
Fungal Whole-Genome Sequencing for Species Identification: From Test Development to Clinical Utilization

 ,
,
Abstract
:1. Introduction
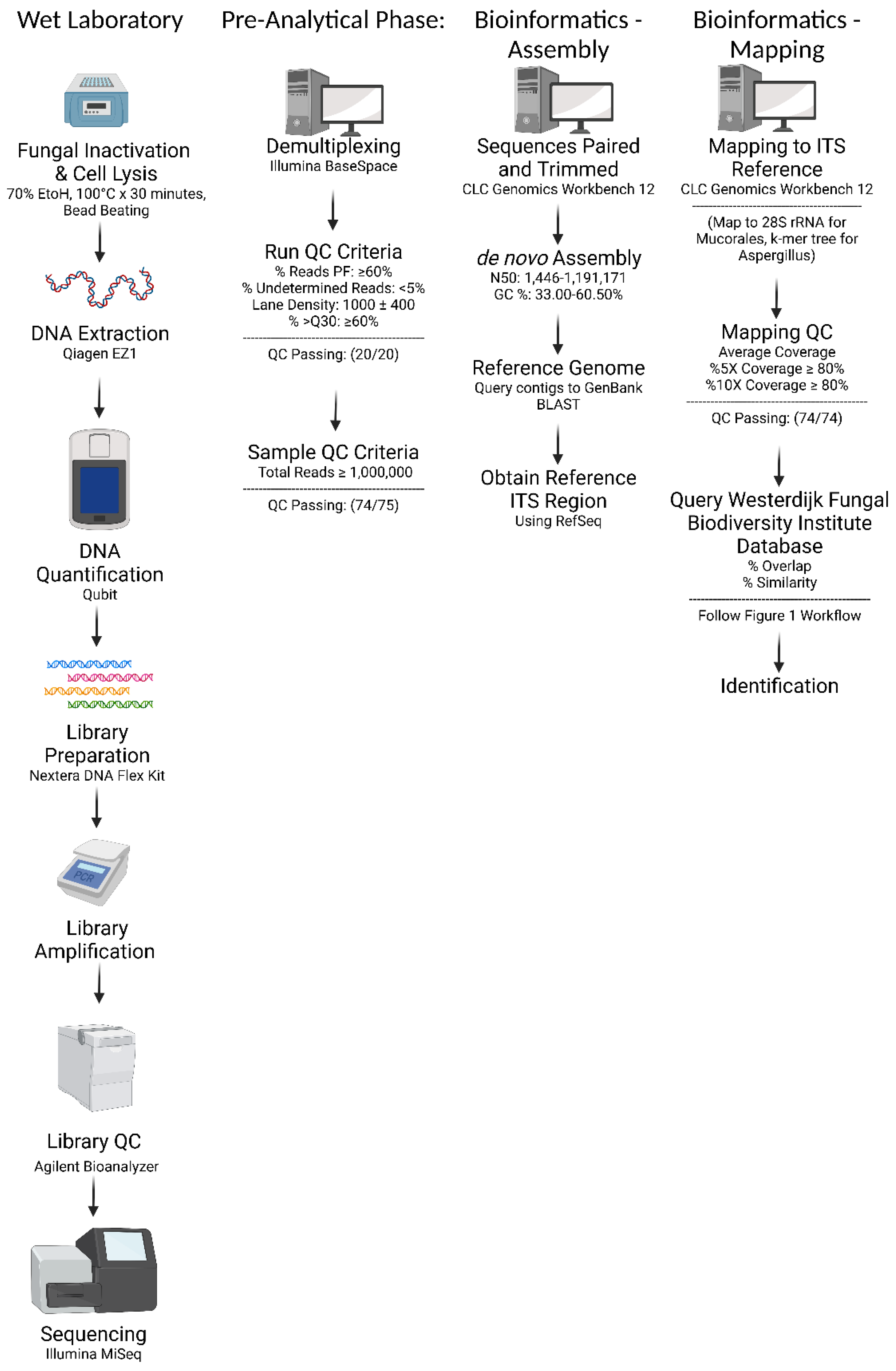
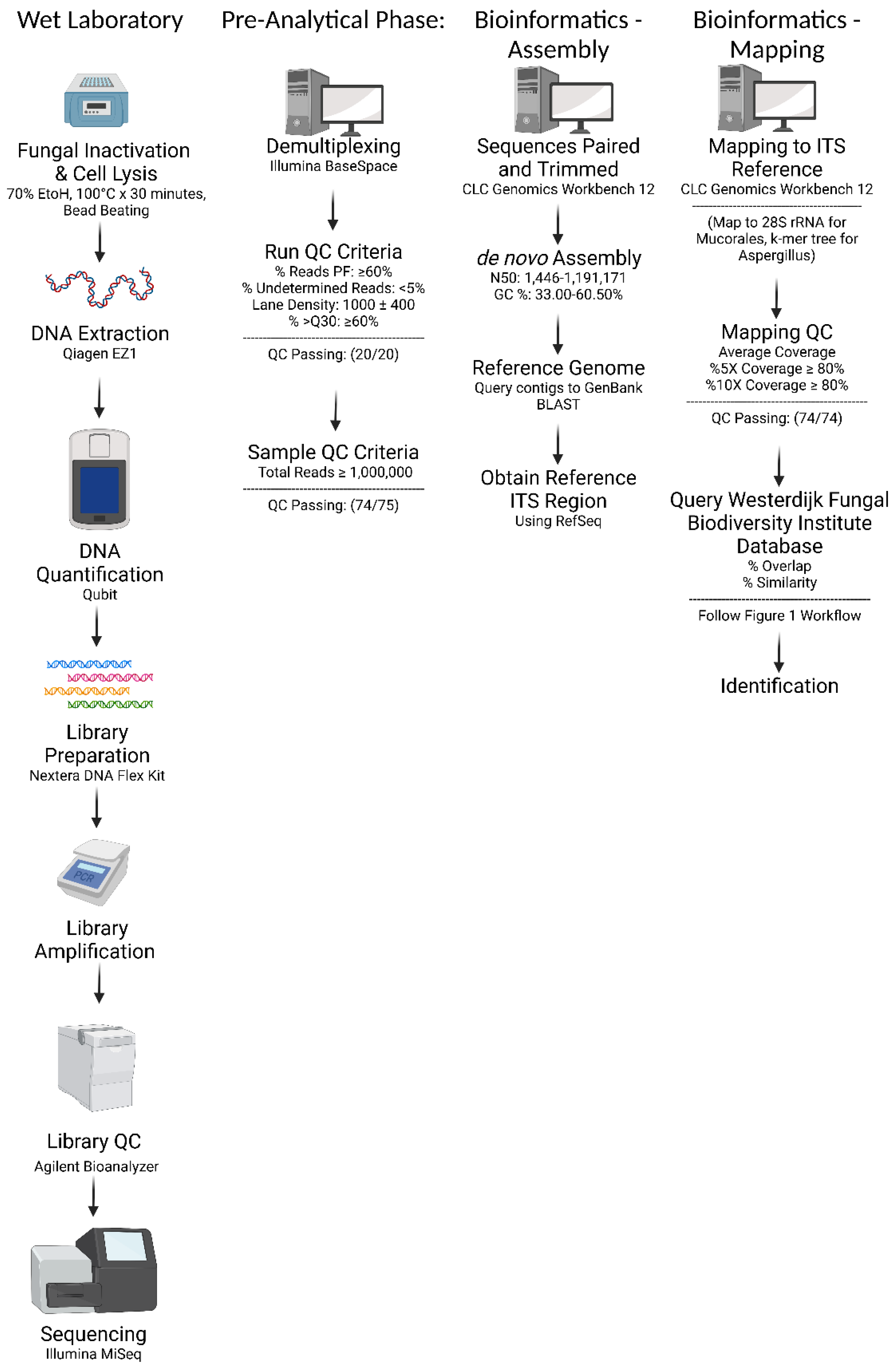
2. Materials and Methods
2.1. Fungal Isolates and Reference Identification Methods
2.2. Fungal Isolate Preparation and DNA Extraction
2.3. Library Preparation and Sequencing
2.4. Bioinformatic Analysis: De novo Assembly and In-House Database Query
2.5. Bioinformatic Analysis: ITS Region Mapping and Consensus Sequence Query
2.6. Bioinformatic Analysis: Additional Steps for Mucorales
2.7. Bioinformatic Analysis: Additional Steps for Aspergillus
2.8. NGS Board
2.9. Validation
2.10. Ethics
3. Results
3.1. Quality Control and Bioinformatics Performance
3.2. Database Validation
3.3. Assay Performance
3.4. Comparison of Beta-tubulin and Calmodulin Genes vs. K-mer Tree Based Analysis for Aspergillus Species Identification
3.5. Concordant Results and Discordant Analysis
3.6. Clinical Utility
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neyrolles, O.; Roetzer, A.; Diel, R.; Kohl, T.A.; Rückert, C.; Nübel, U.; Blom, J.; Wirth, T.; Jaenicke, S.; Schuback, S.; et al. Whole Genome Sequencing versus Traditional Genotyping for Investigation of a Mycobacterium tuberculosis Outbreak: A Longitudinal Molecular Epidemiological Study. PLoS Med. 2013, 10, e1001387. [Google Scholar] [CrossRef]
- McDonnell, J.; Dallman, T.; Atkin, S.; Turbitt, D.A.; Connor, T.R.; Grant, K.A.; Thomson, N.R.; Jenkins, C. Retrospective analysis of whole genome sequencing compared to prospective typing data in further informing the epidemiological investigation of an outbreak of Shigella sonnei in the UK. Epidemiol. Infect. 2013, 141, 2568–2575. [Google Scholar] [CrossRef]
- Bartels, M.D.; Petersen, A.; Worning, P.; Nielsen, J.B.; Larner-Svensson, H.; Johansen, H.K.; Andersen, L.P.; Jarløv, J.O.; Boye, K.; Larsen, A.R.; et al. Comparing Whole-Genome Sequencing with Sanger Sequencing for spa Typing of Methicillin-Resistant Staphylococcus aureus. J. Clin. Microbiol. 2014, 52, 4305–4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dorp, L.; Houldcroft, C.J.; Richard, D.; Balloux, F. COVID-19, the first pandemic in the post-genomic era. Curr. Opin. Virol. 2021, 50, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Balloux, F.; Bronstad Brynildsrud, O.; van Dorp, L.; Shaw, L.P.; Chen, H.; Harris, K.A.; Wang, H.; Eldholm, V. From Theory to Practice: Translating Whole-Genome Sequencing (WGS) into the Clinic. Trends Microbiol. 2018, 26, 1035–1048. [Google Scholar] [CrossRef] [Green Version]
- Rossen, J.W.A.; Friedrich, A.W.; Moran-Gilad, J.; Genomic, E.S.G.f.; Molecular, D. Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin. Microbiol. Infect. 2018, 24, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, T.K.; Realegeno, S.; Mirasol, R.; Tsan, A.; Chandrasekaran, S.; Garner, O.B.; Yang, S. Validation, Implementation, and Clinical Utility of Whole Genome Sequence-Based Bacterial Identification in the Clinical Microbiology Laboratory. J. Mol. Diagn. 2021, 23, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Wickes, B.L.; Romanelli, A.M. Diagnostic Mycology: Xtreme Challenges. J. Clin. Microbiol. 2020, 58, e01345-19. [Google Scholar] [CrossRef]
- White, L.P.; Price, J.S. Recent Advances and Novel Approaches in Laboratory-Based Diagnostic Mycology. J. Fungi 2021, 7, 41. [Google Scholar] [CrossRef]
- Robert, M.G.; Cornet, M.; Hennebique, A.; Rasamoelina, T.; Caspar, Y.; Ponderand, L.; Bidart, M.; Durand, H.; Jacquet, M.; Garnaud, C.; et al. MALDI-TOF MS in a Medical Mycology Laboratory: On Stage and Backstage. Microorganisms 2021, 9, 1283. [Google Scholar] [CrossRef]
- CLSI. Interpretive Criteria for Identification of Bacteria and Fungi by Targeted DNA Sequencing, 2nd ed.; Clinical and Laboratory Standards Institute Guidelines MM18; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- Tam, E.W.T.; Chen, J.H.K.; Lau, E.C.L.; Ngan, A.H.Y.; Fung, K.S.C.; Lee, K.-C.; Lam, C.-W.; Yuen, K.-Y.; Lau, S.K.P.; Woo, P.C.Y.; et al. Misidentification of Aspergillus nomius and Aspergillus tamarii as Aspergillus flavus: Characterization by Internal Transcribed Spacer, β-Tubulin, and Calmodulin Gene Sequencing, Metabolic Fingerprinting, and Matrix-Assisted Laser Desorption Ionization–Time of Flight Mass Spectrometry. J. Clin. Microbiol. 2014, 52, 1153–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irinyi, L.; Serena, C.; Garcia-Hermoso, D.; Arabatzis, M.; Desnos-Ollivier, M.; Vu, D.; Cardinali, G.; Arthur, I.; Normand, A.C.; Giraldo, A.; et al. International Society of Human and Animal Mycology (ISHAM)-ITS reference DNA barcoding database—The quality controlled standard tool for routine identification of human and animal pathogenic Fungi. Med. Mycol. 2015, 53, 313–337. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.T.V.; Irinyi, L.; Chen, S.C.A.; Sorrell, T.C.; Group, I.B.o.M.F.W.; Meyer, W. Dual DNA Barcoding for the Molecular Identification of the Agents of Invasive Fungal Infections. Front. Microbiol. 2019, 10, 1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glockner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Mirhendi, H.; Zarei, F.; Motamedi, M.; Nouripour-Sisakht, S. Aspergillus tubingensis and Aspergillus niger as the dominant black Aspergillus, use of simple PCR-RFLP for preliminary differentiation. J. Mycol. Med. 2016, 26, 9–16. [Google Scholar] [CrossRef]
- Morales-Lopez, S.E.; Garcia-Effron, G. Infections due to Rare Cryptococcus Species. A Literature Review. J. Fungi 2021, 7, 279. [Google Scholar] [CrossRef]
- Glampedakis, E.; Erard, V.; Lamoth, F. Clinical Relevance and Characteristics of Aspergillus calidoustus and Other Aspergillus Species of Section Usti. J. Fungi 2020, 6, 84. [Google Scholar] [CrossRef]
- Nematollahi, S.; Permpalung, N.; Zhang, S.X.; Morales, M.; Marr, K.A. Aspergillus lentulus: An Under-recognized Cause of Antifungal Drug-Resistant Aspergillosis. Open Forum Infect. Dis. 2021, 8, ofab392. [Google Scholar] [CrossRef]
- Imbert, S.; Normand, A.C.; Cassaing, S.; Gabriel, F.; Kristensen, L.; Bonnal, C.; Lachaud, L.; Costa, D.; Guitard, J.; Hasseine, L.; et al. Multicentric Analysis of the Species Distribution and Antifungal Susceptibility of Cryptic Isolates from Aspergillus Section Fumigati. Antimicrob. Agents Chemother. 2020, 64, e01374-20. [Google Scholar] [CrossRef] [PubMed]
- Shivasabesan, G.; Logan, B.; Brennan, X.; Lau, C.; Vaze, A.; Bennett, M.; Gorrie, N.; Mirdad, F.; Deveza, R.; Koo, C.M.; et al. Disseminated Aspergillus lentulus Infection in a Heart Transplant Recipient: A Case Report. Clin. Infect. Dis. 2022, 75, 1235–1238. [Google Scholar] [CrossRef] [PubMed]
- Badali, H.; Khodavaisy, S.; Fakhim, H.; de Hoog, G.S.; Meis, J.F.; Chowdhary, A. In Vitro Susceptibility Profiles of Eight Antifungal Drugs against Clinical and Environmental Strains of Phaeoacremonium. Antimicrob. Agents Chemother. 2015, 59, 7818–7822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.M.; Lee, M.H.; Suh, M.K.; Ha, G.Y.; Kim, H.; Choi, J.S. Onychomycosis Caused by Chaetomium globosum. Ann. Dermatol. 2013, 25, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarro, J.; Soler, L.; Rinaldi, M.G. Pathogenicity and antifungal susceptibility of Chaetomium species. Eur. J. Clin. Microbiol. Infect. Dis. 1995, 14, 613–618. [Google Scholar] [CrossRef]
- Dolatabadi, S.; Najafzadeh, M.J.; Houbraken, J.; Vicente, V.; de Hoog, S.; Meis, J.F. In vitro activity of eight antifungal drugs against Chaetomiaceae. Med. Mycol. 2021, 60, myab074. [Google Scholar] [CrossRef]
- Spettel, K.; Galazka, S.; Kriz, R.; Camp, I.; Willinger, B. Do Candida albicans Isolates with Borderline Resistant Micafungin MICs Always Harbor FKS1 Hot Spot Mutations? J. Fungi 2021, 7, 93. [Google Scholar] [CrossRef]
- Perlin, D.S. Echinocandin Resistance in Candida. Clin. Infect. Dis. 2015, 61 (Suppl. 6), S612–S617. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Effron, G. Molecular Markers of Antifungal Resistance: Potential Uses in Routine Practice and Future Perspectives. J. Fungi 2021, 7, 197. [Google Scholar] [CrossRef]
- Patel, R. A Moldy Application of MALDI: MALDI-ToF Mass Spectrometry for Fungal Identification. J. Fungi 2019, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Araujo, R.; Sampaio-Maia, B. Fungal Genomes and Genotyping. Adv. Appl. Microbiol. 2018, 102, 37–81. [Google Scholar] [CrossRef]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; Riley, R.; Salamov, A.; Zhao, X.; Korzeniewski, F.; et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014, 42, D699–D704. [Google Scholar] [CrossRef] [PubMed]
- Grigoriev, I.V.; Cullen, D.; Goodwin, S.B.; Hibbett, D.; Jeffries, T.W.; Kubicek, C.P.; Kuske, C.; Magnuson, J.K.; Martin, F.; Spatafora, J.W.; et al. Fueling the future with fungal genomics. Mycology 2011, 2, 192–209. [Google Scholar] [CrossRef]
- Price, T.K.; Mirasol, R.; Ward, K.W.; Dayo, A.J.; Hilt, E.E.; Chandrasekaran, S.; Garner, O.B.; de St Maurice, A.; Yang, S. Genomic Characterizations of Clade III Lineage of Candida auris, California, USA. Emerg. Infect. Dis. 2021, 27, 1223–1227. [Google Scholar] [CrossRef]
- de St Maurice, A.; Parti, U.; Anikst, V.E.; Harper, T.; Mirasol, R.; Dayo, A.J.; Garner, O.B.; Prabaker, K.K.; Yang, S. Clinical, microbiological, and genomic characteristics of clade-III Candida auris colonization and infection in southern California, 2019–2022. Infect. Control Hosp. Epidemiol. 2022, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vallabhaneni, S.; Mody, R.K.; Walker, T.; Chiller, T. The Global Burden of Fungal Diseases. Infect. Dis. Clin. North Am. 2016, 30, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Hawkins, N.J.; Sanglard, D.; Gurr, S.J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 2018, 360, 739–742. [Google Scholar] [CrossRef] [Green Version]
- Vitasse, Y.; Ursenbacher, S.; Klein, G.; Bohnenstengel, T.; Chittaro, Y.; Delestrade, A.; Monnerat, C.; Rebetez, M.; Rixen, C.; Strebel, N.; et al. Phenological and elevational shifts of plants, animals and fungi under climate change in the European Alps. Biol. Rev. Camb. Philos. Soc. 2021, 96, 1816–1835. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| ITS Query Result | 28S rRNA Query Result (For Mucorales Only) | ID Chosen | Reporting Scheme | Example |
|---|---|---|---|---|
| ≥98% Overlap AND | ≥98% Overlap AND | Top Result | Genus + Species | Candida albicans |
| ≥98.5% Similarity AND | ≥99.0% Similarity AND | |||
| Only 1 Top Result | Only 1 Top Result | |||
| ≥98% Overlap AND | ≥98% Overlap AND | Top Results | Genus + Species/Species | Candida albicans/dubliniensis |
| ≥98.5% Similarity AND | ≥99.0% Similarity AND | |||
| >1 Top Result | >1 Top Result | |||
| <98–95% Overlap AND/OR | <98–95% Overlap AND/OR | Top Result | Genus only | Candida spp. most closely related to C. albicans |
| <98.5–95% Similarity | <99–95% Similarity | |||
| <95% Overlap AND/OR | <95% Overlap AND/OR | Up to the Order Level | Consult NGS Board | Filamentous basidiomycetes, identified to be in the order of Polyporales, not able to be identified to the genus and species level |
| <95% Similarity | <95% Similarity |
| Sample ID | Total Sequence Reads | Identification Based on K-mer Tree | Beta-Tubulin Gene Coverage (%1X) | Top Beta-Tubulin Hit (%Overlap, %Similarity) | Calmodulin Gene Coverage (%1X) | Top Calmodulin Hit (%Overlap, %Similarity) |
|---|---|---|---|---|---|---|
| UCLA_160 | 2,522,136 | Aspergillus terreus | 99.18 | Aspergillus terreus (100, 100) | 93.56 | Aspergillus felis (59, 79.57) |
| UCLA_261 | 2,182,810 | Aspergillus tubingensis | 100 | Aspergillus tubingensis (100, 100) | 0 | N/A |
| UCLA_262 | 2,638,830 | Aspergillus niger | 100 | Aspergillus niger (100, 100) | 75.56 | Aspergillus niger (87, 99.94) |
| UCLA_274 | 5,580,086 | Aspergillus quadrilineatus | 100 | Aspergillus quadrilineatus (100, 100) | 85.21 | Aspergillus quadrilineatus (100, 96.29) |
| UCLA_280 | 3,325,202 | Aspergillus terreus | 99.18 | Aspergillus terreus (100, 100) | 93.56 | Aspergillus felis (59, 79.42) |
| UCLA_281 | 3,112,042 | Aspergillus fumigatus | 100 | Aspergillus fumigatus (100, 100) | 100 | Aspergillus fumigatus (100, 100) |
| UCLA_289 | 2,775,140 | Aspergillus fumigatus | 100 | Aspergillus fumigatus (100, 100) | 100 | Aspergillus fumigatus (100, 100) |
| UCLA_297 | 3,402,024 | Aspergillus sydowii | 99.64 | Aspergillus sydowii (100, 100) | 83.74 | Aspergillus puulaauensis (100, 94.51) |
| UCLA_305 | 3,501,422 | Aspergillus flavus | 100 | Aspergillus flavus (100, 100) | 75.56 | Aspergillus flavus (100, 99.85) |
| UCLA_306 | 2,043,130 | Aspergillus calidoustus | 100 | Aspergillus calidoustus (100, 100) | 76.6 | Aspergillus calidoustus (98.13, 100) |
| UCLA_312 | 1,778,822 | Aspergillus flavus | 100 | Aspergillus flavus (100, 100) | 75.56 | Aspergillus flavus (100, 99.76) |
| UCLA_417 | 2,696,552 | Aspergillus terreus | 100 | Aspergillus terreus (100, 100) | 93.56 | Aspergillus felis (61, 79.42) |
| UCLA_536 | 1,350,196 | Aspergillus fumigatus | 100 | Aspergillus fumigatus (100, 100) | 94.93 | Aspergillus fumigatus (100, 100) |
| Sample ID | WGS ID | Original ID | Conventional Identification Method |
|---|---|---|---|
| UCLA_24 | Scedosporium dehoogii | Scedosporium apiospermum | Microscopic morphology |
| UCLA_144 | Scedosporium dehoogii | Scedosporium apiospermum | Microscopic morphology |
| UCLA_156 | Candida spp. most closely related to C. haemulonii | Candida haemulonii | Reference Isolate |
| UCLA_261 | Aspergillus tubingensis | Aspergillus niger | Microscopic morphology |
| UCLA_274 | Aspergillus quadrilineatus | Aspergillus nidulans | Microscopic morphology |
| UCLA_291 | Coprinellus spp. most closely related to C. micaceus | Coprinellus micaceus | MALDI-TOF MS |
| UCLA_306 | Aspergillus calidoustus | Aspergillus ustus | Reference Isolate |
| UCLA_415 | Cryptococcus liquefaciens | Cryptococcus albidus | API 20C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem-Bango, Z.; Price, T.K.; Chan, J.L.; Chandrasekaran, S.; Garner, O.B.; Yang, S. Fungal Whole-Genome Sequencing for Species Identification: From Test Development to Clinical Utilization. J. Fungi 2023, 9, 183. https://doi.org/10.3390/jof9020183
Salem-Bango Z, Price TK, Chan JL, Chandrasekaran S, Garner OB, Yang S. Fungal Whole-Genome Sequencing for Species Identification: From Test Development to Clinical Utilization. Journal of Fungi. 2023; 9(2):183. https://doi.org/10.3390/jof9020183
Chicago/Turabian StyleSalem-Bango, Zackary, Travis K Price, June L Chan, Sukantha Chandrasekaran, Omai B Garner, and Shangxin Yang. 2023. "Fungal Whole-Genome Sequencing for Species Identification: From Test Development to Clinical Utilization" Journal of Fungi 9, no. 2: 183. https://doi.org/10.3390/jof9020183
APA StyleSalem-Bango, Z., Price, T. K., Chan, J. L., Chandrasekaran, S., Garner, O. B., & Yang, S. (2023). Fungal Whole-Genome Sequencing for Species Identification: From Test Development to Clinical Utilization. Journal of Fungi, 9(2), 183. https://doi.org/10.3390/jof9020183