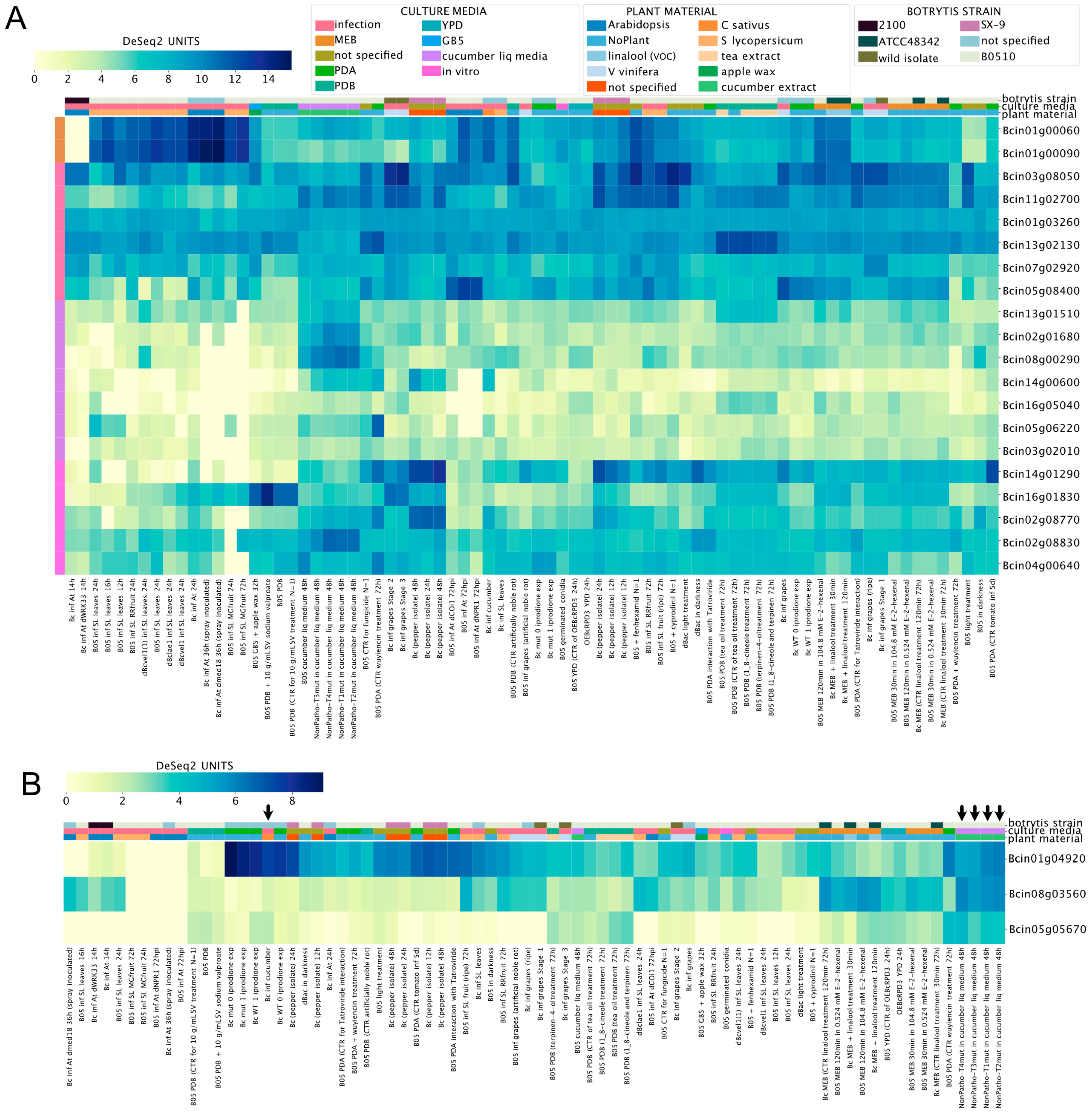
3.5. Inspecting the Expression of Virulence Factors Detected in Proteomics Studies
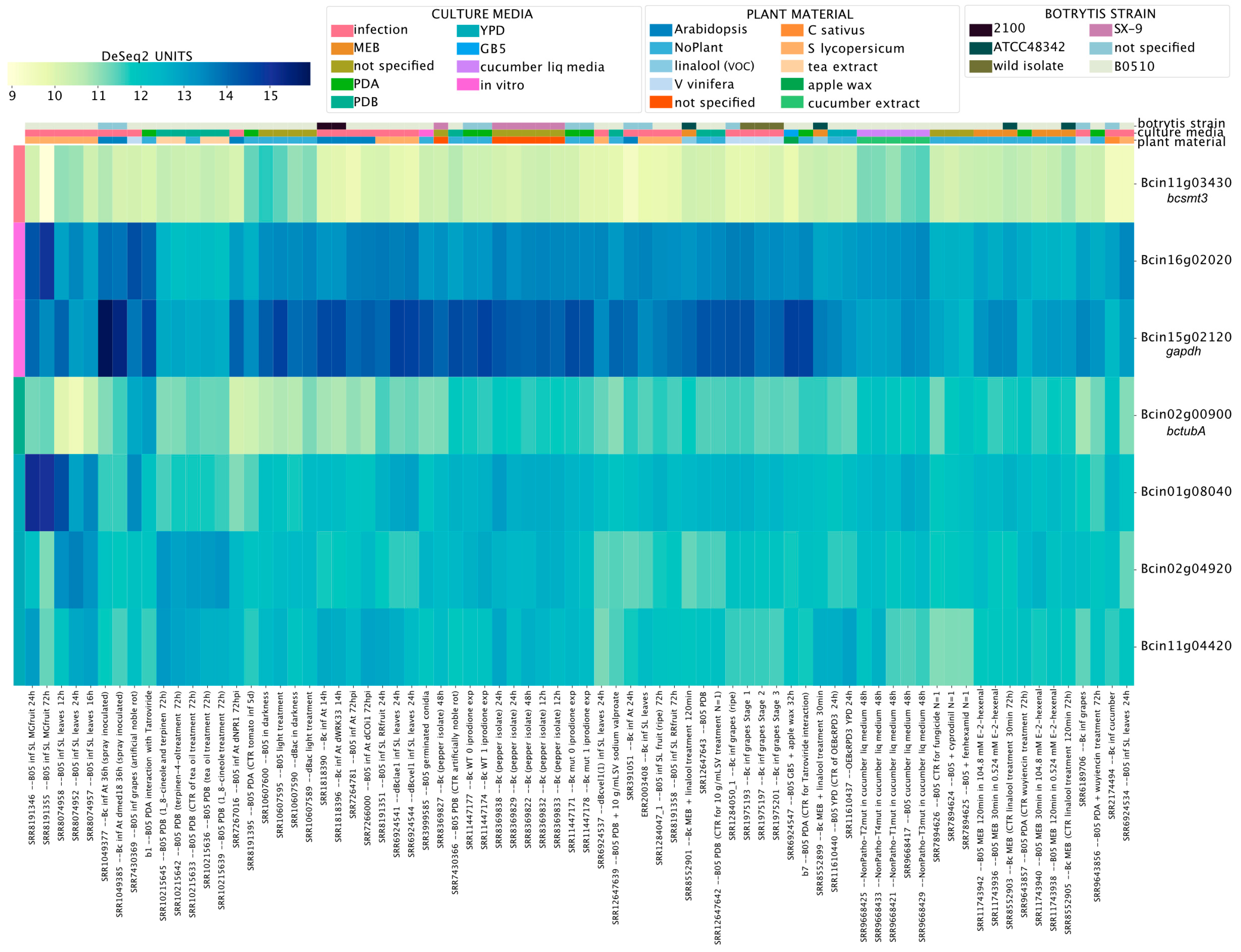
3.6. Revisiting the Expression of Known Genes and Proposing New Reference Genes for Transcript-Level Analyses in B. cinerea
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef]
- Sheng, Q.; Vickers, K.; Zhao, S.; Wang, J.; Samuels, D.C.; Koues, O.; Shyr, Y.; Guo, Y. Multi-perspective quality control of Illumina RNA sequencing data analysis. Brief. Funct. Genom. 2017, 16, 194–204. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Chen, Y.; Lun, A.T.; Smyth, G.K. From reads to genes to pathways: Differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research 2016, 5, 1438. [Google Scholar]
- Dorado, G.; Gálvez, S.; Rosales, T.E.; Vásquez, V.F.; Hernández, P. Analyzing Modern Biomolecules: The Revolution of Nucleic-Acid Sequencing—Review. Biomolecules 2021, 11, 1111. [Google Scholar] [CrossRef] [PubMed]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef] [PubMed]
- Bayega, A.; Fahiminiya, S.; Oikonomopoulos, S.; Ragoussis, J. Current and Future Methods for mRNA Analysis: A Drive Toward Single Molecule Sequencing. Methods Mol. Biol. 2018, 1783, 209–241. [Google Scholar]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef]
- Sullivan, A.; Purohit, P.K.; Freese, N.H.; Pasha, A.; Esteban, E.; Waese, J.; Wu, A.; Chen, M.; Chin, C.Y.; Song, R.; et al. An ’eFP-Seq Browser’ for visualizing and exploring RNA sequencing data. Plant J. 2019, 100, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.J.; Tamiru, M.; Salby, R.; Bolitho, C.; Williams, A.; Huggard, S.; Fisch, E.; Unsworth, K.; Whelan, J.; Lewsey, M.G. AgriSeqDB: An online RNA-Seq database for functional studies of agriculturally relevant plant species. BMC Plant Biol. 2018, 18, 200. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, I.; Moreno, P.; Manning, J.; Fuentes, A.M.-P.; George, N.; Fexova, S.; Fonseca, N.A.; Füllgrabe, A.; Green, M.; Huang, N.; et al. Expression Atlas update: From tissues to single cells. Nucleic Acids Res. 2020, 48, D77–D83. [Google Scholar] [CrossRef]
- Case, N.T.; Heitman, J.; Cowen, L.E. The Rise of Fungi: A Report on the CIFAR Program Fungal Kingdom: Threats & Opportunities Inaugural Meeting. G3 Genes|Genomes|Genet. 2020, 10, 1837–1842. [Google Scholar]
- Adams, T.M.; Olsson, T.S.G.; Ramírez-González, R.H.; Bryant, R.; Bryson, R.; Campos, P.E.; Fenwick, P.; Feuerhelm, D.; Hayes, C.; Henriksson, T.; et al. Rust expression browser: An open source database for simultaneous analysis of host and pathogen gene expression profiles with expVIP. BMC Genom. 2021, 22, 166. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Gurr, S.J.; Cuomo, C.A.; Blehert, D.S.; Jin, H.; Stukenbrock, E.H.; Stajich, J.E.; Kahmann, R.; Boone, C.; Denning, D.W.; et al. Threats Posed by the Fungal Kingdom to Humans, Wildlife, and Agriculture. mBio 2020, 11, e00449-20. [Google Scholar] [CrossRef]
- Couch, B.C.; Fudal, I.; Lebrun, M.H.; Tharreau, D.; Valent, B.; van Kim, P.; Nottéghem, J.L.; Kohn, L.M. Origins of host-specific populations of the blast pathogen Magnaporthe oryzae in crop domestication with subsequent expansion of pandemic clones on rice and weeds of rice. Genetics 2005, 170, 613–630. [Google Scholar] [CrossRef]
- Dean, R.; Van, K.J.A.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di, P.A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Weiberg, A.; Wang, M.; Lin, F.M.; Zhao, H.; Zhang, Z.; Kaloshian, I.; Huang, H.D.; Jin, H. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 2013, 342, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Staats, M.; van Baarlen, P.; van Kan, J.A.L. Molecular phylogeny of the plant pathogenic genus Botrytis and the evolution of host specificity. Mol. Biol. Evol. 2005, 22, 333–346. [Google Scholar] [CrossRef]
- Veloso, J.; van Kan, J.A.L. Many Shades of Grey in Botrytis-Host Plant Interactions. Trends Plant Sci 2018, 23, 613–622. [Google Scholar] [CrossRef]
- van Kan, J.A.L. Licensed to kill: The lifestyle of a necrotrophic plant pathogen. Trends Plant Sci. 2006, 11, 247–253. [Google Scholar] [CrossRef]
- Choquer, M.; Fournier, E.; Kunz, C.; Levis, C.; Pradier, J.M.; Simon, A.; Viaud, M. Botrytis cinerea virulence factors: New insights into a necrotrophic and polyphageous pathogen. FEMS Microbiol. Lett. 2007, 277, 1–10. [Google Scholar] [CrossRef]
- Shlezinger, N.; Doron, A.; Sharon, A. Apoptosis-like programmed cell death in the grey mould fungus Botrytis cinerea: Genes and their role in pathogenicity. Biochem. Soc. Trans. 2011, 39, 1493–1498. [Google Scholar] [CrossRef]
- Mbengue, M.; Navaud, O.; Peyraud, R.; Barascud, M.; Badet, T.; Vincent, R.; Barbacci, A.; Raffaele, S. Emerging Trends in Molecular Interactions between Plants and the Broad Host Range Fungal Pathogens Botrytis cinerea and Sclerotinia sclerotiorum. Front. Plant Sci. 2016, 7, 422. [Google Scholar] [CrossRef]
- Castillo, L.; Plaza, V.; Larrondo, L.F.; Canessa, P. Recent Advances in the Study of the Plant Pathogenic Fungus Botrytis cinerea and its Interaction with the Environment. Curr. Protein Pept. Sci. 2017, 18, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J. How light affects the life of Botrytis. Fungal Genet. Biol. 2017, 106, 26–41. [Google Scholar] [CrossRef]
- Larrondo, L.F.; Canessa, P. The Clock Keeps on Ticking: Emerging Roles for Circadian Regulation in the Control of Fungal Physiology and Pathogenesis. Curr. Top. Microbiol. Immunol. 2019, 422, 121–156. [Google Scholar] [PubMed]
- Cheung, N.; Tian, L.; Liu, X.; Li, X. The Destructive Fungal Pathogen Botrytis cinerea-Insights from Genes Studied with Mutant Analysis. Pathogens 2020, 9, 923. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T.; Sherif, S.M. RNAi-Based Biofungicides as a Promising Next-Generation Strategy for Controlling Devastating Gray Mold Diseases. Int. J. Mol. Sci. 2020, 21, 2072. [Google Scholar] [CrossRef]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.L.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef]
- Staats, M.; van Kan, J.A.L. Genome update of Botrytis cinerea strains B05.10 and T4. Eukaryot. Cell 2012, 11, 1413–1414. [Google Scholar] [CrossRef]
- Van, K.J.A.; Stassen, J.H.; Mosbach, A.; Van, D.L.T.A.; Faino, L.; Farmer, A.D.; Papasotiriou, D.G.; Zhou, S.; Seidl, M.F.; Cottam, E.; et al. A gapless genome sequence of the fungus Botrytis cinerea. Mol. Plant Pathol. 2017, 18, 75–89. [Google Scholar] [CrossRef]
- Isolation and identification of the principal siderophore of the plant pathogenic fungus Botrytis cinerea. Biol. Met. 1998, 1, 90–98.
- Vasquez-Montaño, E.; Hoppe, G.; Vega, A.; Olivares-Yañez, C.; Canessa, P. Defects in the Ferroxidase That Participates in the Reductive Iron Assimilation System Results in Hypervirulence in Botrytis cinerea. mBio 2020, 11, e01379-20. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Contreras-Moreira, B.; De Silva, N.; Maslen, G.; Akanni, W.; Allen, J.; Alvarez-Jarreta, J.; Barba, M.; Bolser, D.M.; Cambell, L.; et al. Ensembl Genomes 2020 enabling non-vertebrate genomic research. Nucleic Acids Res. 2019, 48, D689–D695. [Google Scholar] [CrossRef] [PubMed]
- Sarantopoulou, D.; Brooks, T.G.; Nayak, S.; Mrčela, A.; Lahens, N.F.; Grant, G.R. Comparative evaluation of full-length isoform quantification from RNA-Seq. BMC Bioinform. 2021, 22, 266. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Merkel, D. Docker: Lightweight linux containers for consistent development and deployment. Linux J. 2014, 2014, 2. [Google Scholar]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- McKinney, W. Data Structures for Statistical Computing in Python. Proc. 9th Python Sci. Conf. 2010, 445, 51–56. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Carmona, R.; Arroyo, M.; Jiménez-Quesada, M.J.; Seoane, P.; Zafra, A.; Larrosa, R.; Alché, J.D.; Claros, M.G. Automated identification of reference genes based on RNA-seq data. Biomed. Eng. Online 2017, 16, 65. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pombo, M.A.; Zheng, Y.; Fei, Z.; Martin, G.B.; Rosli, H.G. Use of RNA-seq data to identify and validate RT-qPCR reference genes for studying the tomato-Pseudomonas pathosystem. Sci. Rep. 2017, 7, 44905. [Google Scholar] [CrossRef]
- Tilli, T.M.; Castro, C.S.; Tuszynski, J.A.; Carels, N. A strategy to identify housekeeping genes suitable for analysis in breast cancer diseases. BMC Genom. 2016, 17, 639. [Google Scholar] [CrossRef]
- Winter, D.; Vinegar, B.; Nahal, H.; Ammar, R.; Wilson, G.V.; Provart, N.J. An Electronic Fluorescent Pictograph browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2007, 2, e718. [Google Scholar] [CrossRef]
- Basenko, E.Y.; Pulman, J.A.; Shanmugasundram, A.; Harb, O.S.; Crouch, K.; Starns, D.; Warrenfeltz, S.; Aurrecoechea, C.; Stoeckert, C.J.J.; Kissinger, J.C.; et al. FungiDB: An Integrated Bioinformatic Resource for Fungi and Oomycetes. J. Fungi 2018, 4, 39. [Google Scholar] [CrossRef]
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.J.; Smirnov, S.; Purcell, S.; et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef]
- Lohse, M.; Bolger, A.M.; Nagel, A.; Fernie, A.R.; Lunn, J.E.; Stitt, M.; Usadel, B. RobiNA: A user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 2012, 40, W622–W627. [Google Scholar] [CrossRef]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef] [PubMed]
- Dalmais, B.; Schumacher, J.; Moraga, J.; LE, P.P.; Tudzynski, B.; Collado, I.G.; Viaud, M. The Botrytis cinerea phytotoxin botcinic acid requires two polyketide synthases for production and has a redundant role in virulence with botrydial. Mol. Plant Pathol. 2011, 12, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Porquier, A.; Moraga, J.; Morgant, G.; Dalmais, B.; Simon, A.; Sghyer, H.; Collado, I.G.; Viaud, M. Botcinic acid biosynthesis in Botrytis cinerea relies on a subtelomeric gene cluster surrounded by relics of transposons and is regulated by the Zn2Cys6 transcription factor BcBoa13. Curr. Genet. 2019, 65, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Porquier, A.; Morgant, G.; Moraga, J.; Dalmais, B.; Luyten, I.; Simon, A.; Pradier, J.M.; Amselem, J.; Collado, I.G.; Viaud, M. The botrydial biosynthetic gene cluster of Botrytis cinerea displays a bipartite genomic structure and is positively regulated by the putative Zn(II) 2Cys6 transcription factor BcBot6. Fungal Genet. Biol. 2016, 96, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Sabine Fillinger, Y.E. Botrytis—The Fungus, the Pathogen and Its Management in Agricultural Systems; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Olivares-Yañez, C.; Sánchez, E.; Pérez-Lara, G.; Seguel, A.; Camejo, P.Y.; Larrondo, L.F.; Vidal, E.A.; Canessa, P. A comprehensive transcription factor and DNA-binding motif resource for the construction of gene regulatory networks in Botrytis cinerea and Trichoderma atroviride. Comput. Struct. Biotechnol. J. 2021, 19, 6212–6228. [Google Scholar] [CrossRef]
- Canessa, P.; Larrondo, L.F. Environmental responses and the control of iron homeostasis in fungal systems. Appl. Microbiol. Biotechnol. 2013, 97, 939–955. [Google Scholar] [CrossRef]
- Bushley, K.E.; Turgeon, B.G. Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol. Biol. 2010, 10, 26. [Google Scholar] [CrossRef]
- Ma, L.J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Di, P.A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef]
- van Dam, P.; Fokkens, L.; Ayukawa, Y.; van der Gragt, M.; ter Horst, A.; Brankovics, B.; Houterman, P.M.; Arie, T.; Rep, M. A mobile pathogenicity chromosome in Fusarium oxysporum for infection of multiple cucurbit species. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Li, J.; Fokkens, L.; Conneely, L.J.; Rep, M. Partial pathogenicity chromosomes in Fusarium oxysporum are sufficient to cause disease and can be horizontally transferred. Environ. Microbiol. 2020, 22, 4985–5004. [Google Scholar] [CrossRef]
- Lind, A.L.; Smith, T.D.; Saterlee, T.; Calvo, A.M.; Rokas, A. Regulation of Secondary Metabolism by the Velvet Complex Is Temperature-Responsive in Aspergillus. G3 Genes|Genomes|Genet. 2016, 6, 4023–4033. [Google Scholar] [CrossRef] [PubMed]
- Pontes, J.G.M.; Fernandes, L.S.; Dos, S.R.V.; Tasic, L.; Fill, T.P. Virulence Factors in the Phytopathogen-Host Interactions: An Overview. J. Agric. Food Chem. 2020, 68, 7555–7570. [Google Scholar] [CrossRef] [PubMed]
- Espino, J.J.; Gutiérrez-Sánchez, G.; Brito, N.; Shah, P.; Orlando, R.; González, C. The Botrytis cinerea early secretome. Proteomics 2010, 10, 3020–3034. [Google Scholar] [CrossRef]
- Fernández-Acero, F.J.; Colby, T.; Harzen, A.; Carbú, M.; Wieneke, U.; Cantoral, J.M.; Schmidt, J. 2-DE proteomic approach to the Botrytis cinerea secretome induced with different carbon sources and plant-based elicitors. Proteomics 2010, 10, 2270–2280. [Google Scholar] [CrossRef]
- Shah, P.; Atwood, J.A.; Orlando, R.; El, M.H.; Podila, G.K.; Davis, M.R. Comparative proteomic analysis of Botrytis cinerea secretome. J. Proteome Res. 2009, 8, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Gutierrez-Sanchez, G.; Orlando, R.; Bergmann, C. A proteomic study of pectin-degrading enzymes secreted by Botrytis cinerea grown in liquid culture. Proteomics 2009, 9, 3126–3135. [Google Scholar] [CrossRef]
- ten Have, A.; Mulder, W.; Visser, J.; van Kan, J.A. The endopolygalacturonase gene Bcpg1 is required for full virulence of Botrytis cinerea. Mol. Plant-Microbe Interact. 1998, 11, 1009–1016. [Google Scholar] [CrossRef]
- Chagué, V.; Danit, L.V.; Siewers, V.; Schulze-Gronover, C.; Tudzynski, P.; Tudzynski, B.; Sharon, A. Ethylene sensing and gene activation in Botrytis cinerea: A missing link in ethylene regulation of fungus-plant interactions? Mol. Plant-Microbe Interact. 2006, 19, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Canessa, P.; Schumacher, J.; Hevia, M.A.; Tudzynski, P.; Larrondo, L.F. Assessing the effects of light on differentiation and virulence of the plant pathogen Botrytis cinerea: Characterization of the White Collar Complex. PLoS ONE 2013, 8, e84223. [Google Scholar] [CrossRef]
- Ren, H.; Wu, X.; Lyu, Y.; Zhou, H.; Xie, X.; Zhang, X.; Yang, H. Selection of reliable reference genes for gene expression studies in Botrytis cinerea. J. Microbiol. Methods 2017, 142, 71–75. [Google Scholar] [CrossRef]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005, 6, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De, P.K.; Pattyn, F.; Poppe, B.; Van, R.N.; De, P.A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, J.; Vandesompele, J. Selection of reliable reference genes for RT-qPCR analysis. Methods Mol. Biol. 2014, 1160, 19–26. [Google Scholar] [PubMed]
- Shinohara, M.L.; Loros, J.J.; Dunlap, J.C. Glyceraldehyde-3-phosphate dehydrogenase is regulated on a daily basis by the circadian clock. J. Biol. Chem. 1998, 273, 446–452. [Google Scholar] [CrossRef]
- Hevia, M.A.; Canessa, P.; Müller-Esparza, H.; Larrondo, L.F. A circadian oscillator in the fungus Botrytis cinerea regulates virulence when infecting Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2015, 112, 8744–8749. [Google Scholar] [CrossRef]
- Henríquez-Urrutia, M.; Spanner, R.; Olivares-Yánez, C.; Seguel-Avello, A.; Pérez-Lara, R.; Guillén-Alonso, H.; Winkler, R.; Herrera-Estrella, A.; Canessa, P.; Larrondo, L.F. Circadian oscillations in Trichoderma atroviride and the role of core clock components in secondary metabolism, development, and mycoparasitism against the phytopathogen Botrytis cinerea. eLife 2022, 11, e71358. [Google Scholar] [CrossRef] [PubMed]
- Pinedo, C.; Wang, C.M.; Pradier, J.M.; Dalmais, B.; Choquer, M.; Le, P.P.; Morgant, G.; Collado, I.G.; Cane, D.E.; Viaud, M. Sesquiterpene synthase from the botrydial biosynthetic gene cluster of the phytopathogen Botrytis cinerea. ACS Chem. Biol. 2008, 3, 791–801. [Google Scholar] [CrossRef]
- Schumacher, J.; Gautier, A.; Morgant, G.; Studt, L.; Ducrot, P.H.; Le, P.P.; Azeddine, S.; Fillinger, S.; Leroux, P.; Tudzynski, B.; et al. A functional bikaverin biosynthesis gene cluster in rare strains of Botrytis cinerea is positively controlled by VELVET. PLoS ONE 2013, 8, e53729. [Google Scholar] [CrossRef]
- Schumacher, J.; Simon, A.; Cohrs, K.C.; Viaud, M.; Tudzynski, P. The transcription factor BcLTF1 regulates virulence and light responses in the necrotrophic plant pathogen Botrytis cinerea. PLoS Genet. 2014, 10, e1004040. [Google Scholar] [CrossRef]
- Jeya, M.; Kim, T.S.; Tiwari, M.K.; Li, J.; Zhao, H.; Lee, J.K. The Botrytis cinerea type III polyketide synthase shows unprecedented high catalytic efficiency toward long chain acyl-CoAs. Mol. Biosyst. 2012, 8, 2864–2867. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fungal Species | SRA Experiments | SRA Studies |
|---|---|---|
| Magnaporthe oryzae | 1714 | 125 |
| Botrytis cinerea | 2403 | 89 |
| Puccinia spp. | 3847 | 195 |
| Fusarium graminearum | 2141 | 177 |
| Fusarium oxysporum | 2700 | 188 |
| Blumeria graminis | 1057 | 43 |
| Mycosphaerella graminicola | 2095 | 230 |
| Colletotrichum spp. | 1752 | 219 |
| Ustilago maydis | 538 | 38 |
| Melampsora lini | 205 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Lara, G.; Moyano, T.C.; Vega, A.; Larrondo, L.F.; Polanco, R.; Álvarez, J.M.; Aguayo, D.; Canessa, P. The Botrytis cinerea Gene Expression Browser. J. Fungi 2023, 9, 84. https://doi.org/10.3390/jof9010084
Pérez-Lara G, Moyano TC, Vega A, Larrondo LF, Polanco R, Álvarez JM, Aguayo D, Canessa P. The Botrytis cinerea Gene Expression Browser. Journal of Fungi. 2023; 9(1):84. https://doi.org/10.3390/jof9010084
Chicago/Turabian StylePérez-Lara, Gabriel, Tomás C. Moyano, Andrea Vega, Luis F. Larrondo, Rubén Polanco, José M. Álvarez, Daniel Aguayo, and Paulo Canessa. 2023. "The Botrytis cinerea Gene Expression Browser" Journal of Fungi 9, no. 1: 84. https://doi.org/10.3390/jof9010084
APA StylePérez-Lara, G., Moyano, T. C., Vega, A., Larrondo, L. F., Polanco, R., Álvarez, J. M., Aguayo, D., & Canessa, P. (2023). The Botrytis cinerea Gene Expression Browser. Journal of Fungi, 9(1), 84. https://doi.org/10.3390/jof9010084