
Development of SNP Markers and Core Collection Construction of Berberis L. Based on SLAF-Seq in Xinjiang, China


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
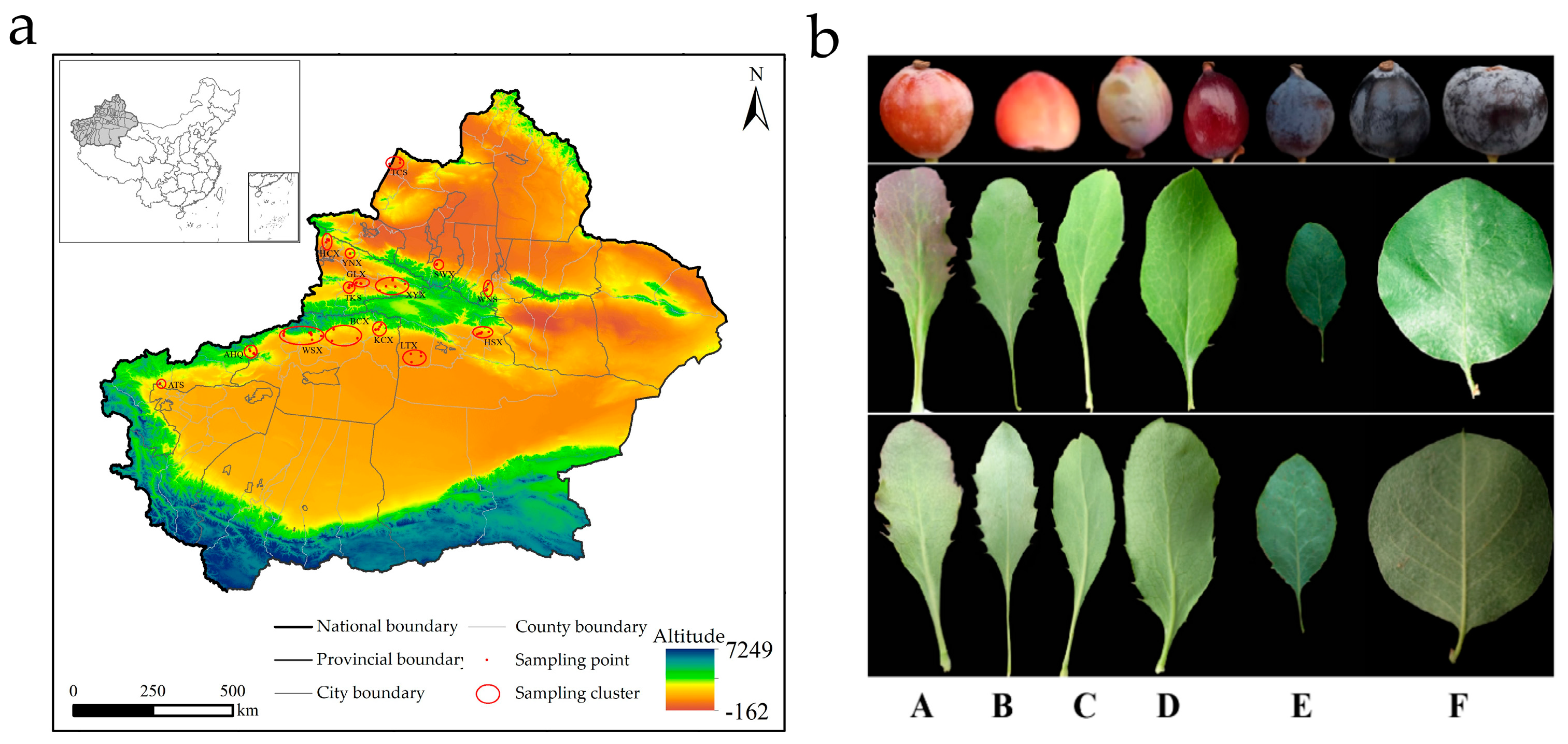
2.1. Plant Materials and Population Sampling
2.2. DNA Extraction and Enzyme Digestion Design
2.3. SLAF Sequencing and Data Evaluation
2.4. Development of SLAF Tags and SNP Markers
2.5. Genetic Relationships Among Samples
2.6. Construction of Core Collection
2.7. Construction of DNA Fingerprints
3. Results
3.1. Sequence and Quality Statistics
3.2. Development of Polymorphic SLAF Tags and Selection of SNP Markers
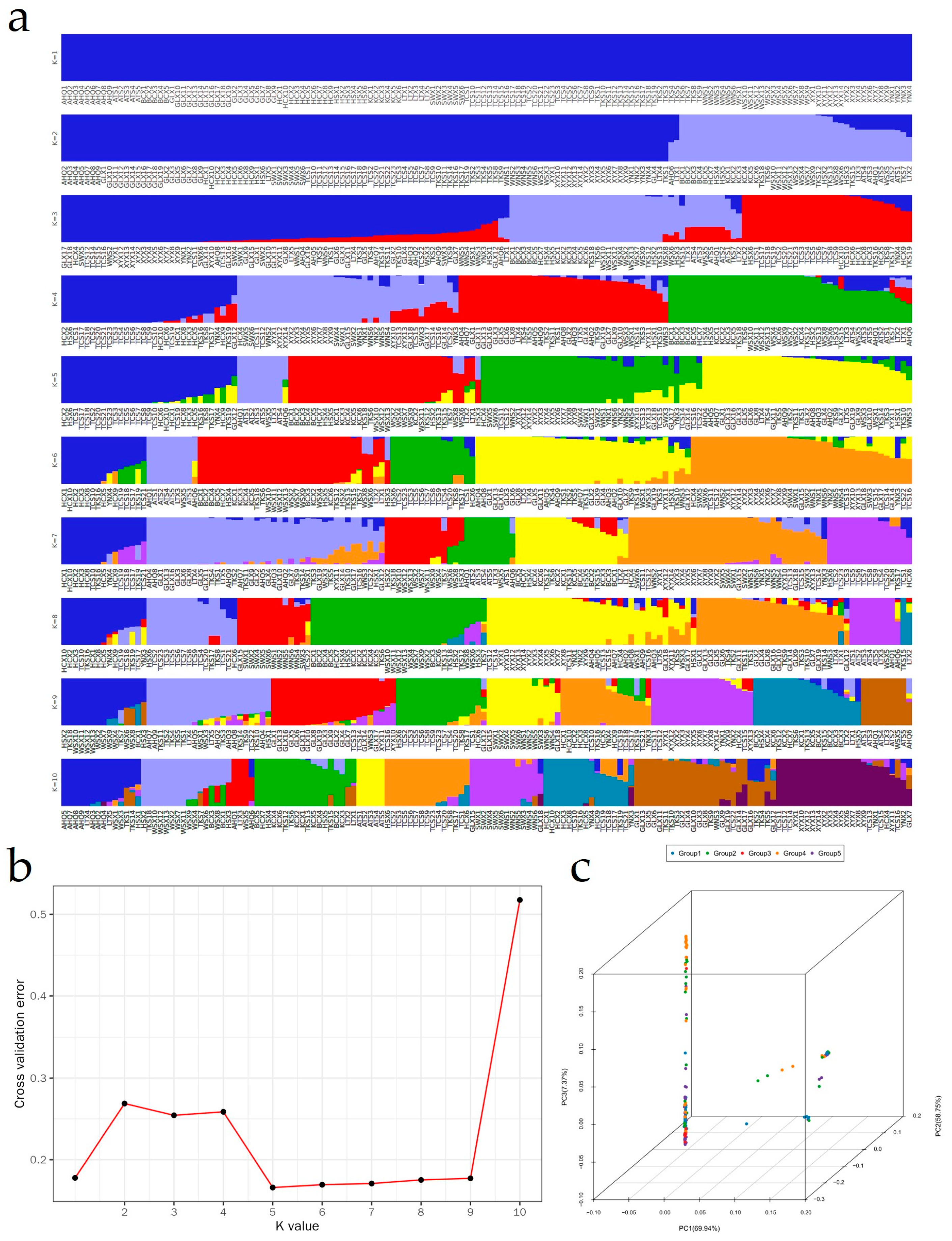
3.3. Genetic Relationships and Phylogenetic Tree Construction
3.4. Population Structure and Principal Components Analysis
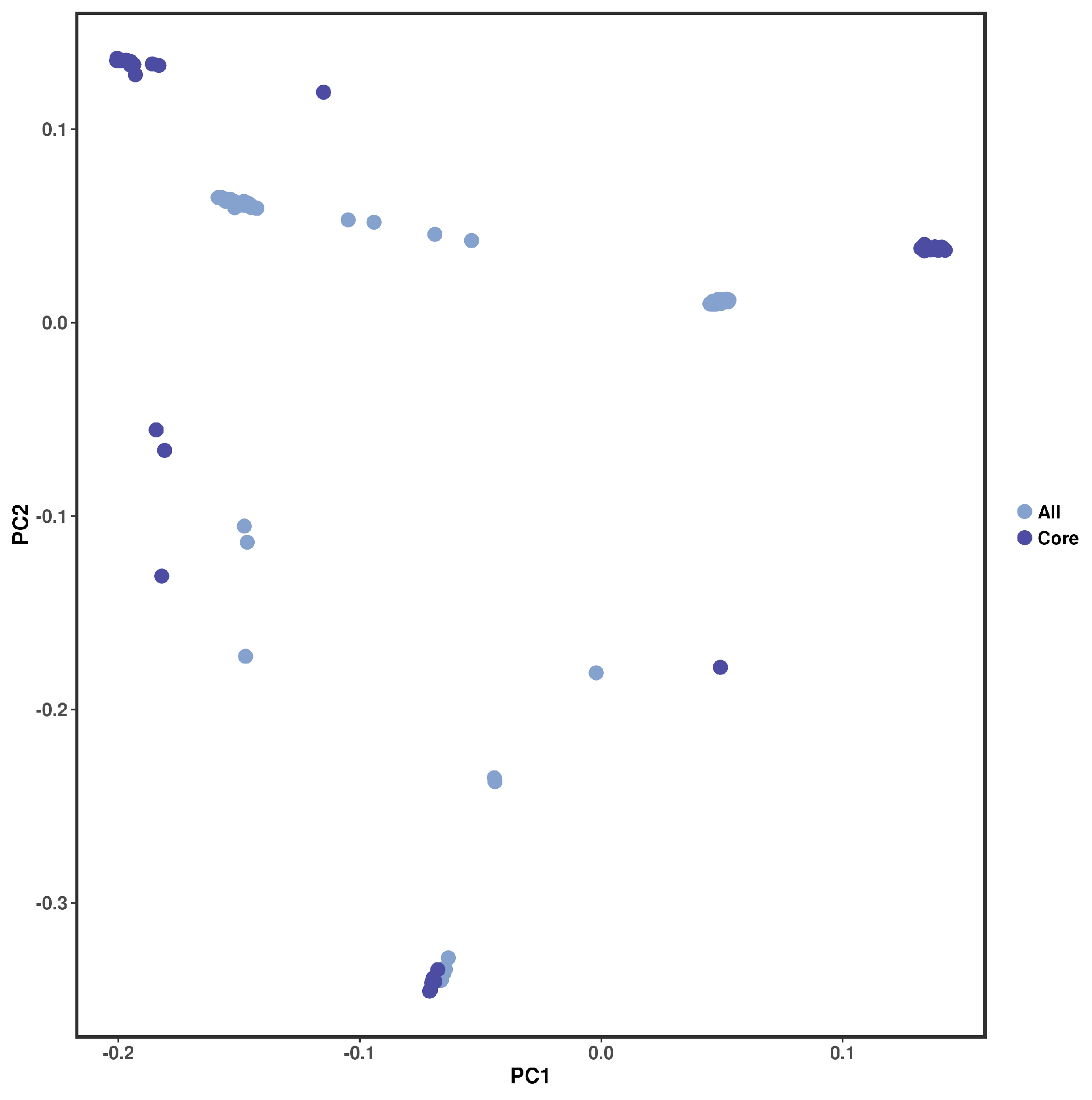
3.5. Construction and Evaluation of Core Collection
3.6. The DNA Fingerprints
4. Discussion
4.1. Optimisation of Molecular Markers Based on SLAF-Seq and SNPs
4.2. Phylogenetic Analysis Based on SNPs
4.3. Construction and Evaluation of Core Collection Based on SNPs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sarraf, M.; Babaei, A.B.; Naji-Tabasi, S. Investigating functional properties of barberry species an overview. J. Sci. Food Agric. 2019, 99, 5255–5269. [Google Scholar] [CrossRef]
- Azim, R.; Awan, S.I.; Tahir, M.M.; Malik, F.A.; Alwahibi, M.S.; Ali, M.A.; Elshiki, M.S.; Kamal, A. Morphological and phylogenetic insights into Berberis species: A tool for species identification and conservation. Genet. Resour. Crop Evol. 2024, 72, 4569–4582. [Google Scholar] [CrossRef]
- Goodarzi, S.; Khadivi, A.; Abbasifar, A.; Akramian, M. Phenotypic, pomological and chemical variations of the seedless barberry (Berberis vulgaris L. var. asperma). Sci. Hortic. 2018, 238, 38–50. [Google Scholar] [CrossRef]
- Ying, J.S.; Editorial Board for Flora of China. Flora of China (Revised English Version); Ying, J.S., Ed.; Science Press: Beijing, China, 2011; Volume 19, pp. 715–771. [Google Scholar]
- Li, Y.-K.; Harber, J.; Peng, C.; Du, Z.-Q.; Xing, Y.-W.; Yu, C.-C. Taxonomic synopsis of Berberis (Berberidaceae) from the northern Hengduan mountains region in China, with descriptions of seven new species. Plant Divers. 2022, 44, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.M.; An, Z.X.; Zhou, G.L.; Yang, C.Y.; Han, Y.L.; Li, X.Y.; Zhang, Y.F. Flora of Xinjiang, Book II; Xinjiang Science &Technology &Hygiene Publishing House: Urumqi, China, 1996; Volume 2, pp. 1–4. [Google Scholar]
- Zhang, X.M. Kuwanisi Pazilijiang hplc determination of berberine in Berberis L. leaf of Vitamin Botrytis cinerea. Cap. Food Med. 2012, 19, 50. [Google Scholar]
- Safari-Khuzani, A.; Rezaei, M.; Khadivi, A. Screening of the superior f1 segregating populations of barberry (Berberis L. spp.) for cultivation based on phenotypic characterizations. Ind. Crops Prod. 2020, 158, 113054. [Google Scholar] [CrossRef]
- Imenshahidi, M.; Hosseinzadeh, H. Berberine and barberry (Berberis L. vulgaris) a clinical review. Phytother. Res. 2019, 33, 504–523. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Z.; Hu, X.; You, L.; Khan, R.A.A.; Yu, Y. Phenolic contents, organic acids, and the antioxidant and bio activity of wild medicinal Berberis plants as sustainable sources of functional food. Molecules 2022, 27, 2497. [Google Scholar] [CrossRef]
- Khan, T.; Khan, I.A.; Rehman, A.; Ahmed, N. Conservation status evaluation of berberis species across the Karakoram Mountain Ranges, Pakistan using iucn red list categories and criteria. J. For. Res. 2016, 6, 172–177. [Google Scholar] [CrossRef]
- Huan, J.; He, Z.; Lei, Y.; Li, W.; Jiang, L.; Luo, X. The genetic diversity of Bletilla spp. Based on SLAF-seq and oligo-FISH. Genes 2022, 13, 1118. [Google Scholar] [CrossRef]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. Slaf-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef]
- Wu, W.; Shao, M.; Qi, J.; Jin, G.; Zhang, R.; Yao, Y.; Jiang, C. Integrating genetic analysis of germplasm wealth for enhanced selection and improvement in olive (Olea europaea L.): Insights from leaves. Plant Cell Rep. 2024, 43, 247. [Google Scholar] [CrossRef]
- Yang, J.; Sun, M.; Ren, X.; Li, P.; Hui, J.; Zhang, J.; Lin, G. Revealing the genetic diversity and population structure of garlic resource cultivars and screening of core cultivars based on specific length amplified fragment sequencing (SLAF-seq). Genes 2024, 15, 1135. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Li, Y.; Sun, X.; Guo, X.; Jiang, N.; Fang, Y.; Chen, J.; Bao, Z.; Ma, F. Association study of SNP locus for color related traits in herbaceous peony (Paeonia lactiflora Pall.) Using slaf-seq. Front. Plant Sci. 2022, 13, 1032449. [Google Scholar] [CrossRef]
- Tian, M.; Li, W.; Luo, P.; He, J.; Zhang, H.; Yan, Q.; Ye, Y. Genetic diversity analysis and core germplasm bank construction in cold resistant germplasm of rubber trees (Hevea brasiliensis). Sci. Rep. 2024, 14, 14533. [Google Scholar] [CrossRef] [PubMed]
- Akishev, Z.D.; Smekenov, I.T.; Usenova, A.N.; Duisenova, A.B.; Smailov, B.B.; Mukhidinov, N.M.; Bissenbaev, A.K. RAPD analysis of genetic diversity in Berberis L. iliensis a species endemic to Kazakhstan. Int. J. Biol. Chem. 2012, 3, 48–53. [Google Scholar]
- Pinar, H.; Yahya, H.N.; Erċışlı, S.; Coskun, O.F.; Yaman, M.; Turgunbaev, K.; Uzun, A. Molecular characterization of barberry genotypes from Turkey and Kyrgyzstan. Erwerbs-Obstbau 2021, 63, 403–407. [Google Scholar] [CrossRef]
- Li, H.; Abdullah, H.; Yang, H.; Guo, H.; Yuan, Y.; Ahmed, I.; Li, G.; Wang, Y.; Chang, Y.; Tian, X. Chloroplast genome evolution of Berberis L. (Berberidaceae): Implications for phylogeny and metabarcoding. Gene 2025, 933, 148959. [Google Scholar] [CrossRef]
- Chen, Z.; He, Y.; Iqbal, Y.; Shi, Y.; Huang, H.; Yi, Z. Investigation of genetic relationships within three miscanthus species using SNP markers identified with SLAF-seq. BMC Genomics 2022, 23, 43. [Google Scholar] [CrossRef]
- Xia, Y.; Xu, J.; Duan, J.; Liu, Q.; Huang, H.; Yi, Z.; Chen, Z. Transgenic Miscanthus lutarioriparius that co-expresses Cry 2Aa# and Bar genes. Can. J. Plant Sci. 2019, 99, 841–851. [Google Scholar]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeqIllumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Mendel, J.M.; Korjani, M.M. On establishing nonlinear combinations of variables from small to big data for use in later processing. Inf. Sci. 2014, 280, 98–110. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows–Wheeler transform. Bioinformatics 2010, 14, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N. The sequence alignmentmap format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. Mega x: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Xia, A.-N.; Yang, A.-A.; Meng, X.-S.; Dong, G.-Z.; Tang, X.-J.; Lei, S.-M.; Liu, Y.-G. Development and application of rose (Rosa chinensis jacq.) SNP markers based on slaf-seq technology. Genet. Resour. Crop Evol. 2021, 69, 173–182. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhou, C.; Zheng, W.; Mason, A.S.; Fan, S.; Wu, C.; Fu, D.; Huang, Y. Genome-wide snp markers based on slaf-seq uncover breeding traces in rapeseed (Brassica napus L.). Front. Plant Sci. 2017, 8, 648. [Google Scholar] [CrossRef]
- Wei, Q.; Wang, W.; Hu, T.; Hu, H.; Wang, J.; Bao, C. Construction of a SNP-based genetic map using SLAF-seq and QTL analysis of morphological traits in eggplant. Front. Genet. 2020, 11, 178. [Google Scholar] [CrossRef]
- Wang, R.J.; Gao, X.F.; Yang, J.; Kong, X.R. Genome-wide association study to identify favorable SNP allelic variations and candidate genes that control the timing of spring bud flush of tea (Camellia sinensis) using SLAF-seq. J. Agric. Food Chem. 2019, 67, 10380–10391. [Google Scholar] [CrossRef]
- Meng, J.; Yang, G.; Li, X.; Zhao, Y.; He, S. Population structure of wild soybean (Glycine soja) based on slaf-seq have implications for its conservation. PeerJ 2023, 11, e16415. [Google Scholar] [CrossRef] [PubMed]
- Nilkanta, H.; Amom, T.; Tikendra, L.; Rahaman, H.; Nongdam, P. ISSR marker based population genetic study of Melocanna baccifera (Roxb.) Kurz: A commercially important bamboo of manipur, north-east india. Scientifica 2017, 2017, 3757238. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, C.; Li, X.; Liang, Y.; Wang, Y.; Li, W. Development of EST-SSR markers and their application in the analysis of the genetic diversity of Sophora japonica linn. Trees Struct. Funct. 2020, 34, 1147–1156. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Zhang, D.; Huang, J.; Shi, W.; Wang, J. Historical spread routes of wild walnuts in central asia shaped by man-made and nature. Front. Plant Sci. 2024, 15, 12. [Google Scholar] [CrossRef]
- Boulnois, L. Silk Road: Monks, Warriors and Merchants on the Silk Road; W. W. Norton & Co. Inc.: New York, NY, USA, 2004; pp. 178–181. [Google Scholar]
- Pengliang, W.; Jiexia, S.; Hongying, W.; Chen, Y.; Xie, Y.; Wang, H.; He, G.; Chen, N.; Wei, C.; Yang, L.; et al. Analysis of germplasm genetic diversity and construction of a core collection in Camellia oleifera C.Abel by integrating novel simple sequence repeat markers. Genet. Resour. Crop Evol. 2023, 70, 1517–1530. [Google Scholar]
- Sun, Y.; Zhang, Y.; Chen, J.; Liu, R.; Wang, S.; Dong, S. Comparison of selection methods to establish a core collection of the Siberian apricot using trait and SSR data. Tree Genet. Genomes 2024, 20, 40. [Google Scholar] [CrossRef]
- Zhu, Y.; An, W.; Peng, J.; Li, J.; Gu, Y.; Jiang, B.; Chen, L.; Zhu, P.; Yang, H. Genetic diversity of nanmu (Phoebe zhennans. Lee. Et f. N. Wei) breeding population and extraction of core collection using nSSR, cpSSR and phenotypic markers. Forests 2022, 13, 1320. [Google Scholar] [CrossRef]
- Liu, Y.; Geng, Y.; Xie, X.; Zhang, P.; Hou, J.; Wang, W. Core collection construction and evaluation of the genetic structure of Glycyrrhiza in China using markers for genomic simple sequence repeats. Genet. Resour. Crop Evol. 2020, 67, 1839–1852. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Song, Y.; Wang, Z.; Zhou, L.; Yang, X.; Aheihati, M. Development of SNP Markers and Core Collection Construction of Berberis L. Based on SLAF-Seq in Xinjiang, China. Horticulturae 2025, 11, 434. https://doi.org/10.3390/horticulturae11040434
Li R, Song Y, Wang Z, Zhou L, Yang X, Aheihati M. Development of SNP Markers and Core Collection Construction of Berberis L. Based on SLAF-Seq in Xinjiang, China. Horticulturae. 2025; 11(4):434. https://doi.org/10.3390/horticulturae11040434
Chicago/Turabian StyleLi, Ruxue, Yan Song, Zilong Wang, Long Zhou, Xiyu Yang, and Meiri Aheihati. 2025. "Development of SNP Markers and Core Collection Construction of Berberis L. Based on SLAF-Seq in Xinjiang, China" Horticulturae 11, no. 4: 434. https://doi.org/10.3390/horticulturae11040434
APA StyleLi, R., Song, Y., Wang, Z., Zhou, L., Yang, X., & Aheihati, M. (2025). Development of SNP Markers and Core Collection Construction of Berberis L. Based on SLAF-Seq in Xinjiang, China. Horticulturae, 11(4), 434. https://doi.org/10.3390/horticulturae11040434