Abstract
Antifungal resistance is a growing concern as it poses a significant threat to public health. Fungal infections are a significant cause of morbidity and mortality, especially in immunocompromised individuals. The limited number of antifungal agents and the emergence of resistance have led to a critical need to understand the mechanisms of antifungal drug resistance. This review provides an overview of the importance of antifungal resistance, the classes of antifungal agents, and their mode of action. It highlights the molecular mechanisms of antifungal drug resistance, including alterations in drug modification, activation, and availability. In addition, the review discusses the response to drugs via the regulation of multidrug efflux systems and antifungal drug–target interactions. We emphasize the importance of understanding the molecular mechanisms of antifungal drug resistance to develop effective strategies to combat the emergence of resistance and highlight the need for continued research to identify new targets for antifungal drug development and explore alternative therapeutic options to overcome resistance. Overall, an understanding of antifungal drug resistance and its mechanisms will be indispensable for the field of antifungal drug development and clinical management of fungal infections.
1. Introduction
Fungi can cause a variety of human diseases [1], including (i) local mycoses of the skin, nails, and hair, (ii) systemic mycoses that spread throughout the body and affect internal organs, such as histoplasmosis, aspergillosis, and candidiasis, (iii) opportunistic infections that occur in people with weakened immune systems, such as Pneumocystis jirovecii and Cryptococcus neoformans, (iv) allergic diseases triggered by an allergic reaction to fungal spores such as bronchopulmonary aspergillosis, or (v) toxigenic infections caused by toxic substances produced by fungi, such as aflatoxicosis and ergotism. Every year, over 150 million severe fungal infections occur globally, leading to around 1.7 million deaths annually. Disturbingly, these numbers are steadily increasing due to social and medical advancements that have facilitated the spread of these infections. Furthermore, the long-term use of antifungal drugs in high-risk patients has resulted in the emergence of drug-resistant fungi, including the highly dangerous Candida auris [2,3]. Very recently, the World Health Organization (WHO) has raised the alert of threatening fungal infections by ranking a total of 19 fungal species as priority pathogens for strategic research, development, and public health actions [4]. The most critical group contains Cryptococcus neoformans, Candida auris, Aspergillus fumigatus, and Candida albicans. According to this ultimate analysis from WHO, research into fungal-invasive diseases still receives insufficient funding, with only 1.5% of the total research budget in infectious diseases, and the approved antifungals and those in clinical trials do not fully solve the challenges posed to healthcare workers due to treatment limitations and increasing resistance. The widespread use of fungicides (azoles) in agriculture additionally exacerbates the issue, thus demanding the development of new types of antifungals with different targets, safer profiles, and mechanisms of action exclusively for human use. Thus, fungal infections are a growing global concern that requires more effective research to understand and combat fungal antimicrobial resistance [3,5]. This article summarizes and highlights the molecular mechanisms underlying the adaptive response and acquired resistance to different classes of antifungal compounds.
2. Antifungal Drugs: Classes and Modes of Action
Currently, only four classes of systemic antifungal treatments are used in clinical practice: azoles, echinocandins, polyenes, and pyrimidines [6] (Figure 1). Azole antifungal drugs, first reported in the late 1960s [7], work by inhibiting the synthesis of ergosterol, an essential component of fungal cell membranes. This results in increased permeability of the fungal cell, leading to its destruction. Azoles are used to treat a variety of fungal infections, including dermatophytosis, candidiasis, and aspergillosis. There are several classes of azole antifungal drugs [8], including (i) imidazoles, such as clotrimazole, econazole, and miconazole, which are commonly used to treat skin and nail infections, (ii) triazoles, such as fluconazole, itraconazole, and voriconazole, which are used to treat systemic fungal infections and (iii) allylamines, such as terbinafine, which is used to treat dermatophyte infections. Ergosterol acts as a control mechanism for the fluidity, asymmetry, and overall stability of the cell membrane in fungal cells [9]. For the cell membrane to maintain its integrity, the incorporated sterols must not have a C-4 methyl group. Azoles primarily impact the heme protein Cyp51 (Erg11), which catalyzes the cytochrome P-450-dependent removal of a methyl group from lanosterol through the process of 14α-demethylation within the ergosterol biosynthesis pathway [10,11]. Azole drugs inhibit the enzyme reaction by non-competitive reversible interaction with the heme and prevent accessing of protons to the active site [12]. When the 14α-demethylase is inhibited, the levels of ergosterol decrease, and the levels of its precursors, such as lanosterol, 4,14-dimethylzymosterol, and 24-methylenedihydrolanosterol, increase, leading to structural and functional changes in the plasma membrane [13].
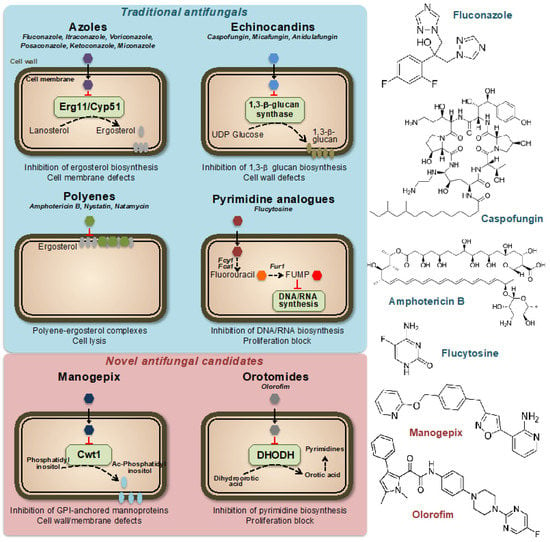
Figure 1.
Mechanisms of action of traditional and novel antifungal drugs. Representative compounds are indicated for each class. FUMP = 5-fluorouridylic acid; Fcy1, Fca1 = cytosine deaminases; Fur1 = Uracil phosphoribosyltransferase; Ac-Phosphatidyl inositol = Acyl-Phosphatidyl inositol; GPI = Glycosylphosphatidyl inositol; DHODH = Dihydroorotate dehydrogenase.
Echinocandins are a class of antifungal agents discovered in the 1980s and 1990s. They work by targeting a key component of the fungal cell wall, β(1,3)-glucan, which is essential for its stability and integrity (Figure 1). The unique composition of the fungal cell wall, which includes compounds such as mannan, chitin, and α- and β-glucans, has made it a prime target for antifungal agents [14]. These components are exclusive to fungi and not present in other organisms, making them a desirable target for drugs to selectively target and kill fungal cells while minimizing damage to other cells [15]. The majority of our understanding of the cell wall composition of fungi that are medically significant comes from research conducted on C. albicans [16]. This yeast species has a complex cell wall structure made up of multiple layers, including chitin, β-glucan, and mannoprotein. These last two components make up approximately 80% of the overall mass of the cell wall, thus highlighting the importance of β-glucan for fungal cell stability [17]. Echinocandins inhibit the synthesis of β(1,3)-glucan by binding to the enzyme β(1,3)-glucan synthase, a large (210 kDa) integral membrane heterodimeric protein, thereby disrupting the fungal cell wall and leading to cell death [18,19]. The mechanism of action of echinocandins makes them effective against a variety of clinically important fungi, including Candida and Aspergillus species. They are typically used to treat serious invasive fungal infections, often in combination with other antifungals.
Polyenes are a class of antifungal agents discovered in the 1950s [20]. Similar to azoles, they target the unique fungal membrane component ergosterol; however, in this case, by direct binding to the sterol, thereby causing a change in the permeability of the membrane and disruption of the membrane potential [21,22] (Figure 1). Polyenes have a broad spectrum of activity against various medically important fungi, as they inactivate all cells with a significant content of sterols in their outer membranes [23]. The most important drawback of the use of polyenes is their intrinsically high host toxicity, which limits their therapeutic index [24,25]. One of the most well-known polyenes is amphotericin B, which was initially derived from the soil bacterium Streptomyces nodosus. Amphotericin B was the first effective antifungal agent and is still widely used today, together with nystatin and natamycin, especially for the treatment of serious or life-threatening fungal infections [20,26].
Pyrimidine analogs are a different class of antifungal agents that act by inhibiting DNA and RNA synthesis [27]. Flucytosine (5FC) is the most commonly used pyrimidine analog for the treatment of fungal infections. The 5FC has been shown to inhibit the growth of various yeasts, including Candida and Cryptococcus neoformans. However, the high prevalence of primary resistance in many fungal species has reduced its initial promise as an antifungal agent [28,29,30]. Nowadays, 5FC is mostly used in combination with other antifungals, such as amphotericin B and fluconazole, and is rarely used as a single agent. The mechanism of action of 5FC involves entering the fungal cell through a permease enzyme and being converted to 5-fluorouracil (5FU) by the enzyme cytosine deaminase. This is followed by the conversion of 5FU into 5-fluorouridylic acid (FUMP) by UMP pyrophosphorylase, which is further phosphorylated and incorporated into RNA, causing disruption of protein synthesis [31]. Additionally, 5FU is converted to 5-fluorodeoxyuridine monophosphate, a potent inhibitor of thymidylate synthase, an enzyme involved in DNA synthesis and nuclear division [32]. Thus, 5FC acts by disrupting pyrimidine metabolism, as well as RNA, DNA, and protein synthesis in the fungal cell (Figure 1).
Although there is a growing need for more antifungal drug options [33,34], no new classes of antifungal drugs have become available in the past 20 years. However, there is some good news, as several new antifungal classes are currently in late-stage clinical development. These include promising drugs currently in clinical trials with previously unexplored modes of action, such as fosmanogepix (a novel inhibitor of the Gwt1 enzyme) or olorofim (a new inhibitor of the dihydroorotate dehydrogenase enzyme) [35,36,37] (Figure 1).
Fosfomanogepix is a N-phosphonooxymethyl prodrug, which after drug administration, is converted into the active form of manogepix by systemic phosphatases [38]. Manogepix has a new mode of inhibition that targets the maturation of glycosylphosphatidylinositol (GPI)-anchored proteins by inhibiting the fungal enzyme Gwt1 at the endoplasmic reticulum [39]. This enzyme, which is crucial for the trafficking and anchoring of mannoproteins to the fungal cell membrane and wall, is an inositol acyltransferase [40]. GPI-anchored mannoproteins act as adhesives, enabling fungi to stick to mucosal and epithelial surfaces in the host before colonization and infection. Several fungal adhesins and virulence factors are derived from GPI-anchored proteins and GPI biosynthesis is essential in fungi [41,42]. The inhibition by manogepix appears to be unique to fungal pathogens because it does not impede human inositol acylation via the nearest mammalian ortholog, PIGW [38,43]. Manogepix has broad-spectrum activity against numerous pathogenic fungi including Candida and Aspergillus spp. [44,45].
Olorofim is a member of a novel class of antifungal drugs named orotomides. Olorofim impairs fungal growth through inhibition of the fungal dihydroorotate dehydrogenase (DHODH) enzyme, which is located in mitochondria and involved in pyrimidine synthesis [46,47] (Figure 1). Olorofim does not show any significant cross-reactivity with the human dihydroorotate dehydrogenase, thus limiting its drug toxicity [48]. While olorofim may not be effective against a wide range of fungal infections, it possesses remarkable novelty and a spectrum of activity that is anticipated to be significant in the future. Olorofim does not demonstrate any efficacy against yeast. Nevertheless, it is effective against various clinically important fungal groups, including dimorphic molds such as Histoplasma and Coccidioides spp. or hyaline hyphomycetes such as Aspergillus spp. [35,46].
3. Impact of Fungal Antimicrobial Resistance
Infections are a leading cause of mortality and disability worldwide, with drug-resistant bacterial infections causing 1.27 million direct deaths and contributing to an estimated 4.95 million deaths annually [49]. This burden is particularly high in resource-limited settings. Fungal diseases cause over 1.5 million deaths and affect over a billion individuals [50]. Serious fungal infections often occur as a consequence of underlying health conditions such as asthma, AIDS, cancer, organ transplantation, and corticosteroid therapies. The WHO recognizes that the incidence of invasive fungal diseases (IFDs) is increasing globally, particularly among immunocompromised populations [4]. However, there are challenges in diagnosing and treating IFDs, including limited access to quality diagnostics and treatments and the emergence of antifungal resistance. Despite the growing concern, fungal infections receive limited attention and resources, resulting in a scarcity of data on their distribution and patterns of antifungal resistance [4,51,52,53]. Thus, it is not possible to accurately estimate their burden.
The acquisition and emergence of antifungal drug resistance can be explained as an evolutionary response to the selective pressure exerted by the drug. The likelihood of resistance emerging due to genetic changes is influenced by several factors, including the number and doubling rate of fungal cells exposed to the drug, the number of different pathways that confer resistance, and the fitness costs associated with each pathway [54]. It is important to note that antifungal drug resistance can originate either in the host or in the environment. In vivo resistance develops in individuals during antifungal therapy, leading to treatment failure for a range of pathogenic fungi [55,56]. This is particularly relevant for Candida spp., which is a major cause of nosocomial bloodstream infections and frequently shows the emergence of resistance to antifungals [57]. For example, azole resistance in C. albicans has been documented in individuals with HIV who were undergoing prolonged fluconazole therapy [58]. The demographics of patients susceptible to IFDs are progressively broadening, with a particular emphasis on populations such as the elderly, individuals with compromised immune systems due to HIV, cancer chemotherapy, or immune-suppression therapy required for transplantation, as well as those suffering from severe viral infections such as influenza virus and COVID-19 [59,60,61,62]. Environmental resistance can also emerge due to prior exposure of human pathogenic fungi to fungicides in nature. Fungicides are commonly used to protect crops and materials against fungal infections, and the resulting selective pressure drives the evolution of resistance against all major classes of fungicides, including azoles [63].
4. Mechanisms of Acquired Antifungal Resistance
At a mechanistic level, antifungal resistance is typically acquired through modifications that impact the interaction between the drug and its target, either directly or indirectly [64,65]. The development of resistance can be attributed to genetic alterations in the binding site of the target, such as mutations in the genes encoding lanosterol demethylase for azoles or β-glucan synthase for echinocandins [6]. Resistance may also result from an increase in the amount of target available [66], or through changes in the effective drug concentration, as a consequence of elevated drug efflux activity for intracellular drugs such as azoles [67,68], or through the inhibition of prodrug activation for flucytosine [69]. In scientific terminology, antifungal resistance is distinct from antifungal tolerance, which refers to the capacity of drug-sensitive cells to survive in the presence of drug concentrations beyond the minimum inhibitory concentration (MIC). This ability is associated with various general stress responses and/or epigenetic pathways, as comprehensively reviewed in [70]. Notably, tolerance is most pronounced when using fungistatic drugs and has been extensively examined in Candida albicans strains treated with fluconazole [71]. Nevertheless, the clinical significance of antifungal tolerance remains uncertain. In this review, we focus on the molecular mechanisms, which enable fungal cells to respond and protect from antibiotic drugs and eventually develop drug resistance.
4.1. Fungal Multidrug Efflux Systems: Components, Regulation, and Impact on Drug Resistance
A notable proportion of observed clinical resistance to antifungal agents can be attributed to the action of ATP-binding cassette (ABC) and major facilitator superfamily (MFS) transport pumps located at the fungal plasma membrane [72,73]. The majority of studies in this field have been conducted with azole drugs [74]; however, multidrug efflux affects resistance to other classes of antifungals [75]. The ABC and MFS transporters are the two most extensively researched transporter families implicated in efflux [73]. Fungi allocate a significant portion of their genomic resources to encoding transporters, with fungal genomes containing approximately 10 to 30 transporter-encoding genes per megabase of genomic DNA [76]. The ABC transporter superfamily comprises primary efflux transporters, which use ATP hydrolysis to export substrates [77,78]. This superfamily can be further divided into five families of transporters (ABCA, ABCB, ABCC, ABCD, and ABCG), with three families being involved in the efflux of toxic compounds. These three families, known as multi-drug resistance (MDR), multi-drug resistance-associated protein (MRP), and pleiotropic drug resistance (PDR), respectively, have been extensively investigated in Saccharomyces cerevisiae, offering insights into their potential roles in pathogenic fungi [79,80]. The PDR family exhibits the lowest level of phylogenetic conservation among the ABC transporter families, with evidence of gene loss and duplication observed in yeasts and filamentous fungi [81]. This implies that the members of the PDR family evolve rapidly in response to external selective pressures.
The conserved structure of ABC transporters comprises a nucleotide-binding domain (NBD) located either preceding or following a transmembrane domain (TMD), which is formed by six transmembrane-spanning helices. This NBD-TMD6 configuration is observed in half ABC transporters, which dimerize to produce a complete functional protein. In contrast, most fungal ABC transporters have undergone evolutionary changes that have led to the fusion of two NBD-TMD6 half transporters, resulting in the formation of a single functional protein instead of a dimeric structure. ABC transporters are a highly complex and fascinating class of proteins. Although some members of this superfamily exhibit high substrate specificity, ABC multidrug transporters are highly promiscuous in their substrate recognition. These proteins harness the energy generated by ATP binding and hydrolysis at the NBDs to induce conformational changes that enable the transporters to bind and release drug substrates [82]. The binding pocket is constructed from the TMDs and can accommodate large substrates. The transporter adopts a high-affinity inward-facing (IF) conformation during the binding phase, while the substrate is released from the outward-facing (OF) structure with lower affinity. As a result, these proteins exhibit polytopic characteristics. The nature of the drug-binding and transport sites is relevant to understanding resistance mechanisms mediated by these transporters. For example, the large drug-binding pockets of P-glycoprotein and Pdr5 contain multiple overlapping sites [83,84]. Notably, it has been observed that a single drug can bind to multiple locations within these sites [85] and that different drugs can simultaneously bind to different locations within the binding pocket [86].
The clinical relevance of efflux pumps extends beyond ABC multidrug transporters, as some members of the MFS family also mediate hyper resistance to drugs [87]. MFS proteins are typically composed of 12 or 14 transmembrane helices, with each transmembrane domain consisting of 6 or 7 helix bundles that pack together to create a cavity lined with amino acids that bind transport substrates. These transporters are generally smaller than ABC transporters and, similar to their counterparts, exhibit considerable substrate promiscuity. However, MFS transporters utilize the energy derived from a proton motive force rather than ATP hydrolysis to facilitate efflux.
Extensive research has been conducted on fungal multidrug transporters, with a particular focus on those found in Saccharomyces cerevisiae, Candida albicans, and Candida glabrata [77,88,89,90]. A group of S. cerevisiae strains was discovered that exhibited resistance to a wide range of drugs and natural toxins, known as pleiotropic drug resistance (PDR) [91]. Subsequent studies identified the primary genes involved in PDR of budding yeast to be ABC and MFS transporters and, importantly, zinc cluster transcription factors (TFs) [92,93,94,95] (Figure 2). The best-characterized PDR ABC transporters, Pdr5, Snq2, and Yor1, each play a role in resistance against hundreds of functionally and structurally unrelated drugs [96]. The most frequently mutated loci in PDR strains were found to encode two paralogous zinc cluster TFs, Pdr1 and Pdr3, which share 36% identity and are an essential part of the PDR network [97]. Recently, it has been shown in yeast that multiple PDR TFs contribute to the overall multidrug response with distinguishable sensitivities towards different xenobiotic molecules [98]. Pdr TFs were found to bind to pleiotropic drug resistance elements (PDREs) within the promoters of different multidrug transporter encoding genes, which is essential for PDR inducibility [99,100,101,102]. Pdr1 and Pdr3 are constitutively bound to DNA, regardless of the presence of drugs, suggesting that their activation is not dependent on changes in nuclear occupancy or DNA-binding [103]. Thakur et al. [104] showed that the regulatory domains XBD (xenobiotic binding domains) of both Pdr1 and Pdr3 bind the antifungal ketoconazole at low micromolar affinity, suggesting that ligand binding may increase their transcriptional activity. Similar to other known zinc cluster proteins, partial deletions or mutations within the regulatory domains of Pdr1 and/or Pdr3 result in constitutively active TFs that induce strong expression of ABC transporters [105,106,107], leading to an instance of up to 10-fold higher minimum inhibitory concentrations of ketoconazole. The pathogenic fungi Candida glabrata and Candida albicans have different mechanisms for controlling the expression of their drug efflux ABC transporters, with Pdr1 regulating Cdr1, Pdh1, and Snq2 transporters in C. glabrata [108,109] and Tac1 and Mrr1 controlling the expression of Cdr1, Cdr2 or Mdr1 transporters in C. albicans [110,111,112,113]. Aspergillus fumigatus lacks a Pdr1 homolog but has intrinsic drug resistance through AbcG1 and its zinc cluster TF regulator AtrR [114,115].
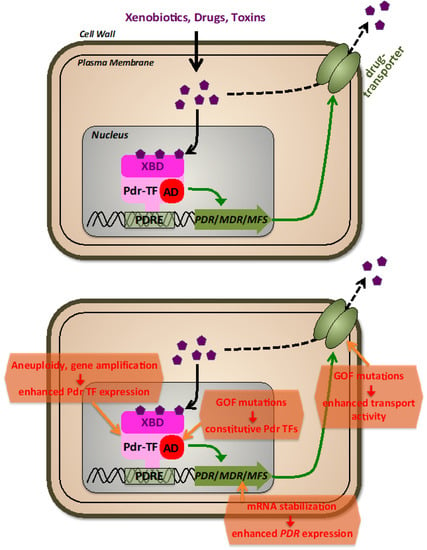
Figure 2.
The fungal pleiotropic drug resistance (PDR). Upper panel: General features of PDR regulation. Different xenobiotic molecules, including antibiotic drugs, are recognized by transcriptional activators (Pdr-TFs) via their xenobiotic binding domains (XBD). Pdr-TFs bound at pleiotropic drug resistance promoter elements (PDRE) stimulate via their transactivation domains (AD) the expression of PDR, MDR, or MFS genes encoding plasma membrane transporters of the pleiotropic drug resistance, multidrug resistance, or major facilitator superfamilies. Lower panel: molecular mechanisms leading to antifungal drug resistance via the PDR system.
The major mechanism of acquired antifungal resistance via drug efflux is the reinforcement of the expression of one or several efflux transporters (Figure 2). This can be achieved by an increase of genomic copies of the relevant PDR genes (gene amplification), the gain of function mutations in PDR TFs, or modification of PDR TFs mRNA stability. Amplification of genes, including ERG11 and TAC1, plays a significant role in the multidrug resistance of Candida species [116,117,118], leading to a linear relationship between gene copy number and the minimum inhibitory concentration of fluconazole. Aneuploidy leading to drug resistance has been observed in C. albicans, C. neoformans, and C. auris [119,120,121]. These findings show that increasing the copy number of genes involved in multidrug efflux, even modestly, creates enough hyper-resistance to have clinical implications. More recently, the fast and reversible amplification of PDR genes leading to drug resistance in C. albicans has been investigated mechanistically, revealing the exchange between long, inverted repeats flanking the amplified region on multiple chromosomes. It is evident from these and other results that hyper resistance by gene duplication is frequent in many fungal species and can be achieved by multiple mechanisms, including aneuploidy, nonhomologous extension after double-strand breaks, and chromosomal exchange between indirect and direct repeats [122]. Because aneuploidities generally reduce fitness, they seem to arise only during severe changes in environment such as drug exposure, and do not persist once the selective pressure is removed [123,124]. Remarkably, aneuploidities were found in a great portion of clinical S. cerevisiae strains [123].
Pleiotropic drug resistance by gain of function (GOF) mutations in TFs is dominated in budding yeast by the zinc cluster factors Pdr1 and Pdr3. It is important to note that, although not understood in detail on many occasions, these GOF mutations generally do not lead to overexpression of the TFs; instead, they create hyperactive TFs, which constitutively overexpress their transporter target genes. Amino acid substitutions in Pdr1, such as M308I in PDR1-2, F815S in PDR1-3, K302Q in PDR1-6, P298A in PDR1-7 and L1036W in PDR1-8 mutants, all result in multidrug-resistant phenotypes [105]. Three regions within the Pdr1 regulator with implications for drug resistance were identified, and mutant alleles PDR1-3, PDR1-6, and PDR1-8 were constructed to investigate their phenotypes, with PDR1-3 being the most effective in activating target promoters [105]. Similar to Pdr1, multiple amino acid substitutions in Pdr3, which cluster in short regions of the TF, can cause drug resistance [106]. According to [125], the presence of GOF mutations isolated from patients suggests that the Candida glabrata homolog Pdr1 plays an important role in the development of clinical antifungal resistance. In this case, Pdr1 expression itself is heavily regulated; thus, even a modest up-regulation of the wild-type Pdr1 copy contributes to drug resistance in this pathogenic fungus [126]. Extensive studies on CgPdr1 have shown an extraordinarily high number of GOF substitutions, which lead to enhanced drug resistance [125,127,128,129]. Recent advances point to the existence of an extensive regulatory domain within CgPdr1, which contributes to the highly flexible transactivation capacity via contacts with other co-activator proteins [126,130]. In Candida albicans, Tac1 is the main contributor to drug resistance via GOF mutations. Many points and small deletion mutations have been correlated in this TF with antifungal resistance by enhancing its transactivation capacity in the context with additional transcriptional co-activator complexes [112,131,132,133]. Additionally, multidrug resistance in C. albicans is caused by the Mrr1 regulator, which resembles Tac1. Mutations in Mrr1 can confer hyper resistance to benomyl, with many GOF mutations found in distinct locations exhibiting a dominant phenotype [113,134]. In the emerging high-risk pathogen Candida auris, two genes similar to C. albicans’ TAC1 were identified, with a majority of GOF mutations reported in one of them, TAC1B, that map to regions analogous to those in the C. albicans homolog [135,136]. This suggests a potential link between the regulatory mechanisms of the two proteins. Rahman et al. [137] identified a novel mechanism of hyper resistance in S. cerevisiae involving a set of serine residues in the Pdr5 linker region. Their study showed that alanine mutations in six different serines exhibited significant hyper resistance, and changing S837 to aspartate produced the same phenotype, which was the result of a 2–3x increase of Pdr5 that was due to an increased Pdr5 mRNA half-life. Mixed effects on both PDR transporter expression and mRNA stabilization had been previously characterized in azole-resistant C. albicans isolates [138]. In any case, it is important to note that overexpression of multidrug efflux transporters interferes with general cell homeostasis, most likely by interfering with proper intracellular metabolites [139], thus only emerges upon a strong selection in the presence of antifungal agents.
A novel mechanism of antifungal resistance has emerged recently by investigating the A666G mutation in the yeast Pdr5 transporter, which was found in several screens for hyper-resistant mutants [140,141]. It conferred 2–5x increased substrate-specific resistance but did not alter susceptibility to clotrimazole. Importantly, the mutant had no difference in the quantity of Pdr5 or the level of ATPase activity compared to the wild type, but it had enhanced cooperativity, which led to higher transport of xenobiotic compounds per molecule of ATP hydrolyzed [82,141].
4.2. Mutational Inhibition of Antifungal Drug–Target Interaction
Several antifungal drugs (azoles, echinocandins, manogepix, or orotomides) directly target specific essential enzymes in the pathogen, such as Erg11/Cyp51, Fks1, Gwt1, or DHODH. Therefore, conformational changes in the target enzyme caused by mutations leading to decreased drug recognition while maintaining sufficient enzyme activity are possible sources of acquired resistance.
Mutations in the ERG11(CYP51) gene are a common azole resistance mechanism in Candida albicans, altering the inhibition of the lanosterol demethylase enzyme [142,143,144]. The effect of these mutations, more than 140 described to date, is not uniform and likely a result of overall reduced azole-binding affinity to the mutant enzyme [145,146,147]. Documentation of new ERG11 mutations in azole-resistant clinical isolates continues to appear, but not all documented mutations were definitively shown to be tied to azole resistance [148,149,150,151,152,153]. Homozygous replacement of the native ERG11 alleles with a mutant ERG11 ORF encoding either a single or double amino acid substitution in the Erg11 protein showed that some substitutions directly influence or interfere with azole binding, while others indirectly affect resistance through, for example, the interaction with cytochrome P450 reductase [154]. The crystal structure of the Erg11 protein of C. albicans has been resolved, leading to further insight into the interactions between azole drugs and their target and the development of 3D models useful for future antifungal discovery [155,156]. Aspergillus spp. has two Cyp51 isoenzymes, Cyp51A, and Cyp51B, that can both act as sterol 14α-demethylase in vivo, resulting in no phenotypic difference in A. fumigatus strains with gene knock-outs of either enzyme [157,158,159]. However, a conditional mutant study showed that growth was abolished in the absence of both CYP51A and CYP51B [160]. The main azole resistance mechanism in A. fumigatus is the development of point mutations in CYP51A, which has less affinity for azole drugs than CYP51B, indicating a molecular basis for this resistance [161]. Lockhart et al. [162] reported that 60% of clinical A. fumigatus isolates had triazole resistance mutations within CYP51A, with the five most frequent mutations being G54, L98, G138, M220, and G448. These mutations have been shown to confer azole resistance in A. fumigatus through gene replacement experiments. Over 40 different CYP51A point mutations have been reported in clinical A. fumigatus isolates, but not all of them confer an azole resistance phenotype, and some can occur alone or in combination with other mutations [158,163,164,165,166,167,168,169]. Azole resistance is an increasing problem in Cryptococcus spp. due to prolonged treatment regimens, with CYP51 mutations identified as one of the main mechanisms [170,171]. Point mutations in CYP51, such as G468S and Y145F, have been shown to confer resistance to fluconazole and voriconazole in C. neoformans [172,173].
The target of echinocandins, β(1,3)-glucan synthase, is encoded by the FKS1 and FKS2 genes in Candida spp. Limited amino acid substitutions in the Fks subunits of glucan synthase are responsible for echinocandin resistance leading to clinical failures [174]. These mutations occur in two highly conserved hot spot regions of FKS1 in C. albicans [175] and in homologous regions of FKS2 in C. glabrata [176], resulting in elevated minimum inhibitory concentration (MIC) values. The most frequent and pronounced resistance phenotype in C. albicans is caused by amino acid changes at S641 and S645, while in C. glabrata, modifications at S663 in Fks2, S629 in Fks1, and F659 in Fks2 are the most prominent substitutions [176,177]. Additionally, in Aspergillus spp., although much less explored, echinocandin resistance due to point-mutated Fks subunits has been reported recently [178].
Point mutations leading to amino acid substitutions within Gwt1, V163A in C. glabrata, and V162A in C. albicans can cause resistance to manogepix but do not affect the activity of other antifungals, including azoles and echinocandins [179,180]. The reduced susceptibility to manogepix of V162A and V168A Gwt1 mutants expressed in C. albicans and Saccharomyces cerevisiae, respectively, indicates the significance of this particular valine residue in manogepix binding across various species.
The olorofim target, dihydroorotate dehydrogenase (DHODH), encoded by the pyrE gene in Aspergillus, also appears to provide a limited resource for mutations leading to olorofim resistance. A recent study applying long-term olorofim treatment in A. fumigatus revealed several amino acid substitutions within PyrE with a hotspot at G119 producing olorofim resistance [181].
Polyene antifungals target an essential membrane compound, ergosterol, instead of an essential cellular enzyme. This different mechanism of action might explain the rare occurrence of polyene resistance among fungi. Nevertheless, the most prominent way to achieve polyene resistance is the alteration of the sterol composition of the plasma membrane [182,183], which comes with a strong fitness trade-off [184]. Several mutations in genes involved in the biosynthesis of ergosterol (ERG genes) have been linked to this mechanism. In C. albicans, when the ERG11 and ERG3 genes, which encode for lanosterol 14α-demethylase and C-5 sterol desaturase, respectively, are dysfunctional, the membrane’s ergosterol is replaced with alternate sterols such as lanosterol, eburicol, and 4,14-dimethyl-zymosterol [183,184]. According to other studies, the substitution in ERG11 and the loss of function of ERG5 (C-22 sterol desaturase) in C. albicans are associated with an alternative membrane sterol composition and amphotericin B resistance [150,184]. In other Candida species, the inactivation of ERG6 (C-24 sterol methyl-transferase) and ERG2 (C-8 sterol isomerase) has a similar effect [185,186]. A mutation in ERG2, which results in its inactivation, is one of the few described mechanisms of polyene resistance in C. neoformans [187].
4.3. Resistance by Overexpression of Antifungal Drug Targets
An important strategy of fungal cells to lower the efficient drug concentration inside the cell is based on titrating the antifungal compound through an increase in the copies of the target enzyme. A recent high-density mutagenesis screen in S. cerevisiae with several antimicrobial agents revealed that the gain of function promoter mutants is enriched for the respective drug target protein on many occasions, pointing to a general strategy to adapt to antifungal treatments [188]. Resistance by overexpression of the drug target is especially relevant for azoles. ERG11 overexpression is a frequently observed mutational adaptation in clinically relevant azole-resistant Candida isolates [189,190,191,192]. One prominent mechanism of constitutive ERG11 expression acts via the gain of function point mutations in the Upc2 TF, which acts as a direct transcriptional activator of ERG11 in Candida spp. [193,194,195] In Aspergillus, different promoter rearrangements in the cyp51A gene leading to robust overexpression have been linked to azole resistance [196].
4.4. Alterations of Drug Modification, Activation, and Availability
Flucytosine (5-FC) stands out among antifungal agents due to its distinctive mode of action that involves inhibiting DNA, RNA, and protein synthesis and the fact that it acts as a prodrug, which means that it requires metabolic conversion through the pyrimidine salvage pathway to become activated [197]. The majority of research on 5-FC resistance mechanisms has concentrated on S. cerevisiae and C. albicans, with particular attention given to identifying mutations in crucial enzymes involved in the pyrimidine salvage pathways. These studies revealed that 5-FC resistance may occur due to the absence or alteration of any of the enzymes responsible for the activity of cytosine permease (FCY2), cytosine deaminase (FCA1, FCY1), or uracil phosphoribosyltransferase (FUR1). In S. cerevisiae, several studies have indicated that mutations in FCY1, FCY2, and FUR1 can result in resistance to 5-FC [198,199]. Disruption of cytosine deaminase is linked to high levels of dose-independent resistance, whereas loss of cytosine permease results in a lower, dose-dependent level of resistance, suggesting the involvement of two additional cytosine permeases, FCY21 and FCY22. Additionally, alternative entry routes of 5-FC exist via other cytosine adenine permease homologs, including TPN1, FUR4, and YOR071c, and can contribute significantly to 5-FC toxicity [200]. Kern et al. [201] have associated phenotypic 5-FC resistance with a single point mutation, R134S, in the FUR1 gene. For C. albicans, acquired 5-FC resistance has been functionally linked to a single R101C amino acid substitution in the Fur1 enzyme [202,203], and several point mutations in the FUR1 gene have been described in clinical 5-FC resistant isolates of C. glabrata [69]. In the emerging multiresistant C. auris, an I211F substitution in the FUR1 gene has been documented in a 5-FC-resistant isolate [204]. Bhattacharya et al. [119] have further exhibited the potential of transient gene duplication in C. auris during replicative aging, leading to the development of tolerance to 5-FC.
Another related mechanism of resistance is the decrease of the effective drug concentration by the selective impairment of azole influx into the fungal cell. This has been recently reported to be relevant for PDR- and ERG11-independent azole-resistant C. glabrata isolates. Here, the molecular key to resistance is mutations in the CgHXT4/6/7 hexose importer, which contributes to azole import across the fungal cell membrane [205].
5. Concluding Remarks
The indiscriminate use of antifungal drugs for prophylactic and empirical treatment of suspected invasive fungal diseases in high-risk individuals such as patients with cystic fibrosis, critically ill, or haemato–oncology patients is a worrying trend. Antifungal stewardship is essential to ensure the appropriate use of these drugs and preserve the limited antifungal options available, particularly in cases of highly transmissible fungal infections such as Candida spp. and Trichophyton spp. [206]. In recent years, systems biology approaches utilizing laboratory evolution techniques have demonstrated the impressive ability of Saccharomyces and Candida species to acquire resistance to existing antifungal agents [207,208,209]. Despite the ongoing threat of drug-resistant fungal infections to human health, there is renewed optimism in academia and industry to enhance our knowledge of the mechanisms underlying antifungal resistance and to develop new therapeutic strategies to combat these pathogens. Combination antimicrobial treatment is an effective strategy to prevent secondary antimicrobial resistance and is established for various bacterial and viral infections. For example, combining antifungal agents such as amphotericin B and flucytosine or fluconazole and flucytosine can prevent the selection of resistant fungal populations and limit the development of clinical antifungal resistance [210,211]. Further investigation is required to unravel the full potential of combinatorial antifungal treatment in the battle against secondary resistance. Another promising approach is to target the resistance mechanisms themselves, with the aim of restoring the efficacy of current antifungals [212,213]. Alternative methods are currently being developed to protect antifungals from resistance, including host-directed approaches such as immunotherapy [214], fungal vaccines [215,216], and improved antifungal targeting [217]. In addition, cell-based therapies, such as dendritic cell transfer and CAR T-cell therapy [218,219,220,221], have demonstrated promising outcomes in vitro but still need to be investigated in clinical trials. These approaches provide potential alternative options for managing antifungal resistance. As such, ongoing and future investigations into the mechanisms of antifungal drug resistance are expected to yield important insights and pave the way for improved clinical outcomes for diverse populations affected by drug-resistant fungal infections worldwide.
Author Contributions
P.O.-T., A.P.-A. and M.P. wrote, revised the manuscript, and agreed on the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Research by the authors was funded by grants from Ministerio de Ciencia, Innovación y Universidades PID2019-104214RB-I00 (A.P.-A., M.P.) and Agencia Valenciana de la Innovación INNEST/2021/266 (A.P.-A.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We apologize to all researchers whose work on related topics was not cited here due to space limitations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [PubMed]
- Sanyaolu, A.; Okorie, C.; Marinkovic, A.; Abbasi, A.F.; Prakash, S.; Mangat, J.; Hosein, Z.; Haider, N.; Chan, J. Candida auris: An Overview of the Emerging Drug-Resistant Fungal Infection. Infect. Chemother. 2022, 54, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Alastruey-Izquierdo, A.; Berman, J.; Bicanic, T.; Bignell, E.M.; Bowyer, P.; Bromley, M.; Brüggemann, R.; Garber, G.; Cornely, O.A.; et al. Tackling the emerging threat of antifungal resistance to human health. Nat. Rev. Microbiol. 2022, 20, 557–571. [Google Scholar] [CrossRef] [PubMed]
- WHO. Fungal Priority Pathogens List to Guide Research, Development and Public Health Action. Available online: https://www.who.int/publications/i/item/9789240060241 (accessed on 7 February 2023).
- Kainz, K.; Bauer, M.A.; Madeo, F.; Carmona-Gutierrez, D. Fungal infections in humans: The silent crisis. Microb. Cell 2020, 7, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Robbins, N.; Caplan, T.; Cowen, L.E. Molecular evolution of antifungal drug resistance. Annu. Rev. Microbiol. 2017, 71, 753–775. [Google Scholar] [CrossRef]
- Sheehan, D.J.; Hitchcock, C.A.; Sibley, C.M. Current and emerging azole antifungal agents. Clin. Microbiol. Rev. 1999, 12, 40–79. [Google Scholar] [CrossRef]
- Shafiei, M.; Peyton, L.; Hashemzadeh, M.; Foroumadi, A. History of the development of antifungal azoles: A review on structures, SAR, and mechanism of action. Bioorg. Chem. 2020, 104, 104240. [Google Scholar] [CrossRef]
- Parks, L.W.; Casey, W.M. Physiological implications of sterol biosynthesis in yeast. Annu. Rev. Microbiol. 1995, 49, 95–116. [Google Scholar] [CrossRef]
- Hitchcock, C.A.; Dickinson, K.; Brown, S.B.; Evans, E.G.; Adams, D.J. Interaction of azole antifungal antibiotics with cytochrome P-450-dependent 14 alpha-sterol demethylase purified from Candida albicans. Biochem. J. 1990, 266, 475–480. [Google Scholar] [CrossRef]
- Zhang, J.; Li, L.; Lv, Q.; Yan, L.; Wang, Y.; Jiang, Y. The fungal cyp51s: Their functions, structures, related drug resistance, and inhibitors. Front. Microbiol. 2019, 10, 691. [Google Scholar] [CrossRef]
- Rosam, K.; Monk, B.C.; Lackner, M. Sterol 14α-Demethylase Ligand-Binding Pocket-Mediated Acquired and Intrinsic Azole Resistance in Fungal Pathogens. J. Fungi 2021, 7, 1. [Google Scholar] [CrossRef]
- Watson, P.F.; Rose, M.E.; Ellis, S.W.; England, H.; Kelly, S.L. Defective sterol C5-6 desaturation and azole resistance: A new hypothesis for the mode of action of azole antifungals. Biochem. Biophys. Res. Commun. 1989, 164, 1170–1175. [Google Scholar] [CrossRef]
- Hector, R.F. Compounds active against cell walls of medically important fungi. Clin. Microbiol. Rev. 1993, 6, 1–21. [Google Scholar] [CrossRef]
- Ibe, C.; Munro, C.A. Fungal cell wall: An underexploited target for antifungal therapies. PLoS Pathog. 2021, 17, e1009470. [Google Scholar] [CrossRef]
- Ruiz-Herrera, J.; Elorza, M.V.; Valentín, E.; Sentandreu, R. Molecular organization of the cell wall of Candida albicans and its relation to pathogenicity. FEMS Yeast Res. 2006, 6, 14–29. [Google Scholar] [CrossRef]
- Sullivan, P.A.; Yin, C.Y.; Molloy, C.; Templeton, M.D.; Shepherd, M.G. An analysis of the metabolism and cell wall composition of Candida albicans during germ-tube formation. Can. J. Microbiol. 1983, 29, 1514–1525. [Google Scholar] [CrossRef]
- Cassone, A.; Mason, R.E.; Kerridge, D. Lysis of growing yeast-form cells of Candida albicans by echinocandin: A cytological study. Sabouraudia 1981, 19, 97–110. [Google Scholar] [CrossRef]
- Walsh, T.J.; Lee, J.W.; Kelly, P.; Bacher, J.; Lecciones, J.; Thomas, V.; Lyman, C.; Coleman, D.; Gordee, R.; Pizzo, P.A. Antifungal effects of the nonlinear pharmacokinetics of cilofungin, a 1,3-beta-glucan synthetase inhibitor, during continuous and intermittent intravenous infusions in treatment of experimental disseminated candidiasis. Antimicrob. Agents Chemother. 1991, 35, 1321–1328. [Google Scholar] [CrossRef]
- Carolus, H.; Pierson, S.; Lagrou, K.; Van Dijck, P. Amphotericin B and Other Polyenes-Discovery, Clinical Use, Mode of Action and Drug Resistance. J. Fungi 2020, 6, 321. [Google Scholar] [CrossRef]
- Odds, F.C.; Brown, A.J.P.; Gow, N.A.R. Antifungal agents: Mechanisms of action. Trends Microbiol. 2003, 11, 272–279. [Google Scholar] [CrossRef]
- Kamiński, D.M. Recent progress in the study of the interactions of amphotericin B with cholesterol and ergosterol in lipid environments. Eur. Biophys. J. 2014, 43, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W.; Demel, R.A.; de Kruyff, B.; Geurts van Kessel, W.S.; van Deenen, L.L. Studies on the biological properties of polyene antibiotics: Comparison of other polyenes with filipin in their ability to interact specifically with sterol. Biochim. Biophys. Acta 1972, 290, 1–14. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zotchev, S.B. Polyene macrolide antibiotics and their applications in human therapy. Curr. Med. Chem. 2003, 10, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D. Amphotericin B: Spectrum and resistance. J. Antimicrob. Chemother. 2002, 49 (Suppl. S1), 7–10. [Google Scholar] [CrossRef]
- Chandrasekar, P. Management of invasive fungal infections: A role for polyenes. J. Antimicrob. Chemother. 2011, 66, 457–465. [Google Scholar] [CrossRef]
- Basha, J.; Goudgaon, N.M. A comprehensive review on pyrimidine analogs-versatile scaffold with medicinal and biological potential. J. Mol. Struct. 2021, 1246, 131168. [Google Scholar] [CrossRef]
- Defever, K.S.; Whelan, W.L.; Rogers, A.L.; Beneke, E.S.; Veselenak, J.M.; Soll, D.R. Candida albicans resistance to 5-fluorocytosine: Frequency of partially resistant strains among clinical isolates. Antimicrob. Agents Chemother. 1982, 22, 810–815. [Google Scholar] [CrossRef]
- Stiller, R.L.; Bennett, J.E.; Scholer, H.J.; Wall, M.; Polak, A.; Stevens, D.A. Susceptibility to 5-fluorocytosine and prevalence of serotype in 402 Candida albicans isolates from the United States. Antimicrob. Agents Chemother. 1982, 22, 482–487. [Google Scholar] [CrossRef]
- Lotfali, E.; Fattahi, A.; Sayyahfar, S.; Ghasemi, R.; Rabiei, M.M.; Fathi, M.; Vakili, K.; Deravi, N.; Soheili, A.; Toreyhi, H.; et al. A Review on Molecular Mechanisms of Antifungal Resistance in Candida glabrata: Update and Recent Advances. Microb. Drug Resist 2021, 27, 1371–1388. [Google Scholar] [CrossRef]
- Diasio, R.B.; Bennett, J.E.; Myers, C.E. Mode of action of 5-fluorocytosine. Biochem. Pharmacol. 1978, 27, 703–707. [Google Scholar] [CrossRef]
- Polak, A.; Scholer, H.J. Mode of action of 5-fluorocytosine and mechanisms of resistance. Chemotherapy 1975, 21, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.; Wolfe, A.; Williamson, J.C. Antifungal resistance and the role of new therapeutic agents. Curr. Infect. Dis. Rep. 2022, 24, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Arastehfar, A.; Gabaldón, T.; Garcia-Rubio, R.; Jenks, J.D.; Hoenigl, M.; Salzer, H.J.F.; Ilkit, M.; Lass-Flörl, C.; Perlin, D.S. Drug-Resistant Fungi: An Emerging Challenge Threatening Our Limited Antifungal Armamentarium. Antibiot. 2020, 9, 877. [Google Scholar] [CrossRef] [PubMed]
- Hoenigl, M.; Sprute, R.; Egger, M.; Arastehfar, A.; Cornely, O.A.; Krause, R.; Lass-Flörl, C.; Prattes, J.; Spec, A.; Thompson, G.R.; et al. The antifungal pipeline: Fosmanogepix, ibrexafungerp, olorofim, opelconazole, and rezafungin. Drugs 2021, 81, 1703–1729. [Google Scholar] [CrossRef]
- Hoenigl, M.; Sprute, R.; Arastehfar, A.; Perfect, J.R.; Lass-Flörl, C.; Bellmann, R.; Prattes, J.; Thompson, G.R.; Wiederhold, N.P.; Al Obaidi, M.M.; et al. Invasive candidiasis: Investigational drugs in the clinical development pipeline and mechanisms of action. Expert Opin. Investig. Drugs 2022, 31, 795–812. [Google Scholar] [CrossRef]
- Mueller, S.W.; Kedzior, S.K.; Miller, M.A.; Reynolds, P.M.; Kiser, T.H.; Krsak, M.; Molina, K.C. An overview of current and emerging antifungal pharmacotherapy for invasive fungal infections. Expert Opin. Pharmacother. 2021, 22, 1355–1371. [Google Scholar] [CrossRef]
- Shaw, K.J.; Ibrahim, A.S. Fosmanogepix: A Review of the First-in-Class Broad Spectrum Agent for the Treatment of Invasive Fungal Infections. J. Fungi 2020, 6, 239. [Google Scholar] [CrossRef]
- Mann, P.A.; McLellan, C.A.; Koseoglu, S.; Si, Q.; Kuzmin, E.; Flattery, A.; Harris, G.; Sher, X.; Murgolo, N.; Wang, H.; et al. Chemical Genomics-Based Antifungal Drug Discovery: Targeting Glycosylphosphatidylinositol (GPI) Precursor Biosynthesis. ACS Infect. Dis. 2015, 1, 59–72. [Google Scholar] [CrossRef]
- Umemura, M.; Okamoto, M.; Nakayama, K.; Sagane, K.; Tsukahara, K.; Hata, K.; Jigami, Y. GWT1 gene is required for inositol acylation of glycosylphosphatidylinositol anchors in yeast. J. Biol. Chem. 2003, 278, 23639–23647. [Google Scholar] [CrossRef]
- Pittet, M.; Conzelmann, A. Biosynthesis and function of GPI proteins in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007, 1771, 405–420. [Google Scholar] [CrossRef]
- Klis, F.M.; Sosinska, G.J.; de Groot, P.W.J.; Brul, S. Covalently linked cell wall proteins of Candida albicans and their role in fitness and virulence. FEMS Yeast Res. 2009, 9, 1013–1028. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.-A.; Miyazaki, M.; Horii, T.; Sagane, K.; Tsukahara, K.; Hata, K. E1210, a new broad-spectrum antifungal, suppresses Candida albicans hyphal growth through inhibition of glycosylphosphatidylinositol biosynthesis. Antimicrob. Agents Chemother. 2012, 56, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Horii, T.; Hata, K.; Watanabe, N.-A.; Nakamoto, K.; Tanaka, K.; Shirotori, S.; Murai, N.; Inoue, S.; Matsukura, M.; et al. In vitro activity of E1210, a novel antifungal, against clinically important yeasts and molds. Antimicrob. Agents Chemother. 2011, 55, 4652–4658. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A.; Huband, M.D.; Flamm, R.K.; Bien, P.A.; Castanheira, M. Antimicrobial activity of manogepix, a first-in-class antifungal, and comparator agents tested against contemporary invasive fungal isolates from an international surveillance programme (2018–2019). J. Glob. Antimicrob. Resist. 2021, 26, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, N.P. Review of the novel investigational antifungal olorofim. J. Fungi 2020, 6, 122. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.D.; Sibley, G.E.M.; Beckmann, N.; Dobb, K.S.; Slater, M.J.; McEntee, L.; du Pré, S.; Livermore, J.; Bromley, M.J.; Wiederhold, N.P.; et al. F901318 represents a novel class of antifungal drug that inhibits dihydroorotate dehydrogenase. Proc. Natl. Acad. Sci. USA 2016, 113, 12809–12814. [Google Scholar] [CrossRef]
- Rauseo, A.M.; Coler-Reilly, A.; Larson, L.; Spec, A. Hope on the horizon: Novel fungal treatments in development. Open Forum Infect. Dis. 2020, 7, ofaa016. [Google Scholar] [CrossRef]
- Antimicrobial Resistance Collaborators Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [CrossRef]
- Bongomin, F.; Gago, S.; Oladele, R.O.; Denning, D.W. Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J. Fungi 2017, 3, 57. [Google Scholar] [CrossRef]
- Fisher, M.C.; Gurr, S.J.; Cuomo, C.A.; Blehert, D.S.; Jin, H.; Stukenbrock, E.H.; Stajich, J.E.; Kahmann, R.; Boone, C.; Denning, D.W.; et al. Threats posed by the fungal kingdom to humans, wildlife, and agriculture. MBio 2020, 11, e00449-20. [Google Scholar] [CrossRef]
- Rodrigues, M.L.; Nosanchuk, J.D. Fungal diseases as neglected pathogens: A wake-up call to public health officials. PLoS Negl. Trop. Dis. 2020, 14, e0007964. [Google Scholar] [CrossRef] [PubMed]
- Arastehfar, A.; Lass-Flörl, C.; Garcia-Rubio, R.; Daneshnia, F.; Ilkit, M.; Boekhout, T.; Gabaldon, T.; Perlin, D.S. The Quiet and Underappreciated Rise of Drug-Resistant Invasive Fungal Pathogens. J. Fungi 2020, 6, 138. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, A.C.; Sethi, P. Experimental evolution of drug resistance in human fungal pathogens. Curr. Opin. Genet. Dev. 2022, 76, 101965. [Google Scholar] [CrossRef]
- Ballard, E.; Melchers, W.J.G.; Zoll, J.; Brown, A.J.P.; Verweij, P.E.; Warris, A. In-host microevolution of Aspergillus fumigatus: A phenotypic and genotypic analysis. Fungal Genet. Biol. 2018, 113, 1–13. [Google Scholar] [CrossRef]
- Shields, R.K.; Nguyen, M.H.; Press, E.G.; Kwa, A.L.; Cheng, S.; Du, C.; Clancy, C.J. The presence of an FKS mutation rather than MIC is an independent risk factor for failure of echinocandin therapy among patients with invasive candidiasis due to Candida glabrata. Antimicrob. Agents Chemother. 2012, 56, 4862–4869. [Google Scholar] [CrossRef] [PubMed]
- Pristov, K.E.; Ghannoum, M.A. Resistance of Candida to azoles and echinocandins worldwide. Clin. Microbiol. Infect. 2019, 25, 792–798. [Google Scholar] [CrossRef]
- Johnson, E.M.; Warnock, D.W.; Luker, J.; Porter, S.R.; Scully, C. Emergence of azole drug resistance in Candida species from HIV-infected patients receiving prolonged fluconazole therapy for oral candidosis. J. Antimicrob. Chemother. 1995, 35, 103–114. [Google Scholar] [CrossRef]
- Arastehfar, A.; Carvalho, A.; Nguyen, M.H.; Hedayati, M.T.; Netea, M.G.; Perlin, D.S.; Hoenigl, M. COVID-19-Associated Candidiasis (CAC): An Underestimated Complication in the Absence of Immunological Predispositions? J. Fungi 2020, 6, 211. [Google Scholar] [CrossRef]
- Garg, D.; Muthu, V.; Sehgal, I.S.; Ramachandran, R.; Kaur, H.; Bhalla, A.; Puri, G.D.; Chakrabarti, A.; Agarwal, R. Coronavirus Disease (Covid-19) Associated Mucormycosis (CAM): Case Report and Systematic Review of Literature. Mycopathologia 2021, 186, 289–298. [Google Scholar] [CrossRef]
- Wauters, J.; Baar, I.; Meersseman, P.; Meersseman, W.; Dams, K.; De Paep, R.; Lagrou, K.; Wilmer, A.; Jorens, P.; Hermans, G. Invasive pulmonary aspergillosis is a frequent complication of critically ill H1N1 patients: A retrospective study. Intensive Care Med. 2012, 38, 1761–1768. [Google Scholar] [CrossRef]
- Janssen, N.A.F.; Nyga, R.; Vanderbeke, L.; Jacobs, C.; Ergün, M.; Buil, J.B.; van Dijk, K.; Altenburg, J.; Bouman, C.S.C.; van der Spoel, H.I.; et al. Multinational Observational Cohort Study of COVID-19-Associated Pulmonary Aspergillosis1. Emerg. Infect. Dis. 2021, 27, 2892–2898. [Google Scholar] [CrossRef]
- Fisher, M.C.; Hawkins, N.J.; Sanglard, D.; Gurr, S.J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 2018, 360, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Wolf, C.R. Molecular mechanisms of drug resistance. Biochem. J. 1990, 272, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Cowen, L.E.; Sanglard, D.; Howard, S.J.; Rogers, P.D.; Perlin, D.S. Mechanisms of Antifungal Drug Resistance. Cold Spring Harb. Perspect. Med. 2014, 5, a019752. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.C.; Kishony, R. Opposing effects of target overexpression reveal drug mechanisms. Nat. Commun. 2014, 5, 4296. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Dias, P.J.; Sá-Correia, I.; Teixeira, M.C. MFS multidrug transporters in pathogenic fungi: Do they have real clinical impact? Front. Physiol. 2014, 5, 197. [Google Scholar] [CrossRef]
- Morschhäuser, J. Regulation of multidrug resistance in pathogenic fungi. Fungal Genet. Biol. 2010, 47, 94–106. [Google Scholar] [CrossRef]
- Edlind, T.D.; Katiyar, S.K. Mutational analysis of flucytosine resistance in Candida glabrata. Antimicrob. Agents Chemother. 2010, 54, 4733–4738. [Google Scholar] [CrossRef]
- Berman, J.; Krysan, D.J. Drug resistance and tolerance in fungi. Nat. Rev. Microbiol. 2020, 18, 319–331. [Google Scholar] [CrossRef]
- Levinson, T.; Dahan, A.; Novikov, A.; Paran, Y.; Berman, J.; Ben-Ami, R. Impact of tolerance to fluconazole on treatment response in Candida albicans bloodstream infection. Mycoses 2021, 64, 78–85. [Google Scholar] [CrossRef]
- Prasad, R.; Rawal, M.K. Efflux pump proteins in antifungal resistance. Front. Pharmacol. 2014, 5, 202. [Google Scholar] [CrossRef]
- Coleman, J.J.; Mylonakis, E. Efflux in fungi: La pièce de résistance. PLoS Pathog. 2009, 5, e1000486. [Google Scholar] [CrossRef]
- Holmes, A.R.; Cardno, T.S.; Strouse, J.J.; Ivnitski-Steele, I.; Keniya, M.V.; Lackovic, K.; Monk, B.C.; Sklar, L.A.; Cannon, R.D. Targeting efflux pumps to overcome antifungal drug resistance. Future Med. Chem. 2016, 8, 1485–1501. [Google Scholar] [CrossRef]
- Sastré-Velásquez, L.E.; Dallemulle, A.; Kühbacher, A.; Baldin, C.; Alcazar-Fuoli, L.; Niedrig, A.; Müller, C.; Gsaller, F. The fungal expel of 5-fluorocytosine derived fluoropyrimidines mitigates its antifungal activity and generates a cytotoxic environment. PLoS Pathog. 2022, 18, e1011066. [Google Scholar] [CrossRef]
- Ren, Q.; Chen, K.; Paulsen, I.T. TransportDB: A comprehensive database resource for cytoplasmic membrane transport systems and outer membrane channels. Nucleic Acids Res. 2007, 35, D274–D279. [Google Scholar] [CrossRef] [PubMed]
- Víglaš, J.; Olejníková, P. An update on ABC transporters of filamentous fungi—From physiological substrates to xenobiotics. Microbiol. Res. 2021, 246, 126684. [Google Scholar] [CrossRef]
- Kovalchuk, A.; Driessen, A.J.M. Phylogenetic analysis of fungal ABC transporters. BMC Genom. 2010, 11, 177. [Google Scholar] [CrossRef] [PubMed]
- Sipos, G.; Kuchler, K. Fungal ATP-binding cassette (ABC) transporters in drug resistance & detoxification. Curr. Drug Targets 2006, 7, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Buechel, E.R.; Pinkett, H.W. Transcription factors and ABC transporters: From pleiotropic drug resistance to cellular signaling in yeast. FEBS Lett. 2020, 594, 3943–3964. [Google Scholar] [CrossRef]
- Gbelska, Y.; Krijger, J.-J.; Breunig, K.D. Evolution of gene families: The multidrug resistance transporter genes in five related yeast species. FEMS Yeast Res. 2006, 6, 345–355. [Google Scholar] [CrossRef]
- Harris, A.; Wagner, M.; Du, D.; Raschka, S.; Nentwig, L.-M.; Gohlke, H.; Smits, S.H.J.; Luisi, B.F.; Schmitt, L. Structure and efflux mechanism of the yeast pleiotropic drug resistance transporter Pdr5. Nat. Commun. 2021, 12, 5254. [Google Scholar] [CrossRef] [PubMed]
- Golin, J.; Ambudkar, S.V.; Gottesman, M.M.; Habib, A.D.; Sczepanski, J.; Ziccardi, W.; May, L. Studies with novel Pdr5p substrates demonstrate a strong size dependence for xenobiotic efflux. J. Biol. Chem. 2003, 278, 5963–5969. [Google Scholar] [CrossRef] [PubMed]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Chufan, E.E.; Kapoor, K.; Sim, H.-M.; Singh, S.; Talele, T.T.; Durell, S.R.; Ambudkar, S.V. Multiple transport-active binding sites are available for a single substrate on human P-glycoprotein (ABCB1). PLoS ONE 2013, 8, e82463. [Google Scholar] [CrossRef]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2003, 278, 39706–39710. [Google Scholar] [CrossRef]
- Drew, D.; North, R.A.; Nagarathinam, K.; Tanabe, M. Structures and general transport mechanisms by the major facilitator superfamily (MFS). Chem. Rev. 2021, 121, 5289–5335. [Google Scholar] [CrossRef]
- Prasad, R.; Banerjee, A.; Khandelwal, N.K.; Dhamgaye, S. The ABCs of Candida albicans Multidrug Transporter Cdr1. Eukaryot. Cell 2015, 14, 1154–1164. [Google Scholar] [CrossRef]
- Moreno, A.; Banerjee, A.; Prasad, R.; Falson, P. PDR-like ABC systems in pathogenic fungi. Res. Microbiol. 2019, 170, 417–425. [Google Scholar] [CrossRef]
- Prasad, R.; Balzi, E.; Banerjee, A.; Khandelwal, N.K. All about CDR transporters: Past, present, and future. Yeast 2019, 36, 223–233. [Google Scholar] [CrossRef]
- Saunders, G.W.; Rank, G.H. Allelism of pleiotropic drug resistance in Saccharomyces cerevisiae. Can. J. Genet. Cytol. 1982, 24, 493–503. [Google Scholar] [CrossRef]
- Nourani, A.; Wesolowski-Louvel, M.; Delaveau, T.; Jacq, C.; Delahodde, A. Multiple-drug-resistance phenomenon in the yeast Saccharomyces cerevisiae: Involvement of two hexose transporters. Mol. Cell. Biol. 1997, 17, 5453–5460. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, D.J.; Hallstrom, T.C.; Voet, M.; Wysock, W.; Golin, J.; Volckaert, G.; Moye-Rowley, W.S. Expression of an ATP-binding cassette transporter-encoding gene (YOR1) is required for oligomycin resistance in Saccharomyces cerevisiae. Mol. Cell. Biol. 1995, 15, 6875–6883. [Google Scholar] [CrossRef] [PubMed]
- Balzi, E.; Chen, W.; Ulaszewski, S.; Capieaux, E.; Goffeau, A. The multidrug resistance gene PDR1 from Saccharomyces cerevisiae. J. Biol. Chem. 1987, 262, 16871–16879. [Google Scholar] [CrossRef]
- Decottignies, A.; Lambert, L.; Catty, P.; Degand, H.; Epping, E.A.; Moye-Rowley, W.S.; Balzi, E.; Goffeau, A. Identification and characterization of SNQ2, a new multidrug ATP binding cassette transporter of the yeast plasma membrane. J. Biol. Chem. 1995, 270, 18150–18157. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowski, M.; Kolaczowska, A.; Luczynski, J.; Witek, S.; Goffeau, A. In vivo characterization of the drug resistance profile of the major ABC transporters and other components of the yeast pleiotropic drug resistance network. Microb. Drug Resist. 1998, 4, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Rogers, B.; Decottignies, A.; Kolaczkowski, M.; Carvajal, E.; Balzi, E.; Goffeau, A. The pleitropic drug ABC transporters from Saccharomyces cerevisiae. J. Mol. Microbiol. Biotechnol. 2001, 3, 207–214. [Google Scholar] [PubMed]
- Vanacloig-Pedros, E.; Lozano-Pérez, C.; Alarcón, B.; Pascual-Ahuir, A.; Proft, M. Live-cell assays reveal selectivity and sensitivity of the multidrug response in budding yeast. J. Biol. Chem. 2019, 294, 12933–12946. [Google Scholar] [CrossRef]
- Katzmann, D.J.; Hallstrom, T.C.; Mahé, Y.; Moye-Rowley, W.S. Multiple Pdr1p/Pdr3p binding sites are essential for normal expression of the ATP binding cassette transporter protein-encoding gene PDR5. J. Biol. Chem. 1996, 271, 23049–23054. [Google Scholar] [CrossRef]
- Katzmann, D.J.; Burnett, P.E.; Golin, J.; Mahé, Y.; Moye-Rowley, W.S. Transcriptional control of the yeast PDR5 gene by the PDR3 gene product. Mol. Cell. Biol. 1994, 14, 4653–4661. [Google Scholar] [CrossRef]
- Meyers, S.; Schauer, W.; Balzi, E.; Wagner, M.; Goffeau, A.; Golin, J. Interaction of the yeast pleiotropic drug resistance genes PDR1 and PDR5. Curr. Genet. 1992, 21, 431–436. [Google Scholar] [CrossRef]
- DeRisi, J.; van den Hazel, B.; Marc, P.; Balzi, E.; Brown, P.; Jacq, C.; Goffeau, A. Genome microarray analysis of transcriptional activation in multidrug resistance yeast mutants. FEBS Lett. 2000, 470, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wang, L.; Milgrom, E.; Shen, W.-C.W. On the mechanism of constitutive Pdr1 activator-mediated PDR5 transcription in Saccharomyces cerevisiae: Evidence for enhanced recruitment of coactivators and altered nucleosome structures. J. Biol. Chem. 2004, 279, 42677–42686. [Google Scholar] [CrossRef] [PubMed]
- Thakur, J.K.; Arthanari, H.; Yang, F.; Pan, S.-J.; Fan, X.; Breger, J.; Frueh, D.P.; Gulshan, K.; Li, D.K.; Mylonakis, E.; et al. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 2008, 452, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, E.; van den Hazel, H.B.; Cybularz-Kolaczkowska, A.; Balzi, E.; Goffeau, A. Molecular and phenotypic characterization of yeast PDR1 mutants that show hyperactive transcription of various ABC multidrug transporter genes. Mol. Gen. Genet. 1997, 256, 406–415. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nourani, A.; Papajova, D.; Delahodde, A.; Jacq, C.; Subik, J. Clustered amino acid substitutions in the yeast transcription regulator Pdr3p increase pleiotropic drug resistance and identify a new central regulatory domain. Mol. Gen. Genet. 1997, 256, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, A.; Kolaczkowski, M.; Delahodde, A.; Goffeau, A. Functional dissection of Pdr1p, a regulator of multidrug resistance in Saccharomyces cerevisiae. Mol. Genet. Genom. 2002, 267, 96–106. [Google Scholar] [CrossRef]
- Vermitsky, J.-P.; Edlind, T.D. Azole resistance in Candida glabrata: Coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob. Agents Chemother. 2004, 48, 3773–3781. [Google Scholar] [CrossRef]
- Whaley, S.G.; Zhang, Q.; Caudle, K.E.; Rogers, P.D. Relative Contribution of the ABC Transporters Cdr1, Pdh1, and Snq2 to Azole Resistance in Candida glabrata. Antimicrob. Agents Chemother. 2018, 62, e01070-18. [Google Scholar] [CrossRef]
- Coste, A.T.; Karababa, M.; Ischer, F.; Bille, J.; Sanglard, D. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot. Cell 2004, 3, 1639–1652. [Google Scholar] [CrossRef]
- Liu, T.T.; Znaidi, S.; Barker, K.S.; Xu, L.; Homayouni, R.; Saidane, S.; Morschhäuser, J.; Nantel, A.; Raymond, M.; Rogers, P.D. Genome-wide expression and location analyses of the Candida albicans Tac1p regulon. Eukaryot. Cell 2007, 6, 2122–2138. [Google Scholar] [CrossRef]
- Coste, A.; Selmecki, A.; Forche, A.; Diogo, D.; Bougnoux, M.-E.; d’Enfert, C.; Berman, J.; Sanglard, D. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot. Cell 2007, 6, 1889–1904. [Google Scholar] [CrossRef] [PubMed]
- Dunkel, N.; Blass, J.; Rogers, P.D.; Morschhäuser, J. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol. Microbiol. 2008, 69, 827–840. [Google Scholar] [CrossRef]
- Hagiwara, D.; Miura, D.; Shimizu, K.; Paul, S.; Ohba, A.; Gonoi, T.; Watanabe, A.; Kamei, K.; Shintani, T.; Moye-Rowley, W.S.; et al. A Novel Zn2-Cys6 Transcription Factor AtrR Plays a Key Role in an Azole Resistance Mechanism of Aspergillus fumigatus by Co-regulating cyp51A and cdr1B Expressions. PLoS Pathog. 2017, 13, e1006096. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Stamnes, M.; Thomas, G.H.; Liu, H.; Hagiwara, D. AtrR is an essential determinant of azole resistance in Aspergillus fumigatus. mBio 2019, 10, e02563-18. [Google Scholar] [CrossRef] [PubMed]
- Selmecki, A.; Forche, A.; Berman, J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 2006, 313, 367–370. [Google Scholar] [CrossRef]
- Selmecki, A.; Gerami-Nejad, M.; Paulson, C.; Forche, A.; Berman, J. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol. Microbiol. 2008, 68, 624–641. [Google Scholar] [CrossRef]
- Selmecki, A.M.; Dulmage, K.; Cowen, L.E.; Anderson, J.B.; Berman, J. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet. 2009, 5, e1000705. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Holowka, T.; Orner, E.P.; Fries, B.C. Gene Duplication Associated with Increased Fluconazole Tolerance in Candida auris cells of Advanced Generational Age. Sci. Rep. 2019, 9, 5052. [Google Scholar] [CrossRef]
- Ngamskulrungroj, P.; Chang, Y.; Hansen, B.; Bugge, C.; Fischer, E.; Kwon-Chung, K.J. Characterization of the chromosome 4 genes that affect fluconazole-induced disomy formation in Cryptococcus neoformans. PLoS ONE 2012, 7, e33022. [Google Scholar] [CrossRef]
- Sionov, E.; Chang, Y.C.; Kwon-Chung, K.J. Azole heteroresistance in Cryptococcus neoformans: Emergence of resistant clones with chromosomal disomy in the mouse brain during fluconazole treatment. Antimicrob. Agents Chemother. 2013, 57, 5127–5130. [Google Scholar] [CrossRef]
- Banerjee, A.; Rahman, H.; Prasad, R.; Golin, J. How fungal multidrug transporters mediate hyper resistance through DNA amplification and mutation. Mol. Microbiol. 2022, 118, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.J.; Nelliat, A. A double-edged sword: Aneuploidy is a prevalent strategy in fungal adaptation. Genes 2019, 10, 787. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Todd, R.T.; Selmecki, A.; Jiang, Y.-Y.; Cao, Y.-B.; Berman, J. The fitness costs and benefits of trisomy of each Candida albicans chromosome. Genetics 2021, 218, iyab056. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Ischer, F.; Calabrese, D.; Posteraro, B.; Sanguinetti, M.; Fadda, G.; Rohde, B.; Bauser, C.; Bader, O.; Sanglard, D. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 2009, 5, e1000268. [Google Scholar] [CrossRef] [PubMed]
- Khakhina, S.; Simonicova, L.; Moye-Rowley, W.S. Positive autoregulation and repression of transactivation are key regulatory features of the Candida glabrata Pdr1 transcription factor. Mol. Microbiol. 2018, 107, 747–764. [Google Scholar] [CrossRef]
- Tsai, H.-F.; Krol, A.A.; Sarti, K.E.; Bennett, J.E. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 2006, 50, 1384–1392. [Google Scholar] [CrossRef]
- Torelli, R.; Posteraro, B.; Ferrari, S.; La Sorda, M.; Fadda, G.; Sanglard, D.; Sanguinetti, M. The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributing to the CgPDR1-dependent azole resistance of Candida glabrata. Mol. Microbiol. 2008, 68, 186–201. [Google Scholar] [CrossRef]
- Vermitsky, J.-P.; Earhart, K.D.; Smith, W.L.; Homayouni, R.; Edlind, T.D.; Rogers, P.D. Pdr1 regulates multidrug resistance in Candida glabrata: Gene disruption and genome-wide expression studies. Mol. Microbiol. 2006, 61, 704–722. [Google Scholar] [CrossRef]
- Simonicova, L.; Moye-Rowley, W.S. Functional information from clinically-derived drug resistant forms of the Candida glabrata Pdr1 transcription factor. PLoS Genet. 2020, 16, e1009005. [Google Scholar] [CrossRef]
- Coste, A.; Turner, V.; Ischer, F.; Morschhäuser, J.; Forche, A.; Selmecki, A.; Berman, J.; Bille, J.; Sanglard, D. A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics 2006, 172, 2139–2156. [Google Scholar] [CrossRef]
- Liu, Z.; Myers, L.C. Mediator Tail Module Is Required for Tac1-Activated CDR1 Expression and Azole Resistance in Candida albicans. Antimicrob. Agents Chemother. 2017, 61, e01342-17. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Myers, L.C. Candida albicans Swi/Snf and Mediator Complexes Differentially Regulate Mrr1-Induced MDR1 Expression and Fluconazole Resistance. Antimicrob. Agents Chemother. 2017, 61, e01344-17. [Google Scholar] [CrossRef]
- Morschhauser, J.; Barker, K.S.; Liu, T.T.; Bla, B.W.J.; Rogers, P.D. The transcription factor Mrr1p controls expression of the MDR1 efflux 7 pump and mediates multidrug resistance in Candida albicans. PLoS Pathog. 2007, 3, e164. [Google Scholar] [CrossRef] [PubMed]
- Rybak, J.M.; Muñoz, J.F.; Barker, K.S.; Parker, J.E.; Esquivel, B.D.; Berkow, E.L.; Lockhart, S.R.; Gade, L.; Palmer, G.E.; White, T.C.; et al. Mutations in TAC1B: A Novel Genetic Determinant of Clinical Fluconazole Resistance in Candida auris. MBio 2020, 11, e00365-20. [Google Scholar] [CrossRef]
- Rybak, J.M.; Cuomo, C.A.; David Rogers, P. The molecular and genetic basis of antifungal resistance in the emerging fungal pathogen Candida auris. Curr. Opin. Microbiol. 2022, 70, 102208. [Google Scholar] [CrossRef] [PubMed]
- Rahman, H.; Rudrow, A.; Carneglia, J.; Joly, S.S.P.; Nicotera, D.; Naldrett, M.; Choy, J.; Ambudkar, S.V.; Golin, J. Nonsynonymous Mutations in Linker-2 of the Pdr5 Multidrug Transporter Identify a New RNA Stability Element. G3 2020, 10, 357–369. [Google Scholar] [CrossRef]
- Manoharlal, R.; Gaur, N.A.; Panwar, S.L.; Morschhäuser, J.; Prasad, R. Transcriptional activation and increased mRNA stability contribute to overexpression of CDR1 in azole-resistant Candida albicans. Antimicrob. Agents Chemother. 2008, 52, 1481–1492. [Google Scholar] [CrossRef]
- Knorre, D.A.; Galkina, K.V.; Shirokovskikh, T.; Banerjee, A.; Prasad, R. Do Multiple Drug Resistance Transporters Interfere with Cell Functioning under Normal Conditions? Biochemistry 2020, 85, 1560–1569. [Google Scholar] [CrossRef]
- Arya, N.; Rahman, H.; Rudrow, A.; Wagner, M.; Schmitt, L.; Ambudkar, S.V.; Golin, J. An A666G mutation in transmembrane helix 5 of the yeast multidrug transporter Pdr5 increases drug efflux by enhancing cooperativity between transport sites. Mol. Microbiol. 2019, 112, 1131–1144. [Google Scholar] [CrossRef]
- Downes, M.T.; Mehla, J.; Ananthaswamy, N.; Wakschlag, A.; Lamonde, M.; Dine, E.; Ambudkar, S.V.; Golin, J. The transmission interface of the Saccharomyces cerevisiae multidrug transporter Pdr5: Val-656 located in intracellular loop 2 plays a major role in drug resistance. Antimicrob. Agents Chemother. 2013, 57, 1025–1034. [Google Scholar] [CrossRef]
- Nishimoto, A.T.; Sharma, C.; Rogers, P.D. Molecular and genetic basis of azole antifungal resistance in the opportunistic pathogenic fungus Candida albicans. J. Antimicrob. Chemother. 2020, 75, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Whaley, S.G.; Berkow, E.L.; Rybak, J.M.; Nishimoto, A.T.; Barker, K.S.; Rogers, P.D. Azole Antifungal Resistance in Candida albicans and Emerging Non-albicans Candida Species. Front. Microbiol. 2016, 7, 2173. [Google Scholar] [CrossRef] [PubMed]
- Morio, F.; Loge, C.; Besse, B.; Hennequin, C.; Le Pape, P. Screening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: New substitutions and a review of the literature. Diagn. Microbiol. Infect. Dis. 2010, 66, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Sanglard, D.; Ischer, F.; Koymans, L.; Bille, J. Amino acid substitutions in the cytochrome P-450 lanosterol 14alpha-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob. Agents Chemother. 1998, 42, 241–253. [Google Scholar] [CrossRef]
- Kelly, S.L.; Lamb, D.C.; Kelly, D.E. Y132H substitution in Candida albicans sterol 14alpha-demethylase confers fluconazole resistance by preventing binding to haem. FEMS Microbiol. Lett. 1999, 180, 171–175. [Google Scholar] [CrossRef][Green Version]
- Kelly, S.L.; Lamb, D.C.; Loeffler, J.; Einsele, H.; Kelly, D.E. The G464S amino acid substitution in Candida albicans sterol 14alpha-demethylase causes fluconazole resistance in the clinic through reduced affinity. Biochem. Biophys. Res. Commun. 1999, 262, 174–179. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, L.; Li, C. Susceptibility of clinical isolates of Candida species to fluconazole and detection of Candida albicans ERG11 mutations. J. Antimicrob. Chemother. 2008, 61, 798–804. [Google Scholar] [CrossRef]
- Wang, H.; Kong, F.; Sorrell, T.C.; Wang, B.; McNicholas, P.; Pantarat, N.; Ellis, D.; Xiao, M.; Widmer, F.; Chen, S.C. Rapid detection of ERG11 gene mutations in clinical Candida albicans isolates with reduced susceptibility to fluconazole by rolling circle amplification and DNA sequencing. BMC Microbiol. 2009, 9, 167. [Google Scholar] [CrossRef]
- Martel, C.M.; Parker, J.E.; Bader, O.; Weig, M.; Gross, U.; Warrilow, A.G.S.; Kelly, D.E.; Kelly, S.L. A clinical isolate of Candida albicans with mutations in ERG11 (encoding sterol 14alpha-demethylase) and ERG5 (encoding C22 desaturase) is cross resistant to azoles and amphotericin B. Antimicrob. Agents Chemother. 2010, 54, 3578–3583. [Google Scholar] [CrossRef]
- Xiang, M.-J.; Liu, J.-Y.; Ni, P.-H.; Wang, S.; Shi, C.; Wei, B.; Ni, Y.-X.; Ge, H.-L. Erg11 mutations associated with azole resistance in clinical isolates of Candida albicans. FEMS Yeast Res. 2013, 13, 386–393. [Google Scholar] [CrossRef]
- Ying, Y.; Zhao, Y.; Hu, X.; Cai, Z.; Liu, X.; Jin, G.; Zhang, J.; Zhang, J.; Liu, J.; Huang, X. In vitro fluconazole susceptibility of 1,903 clinical isolates of Candida albicans and the identification of ERG11 mutations. Microb. Drug Resist. 2013, 19, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, H.-F.; Liu, Y.-Y.; Xu, X.-H.; Ye, Y.; Li, J.-B. Reduced susceptibility of Candida albicans clinical isolates to azoles and detection of mutations in the ERG11 gene. Diagn. Microbiol. Infect. Dis. 2013, 77, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Flowers, S.A.; Colón, B.; Whaley, S.G.; Schuler, M.A.; Rogers, P.D. Contribution of clinically derived mutations in ERG11 to azole resistance in Candida albicans. Antimicrob. Agents Chemother. 2015, 59, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Friggeri, L.; Wawrzak, Z.; Qi, A.; Hoekstra, W.J.; Schotzinger, R.J.; York, J.D.; Guengerich, F.P.; Lepesheva, G.I. Structural analyses of Candida albicans sterol 14α-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J. Biol. Chem. 2017, 292, 6728–6743. [Google Scholar] [CrossRef]
- Keniya, M.V.; Sabherwal, M.; Wilson, R.K.; Woods, M.A.; Sagatova, A.A.; Tyndall, J.D.A.; Monk, B.C. Crystal Structures of Full-Length Lanosterol 14α-Demethylases of Prominent Fungal Pathogens Candida albicans and Candida glabrata Provide Tools for Antifungal Discovery. Antimicrob. Agents Chemother. 2018, 62, e01134-18. [Google Scholar] [CrossRef]
- Mellado, E.; Garcia-Effron, G.; Buitrago, M.J.; Alcazar-Fuoli, L.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L. Targeted gene disruption of the 14-alpha sterol demethylase (cyp51A) in Aspergillus fumigatus and its role in azole drug susceptibility. Antimicrob. Agents Chemother. 2005, 49, 2536–2538. [Google Scholar] [CrossRef]
- Garcia-Effron, G.; Mellado, E.; Gomez-Lopez, A.; Alcazar-Fuoli, L.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L. Differences in interactions between azole drugs related to modifications in the 14-alpha sterol demethylase gene (cyp51A) of Aspergillus fumigatus. Antimicrob. Agents Chemother. 2005, 49, 2119–2121. [Google Scholar] [CrossRef]
- Martel, C.M.; Parker, J.E.; Warrilow, A.G.S.; Rolley, N.J.; Kelly, S.L.; Kelly, D.E. Complementation of a Saccharomyces cerevisiae ERG11/CYP51 (sterol 14α-demethylase) doxycycline-regulated mutant and screening of the azole sensitivity of Aspergillus fumigatus isoenzymes CYP51A and CYP51B. Antimicrob. Agents Chemother. 2010, 54, 4920–4923. [Google Scholar] [CrossRef]
- Hu, W.; Sillaots, S.; Lemieux, S.; Davison, J.; Kauffman, S.; Breton, A.; Linteau, A.; Xin, C.; Bowman, J.; Becker, J.; et al. Essential gene identification and drug target prioritization in Aspergillus fumigatus. PLoS Pathog. 2007, 3, e24. [Google Scholar] [CrossRef]
- Warrilow, A.G.S.; Melo, N.; Martel, C.M.; Parker, J.E.; Nes, W.D.; Kelly, S.L.; Kelly, D.E. Expression, purification, and characterization of Aspergillus fumigatus sterol 14-alpha demethylase (CYP51) isoenzymes A and B. Antimicrob. Agents Chemother. 2010, 54, 4225–4234. [Google Scholar] [CrossRef]
- Lockhart, S.R.; Frade, J.P.; Etienne, K.A.; Pfaller, M.A.; Diekema, D.J.; Balajee, S.A. Azole resistance in Aspergillus fumigatus isolates from the ARTEMIS global surveillance study is primarily due to the TR/L98H mutation in the cyp51A gene. Antimicrob. Agents Chemother. 2011, 55, 4465–4468. [Google Scholar] [CrossRef] [PubMed]
- Albarrag, A.M.; Anderson, M.J.; Howard, S.J.; Robson, G.D.; Warn, P.A.; Sanglard, D.; Denning, D.W. Interrogation of related clinical pan-azole-resistant Aspergillus fumigatus strains: G138C, Y431C, and G434C single nucleotide polymorphisms in cyp51A, upregulation of cyp51A, and integration and activation of transposon Atf1 in the cyp51A promoter. Antimicrob. Agents Chemother. 2011, 55, 5113–5121. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Ferreira, M.E.; Capellaro, J.L.; dos Reis Marques, E.; Malavazi, I.; Perlin, D.; Park, S.; Anderson, J.B.; Colombo, A.L.; Arthington-Skaggs, B.A.; Goldman, M.H.S.; et al. In vitro evolution of itraconazole resistance in Aspergillus fumigatus involves multiple mechanisms of resistance. Antimicrob. Agents Chemother. 2004, 48, 4405–4413. [Google Scholar] [CrossRef]
- Mann, P.A.; Parmegiani, R.M.; Wei, S.-Q.; Mendrick, C.A.; Li, X.; Loebenberg, D.; DiDomenico, B.; Hare, R.S.; Walker, S.S.; McNicholas, P.M. Mutations in Aspergillus fumigatus resulting in reduced susceptibility to posaconazole appear to be restricted to a single amino acid in the cytochrome P450 14alpha-demethylase. Antimicrob. Agents Chemother. 2003, 47, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Mellado, E.; Garcia-Effron, G.; Alcazar-Fuoli, L.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L. Substitutions at methionine 220 in the 14alpha-sterol demethylase (Cyp51A) of Aspergillus fumigatus are responsible for resistance in vitro to azole antifungal drugs. Antimicrob. Agents Chemother. 2004, 48, 2747–2750. [Google Scholar] [CrossRef]
- Snelders, E.; Karawajczyk, A.; Verhoeven, R.J.A.; Venselaar, H.; Schaftenaar, G.; Verweij, P.E.; Melchers, W.J.G. The structure–function relationship of the Aspergillus fumigatus cyp51A L98H conversion by site-directed mutagenesis: The mechanism of L98H azole resistance. Fungal Genet. Biol. 2011, 48, 1062–1070. [Google Scholar] [CrossRef]
- Krishnan Natesan, S.; Wu, W.; Cutright, J.L.; Chandrasekar, P.H. In vitro-in vivo correlation of voriconazole resistance due to G448S mutation (cyp51A gene) in Aspergillus fumigatus. Diagn. Microbiol. Infect. Dis. 2012, 74, 272–277. [Google Scholar] [CrossRef]
- Escribano, P.; Recio, S.; Peláez, T.; Bouza, E.; Guinea, J. Aspergillus fumigatus strains with mutations in the cyp51A gene do not always show phenotypic resistance to itraconazole, voriconazole, or posaconazole. Antimicrob. Agents Chemother. 2011, 55, 2460–2462. [Google Scholar] [CrossRef]
- Lamb, D.C.; Corran, A.; Baldwin, B.C.; Kwon-Chung, J.; Kelly, S.L. Resistant P45051A1 activity in azole antifungal tolerant Cryptococcus neoformans from AIDS patients. FEBS Lett. 1995, 368, 326–330. [Google Scholar] [CrossRef]
- Perfect, J.R.; Cox, G.M. Drug resistance in Cryptococcus neoformans. Drug Resist Updat. 1999, 2, 259–269. [Google Scholar] [CrossRef]
- Sionov, E.; Chang, Y.C.; Garraffo, H.M.; Dolan, M.A.; Ghannoum, M.A.; Kwon-Chung, K.J. Identification of a Cryptococcus neoformans cytochrome P450 lanosterol 14α-demethylase (Erg11) residue critical for differential susceptibility between fluconazole/voriconazole and itraconazole/posaconazole. Antimicrob. Agents Chemother. 2012, 56, 1162–1169. [Google Scholar] [CrossRef]
- Rodero, L.; Mellado, E.; Rodriguez, A.C.; Salve, A.; Guelfand, L.; Cahn, P.; Cuenca-Estrella, M.; Davel, G.; Rodriguez-Tudela, J.L. G484S amino acid substitution in lanosterol 14-alpha demethylase (ERG11) is related to fluconazole resistance in a recurrent Cryptococcus neoformans clinical isolate. Antimicrob. Agents Chemother. 2003, 47, 3653–3656. [Google Scholar] [CrossRef] [PubMed]
- Perlin, D.S. Current perspectives on echinocandin class drugs. Future Microbiol. 2011, 6, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kelly, R.; Kahn, J.N.; Robles, J.; Hsu, M.J.; Register, E.; Li, W.; Vyas, V.; Fan, H.; Abruzzo, G.; et al. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob. Agents Chemother. 2005, 49, 3264–3273. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Effron, G.; Lee, S.; Park, S.; Cleary, J.D.; Perlin, D.S. Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-beta-D-glucan synthase: Implication for the existing susceptibility breakpoint. Antimicrob. Agents Chemother. 2009, 53, 3690–3699. [Google Scholar] [CrossRef]
- Garcia-Effron, G.; Park, S.; Perlin, D.S. Correlating echinocandin MIC and kinetic inhibition of fks1 mutant glucan synthases for Candida albicans: Implications for interpretive breakpoints. Antimicrob. Agents Chemother. 2009, 53, 112–122. [Google Scholar] [CrossRef]
- Jiménez-Ortigosa, C.; Moore, C.; Denning, D.W.; Perlin, D.S. Emergence of Echinocandin Resistance Due to a Point Mutation in the fks1 Gene of Aspergillus fumigatus in a Patient with Chronic Pulmonary Aspergillosis. Antimicrob. Agents Chemother. 2017, 61, e01277-17. [Google Scholar] [CrossRef]
- Wiederhold, N.P. Pharmacodynamics, mechanisms of action and resistance, and spectrum of activity of new antifungal agents. J. Fungi 2022, 8, 857. [Google Scholar] [CrossRef]
- Kapoor, M.; Moloney, M.; Soltow, Q.A.; Pillar, C.M.; Shaw, K.J. Evaluation of resistance development to the gwt1 inhibitor manogepix (APX001A) in candida species. Antimicrob. Agents Chemother. 2019, 64, e01387-19. [Google Scholar] [CrossRef]
- Buil, J.B.; Oliver, J.D.; Law, D.; Baltussen, T.; Zoll, J.; Hokken, M.W.J.; Tehupeiory-Kooreman, M.; Melchers, W.J.G.; Birch, M.; Verweij, P.E. Resistance profiling of Aspergillus fumigatus to olorofim indicates absence of intrinsic resistance and unveils the molecular mechanisms of acquired olorofim resistance. Emerg. Microbes. Infect. 2022, 11, 703–714. [Google Scholar] [CrossRef]
- Geber, A.; Hitchcock, C.A.; Swartz, J.E.; Pullen, F.S.; Marsden, K.E.; Kwon-Chung, K.J.; Bennett, J.E. Deletion of the Candida glabrata ERG3 and ERG11 genes: Effect on cell viability, cell growth, sterol composition, and antifungal susceptibility. Antimicrob. Agents Chemother. 1995, 39, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Sanglard, D.; Ischer, F.; Parkinson, T.; Falconer, D.; Bille, J. Candida albicans mutations in the ergosterol biosynthetic pathway and resistance to several antifungal agents. Antimicrob. Agents Chemother. 2003, 47, 2404–2412. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.M.; Lancaster, A.K.; Scherz-Shouval, R.; Whitesell, L.; Lindquist, S. Fitness trade-offs restrict the evolution of resistance to amphotericin B. PLoS Biol. 2013, 11, e1001692. [Google Scholar] [CrossRef]
- Ahmad, S.; Joseph, L.; Parker, J.E.; Asadzadeh, M.; Kelly, S.L.; Meis, J.F.; Khan, Z. ERG6 and ERG2 Are Major Targets Conferring Reduced Susceptibility to Amphotericin B in Clinical Candida glabrata Isolates in Kuwait. Antimicrob. Agents Chemother. 2019, 63, e01900-18. [Google Scholar] [CrossRef] [PubMed]
- Young, L.Y.; Hull, C.M.; Heitman, J. Disruption of ergosterol biosynthesis confers resistance to amphotericin B in Candida lusitaniae. Antimicrob. Agents Chemother. 2003, 47, 2717–2724. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.L.; Lamb, D.C.; Taylor, M.; Corran, A.J. Resistance to amphotericin B associated with defective sterol Δ8→7 isomerase in a Cryptococcus neoformans strain from an AIDS patient. FEMS Microbiol. 1994, 122, 39–42. [Google Scholar] [CrossRef]
- Michel, A.H.; van Schie, S.; Mosbach, A.; Scalliet, G.; Kornmann, B. Exploiting homologous recombination increases SATAY efficiency for loss- and gain-of-function screening. BioRxiv 2019, 866483. [Google Scholar] [CrossRef]
- Feng, W.; Yang, J.; Xi, Z.; Qiao, Z.; Lv, Y.; Wang, Y.; Ma, Y.; Wang, Y.; Cen, W. Mutations and/or Overexpressions of ERG4 and ERG11 Genes in Clinical Azoles-Resistant Isolates of Candida albicans. Microb. Drug Resist. 2017, 23, 563–570. [Google Scholar] [CrossRef]
- Morais Vasconcelos Oliveira, J.; Conceição Oliver, J.; Latércia Tranches Dias, A.; Barbosa Padovan, A.C.; Siqueira Caixeta, E.; Caixeta Franco Ariosa, M. Detection of ERG11 Overexpression in Candida albicans isolates from environmental sources and clinical isolates treated with inhibitory and subinhibitory concentrations of fluconazole. Mycoses 2021, 64, 220–227. [Google Scholar] [CrossRef]
- Rosana, Y.; Yasmon, A.; Lestari, D.C. Overexpression and mutation as a genetic mechanism of fluconazole resistance in Candida albicans isolated from human immunodeficiency virus patients in Indonesia. J. Med. Microbiol. 2015, 64, 1046–1052. [Google Scholar] [CrossRef]
- He, X.; Zhao, M.; Chen, J.; Wu, R.; Zhang, J.; Cui, R.; Jiang, Y.; Chen, J.; Cao, X.; Xing, Y.; et al. Overexpression of Both ERG11 and ABC2 Genes Might Be Responsible for Itraconazole Resistance in Clinical Isolates of Candida krusei. PLoS ONE 2015, 10, e0136185. [Google Scholar] [CrossRef] [PubMed]
- Dunkel, N.; Liu, T.T.; Barker, K.S.; Homayouni, R.; Morschhäuser, J.; Rogers, P.D. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot. Cell 2008, 7, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Morio, F.; Pagniez, F.; Besse, M.; Gay-andrieu, F.; Miegeville, M.; Le Pape, P. Deciphering azole resistance mechanisms with a focus on transcription factor-encoding genes TAC1, MRR1 and UPC2 in a set of fluconazole-resistant clinical isolates of Candida albicans. Int. J. Antimicrob. Agents 2013, 42, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Hoot, S.J.; Smith, A.R.; Brown, R.P.; White, T.C. An A643V amino acid substitution in Upc2p contributes to azole resistance in well-characterized clinical isolates of Candida albicans. Antimicrob. Agents Chemother. 2011, 55, 940–942. [Google Scholar] [CrossRef] [PubMed]
- Mellado, E.; Garcia-Effron, G.; Alcázar-Fuoli, L.; Melchers, W.J.G.; Verweij, P.E.; Cuenca-Estrella, M.; Rodríguez-Tudela, J.L. A new Aspergillus fumigatus resistance mechanism conferring in vitro cross-resistance to azole antifungals involves a combination of cyp51A alterations. Antimicrob. Agents Chemother. 2007, 51, 1897–1904. [Google Scholar] [CrossRef]
- Delma, F.Z.; Al-Hatmi, A.M.S.; Brüggemann, R.J.M.; Melchers, W.J.G.; de Hoog, S.; Verweij, P.E.; Buil, J.B. Molecular Mechanisms of 5-Fluorocytosine Resistance in Yeasts and Filamentous Fungi. J. Fungi 2021, 7, 909. [Google Scholar] [CrossRef]
- Jund, R.; Lacroute, F. Genetic and physiological aspects of resistance to 5-fluoropyrimidines in Saccharomyces cerevisiae. J. Bacteriol. 1970, 102, 607–615. [Google Scholar] [CrossRef]
- Chevallier, M.R.; Jund, R.; Lacroute, F. Characterization of cytosine permeation in Saccharomyces cerevisiae. J. Bacteriol. 1975, 122, 629–641. [Google Scholar] [CrossRef]
- Paluszynski, J.P.; Klassen, R.; Rohe, M.; Meinhardt, F. Various cytosine/adenine permease homologues are involved in the toxicity of 5-fluorocytosine in Saccharomyces cerevisiae. Yeast 2006, 23, 707–715. [Google Scholar] [CrossRef]
- Kern, L.; de Montigny, J.; Lacroute, F.; Jund, R. Regulation of the pyrimidine salvage pathway by the FUR1 gene product of Saccharomyces cerevisiae. Curr. Genet. 1991, 19, 333–337. [Google Scholar] [CrossRef]
- Dodgson, A.R.; Dodgson, K.J.; Pujol, C.; Pfaller, M.A.; Soll, D.R. Clade-specific flucytosine resistance is due to a single nucleotide change in the FUR1 gene of Candida albicans. Antimicrob. Agents Chemother. 2004, 48, 2223–2227. [Google Scholar] [CrossRef] [PubMed]
- Hope, W.W.; Tabernero, L.; Denning, D.W.; Anderson, M.J. Molecular mechanisms of primary resistance to flucytosine in Candida albicans. Antimicrob. Agents Chemother. 2004, 48, 4377–4386. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.; Abdolrasouli, A.; Farrer, R.A.; Cuomo, C.A.; Aanensen, D.M.; Armstrong-James, D.; Fisher, M.C.; Schelenz, S. Genomic epidemiology of the UK outbreak of the emerging human fungal pathogen Candida auris. Emerg. Microbes. Infect. 2018, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Galocha, M.; Viana, R.; Pais, P.; Silva-Dias, A.; Cavalheiro, M.; Miranda, I.M.; Van Ende, M.; Souza, C.S.; Costa, C.; Branco, J.; et al. Genomic evolution towards azole resistance in Candida glabrata clinical isolates unveils the importance of CgHxt4/6/7 in azole accumulation. Commun. Biol. 2022, 5, 1118. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Mohamed, N.; Capparella, M.R.; Townsend, A.; Sung, A.H.; Yura, R.; Muñoz, P. The role of diagnostics-driven antifungal stewardship in the management of invasive fungal infections: A systematic literature review. Open Forum Infect. Dis. 2022, 9, ofac234. [Google Scholar] [CrossRef] [PubMed]
- Ottilie, S.; Luth, M.R.; Hellemann, E.; Goldgof, G.M.; Vigil, E.; Kumar, P.; Cheung, A.L.; Song, M.; Godinez-Macias, K.P.; Carolino, K.; et al. Adaptive laboratory evolution in S. cerevisiae highlights role of transcription factors in fungal xenobiotic resistance. Commun. Biol. 2022, 5, 128. [Google Scholar] [CrossRef]
- Narayanan, A.; Kumar, P.; Chauhan, A.; Kumar, M.; Yadav, K.; Banerjee, A.; Sharma, R.D.; Rudramurthy, S.M.; Chakrabarti, A.; Sanyal, K.; et al. Directed Evolution Detects Supernumerary Centric Chromosomes Conferring Resistance to Azoles in Candida auris. MBio 2022, 13, e0305222. [Google Scholar] [CrossRef]
- Ksiezopolska, E.; Schikora-Tamarit, M.À.; Beyer, R.; Nunez-Rodriguez, J.C.; Schüller, C.; Gabaldón, T. Narrow mutational signatures drive acquisition of multidrug resistance in the fungal pathogen Candida glabrata. Curr. Biol. 2021, 31, 5314–5326.e10. [Google Scholar] [CrossRef]
- Marr, K.A.; Schlamm, H.T.; Herbrecht, R.; Rottinghaus, S.T.; Bow, E.J.; Cornely, O.A.; Heinz, W.J.; Jagannatha, S.; Koh, L.P.; Kontoyiannis, D.P.; et al. Combination antifungal therapy for invasive aspergillosis: A randomized trial. Ann. Intern. Med. 2015, 162, 81–89. [Google Scholar] [CrossRef]
- Molloy, S.F.; Kanyama, C.; Heyderman, R.S.; Loyse, A.; Kouanfack, C.; Chanda, D.; Mfinanga, S.; Temfack, E.; Lakhi, S.; Lesikari, S.; et al. ACTA Trial Study Team Antifungal combinations for treatment of cryptococcal meningitis in Africa. N. Engl. J. Med. 2018, 378, 1004–1017. [Google Scholar] [CrossRef]
- Nishikawa, J.L.; Boeszoermenyi, A.; Vale-Silva, L.A.; Torelli, R.; Posteraro, B.; Sohn, Y.-J.; Ji, F.; Gelev, V.; Sanglard, D.; Sanguinetti, M.; et al. Inhibiting fungal multidrug resistance by disrupting an activator-Mediator interaction. Nature 2016, 530, 485–489. [Google Scholar] [CrossRef]
- Chang, W.; Liu, J.; Zhang, M.; Shi, H.; Zheng, S.; Jin, X.; Gao, Y.; Wang, S.; Ji, A.; Lou, H. Efflux pump-mediated resistance to antifungal compounds can be prevented by conjugation with triphenylphosphonium cation. Nat. Commun. 2018, 9, 5102. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.J.; Harvey, S.; Armstrong-James, D. Immunotherapeutic approaches for fungal infections. Curr. Opin. Microbiol. 2020, 58, 130–137. [Google Scholar] [CrossRef]
- Rayens, E.; Rabacal, W.; Willems, H.M.E.; Kirton, G.M.; Barber, J.P.; Mousa, J.J.; Celia-Sanchez, B.N.; Momany, M.; Norris, K.A. Immunogenicity and protective efficacy of a pan-fungal vaccine in preclinical models of aspergillosis, candidiasis, and pneumocystosis. PNAS Nexus 2022, 1, pgac248. [Google Scholar] [CrossRef]
- Oliveira, L.V.N.; Wang, R.; Specht, C.A.; Levitz, S.M. Vaccines for human fungal diseases: Close but still a long way to go. Npj. Vaccines 2021, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Ambati, S.; Ellis, E.C.; Pham, T.; Lewis, Z.A.; Lin, X.; Meagher, R.B. Antifungal Liposomes Directed by Dectin-2 Offer a Promising Therapeutic Option for Pulmonary Aspergillosis. MBio 2021, 12, e00030-21. [Google Scholar] [CrossRef] [PubMed]
- Kumaresan, P.R.; Manuri, P.R.; Albert, N.D.; Maiti, S.; Singh, H.; Mi, T.; Roszik, J.; Rabinovich, B.; Olivares, S.; Krishnamurthy, J.; et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc. Natl. Acad. Sci. USA 2014, 111, 10660–10665. [Google Scholar] [CrossRef]
- Roy, R.M.; Klein, B.S. Dendritic cells in antifungal immunity and vaccine design. Cell Host Microbe 2012, 11, 436–446. [Google Scholar] [CrossRef]
- Da Silva, T.A.; Hauser, P.J.; Bandey, I.; Laskowski, T.; Wang, Q.; Najjar, A.M.; Kumaresan, P.R. Glucuronoxylomannan in the Cryptococcus species capsule as a target for Chimeric Antigen Receptor T-cell therapy. Cytotherapy 2021, 23, 119–130. [Google Scholar] [CrossRef]
- Seif, M.; Kakoschke, T.K.; Ebel, F.; Bellet, M.M.; Trinks, N.; Renga, G.; Pariano, M.; Romani, L.; Tappe, B.; Espie, D.; et al. CAR T cells targeting Aspergillus fumigatus are effective at treating invasive pulmonary aspergillosis in preclinical models. Sci. Transl. Med. 2022, 14, eabh1209. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).