
Additive Manufacturing of Ti3AlC2/TiC and Ti3AlC2/SiC Ceramics Using the Fused Granules Fabrication Technique


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Feedstock Preparation
2.2. Fabrication of Ceramic Composites
2.3. Characterization
3. Results and Discussion
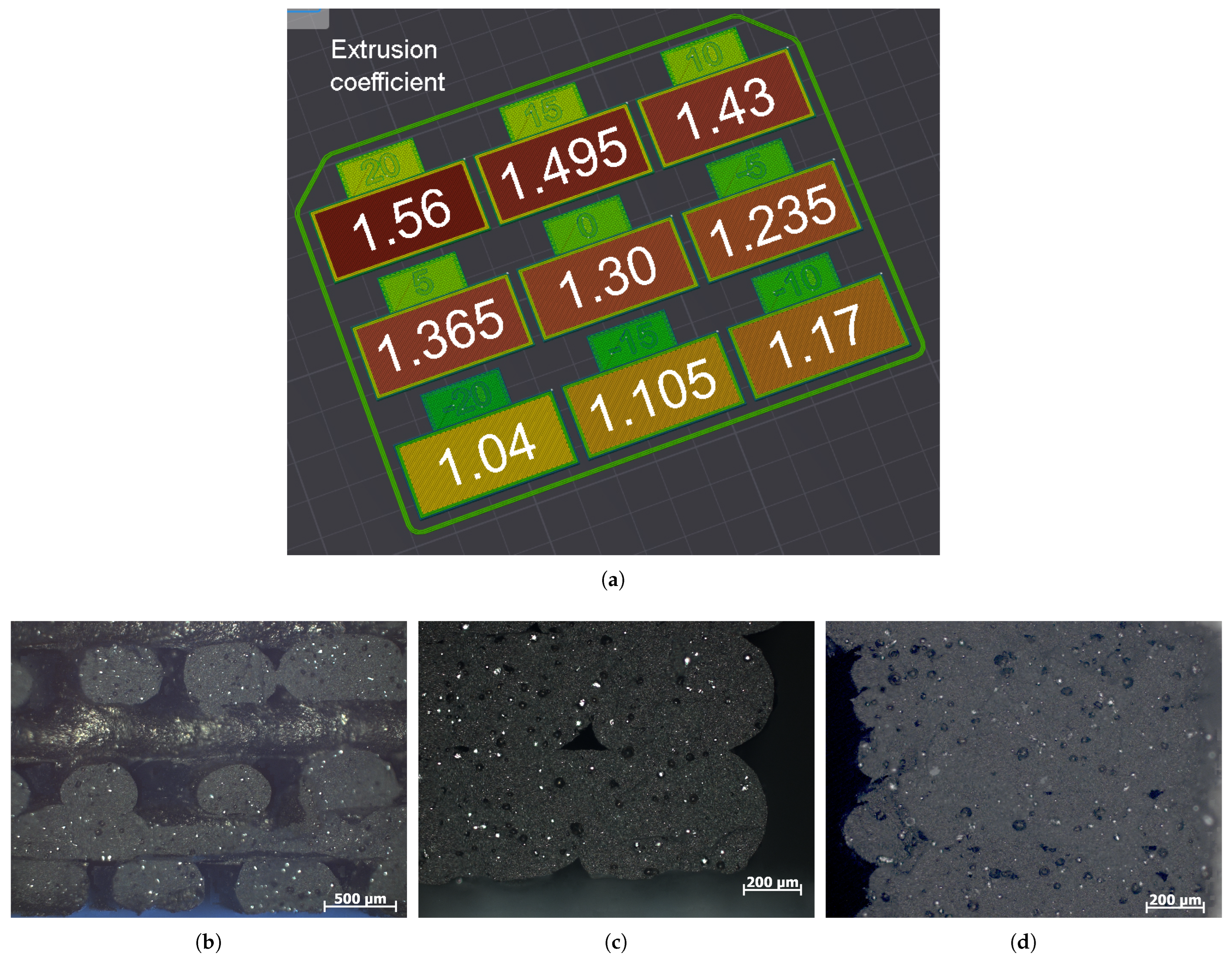
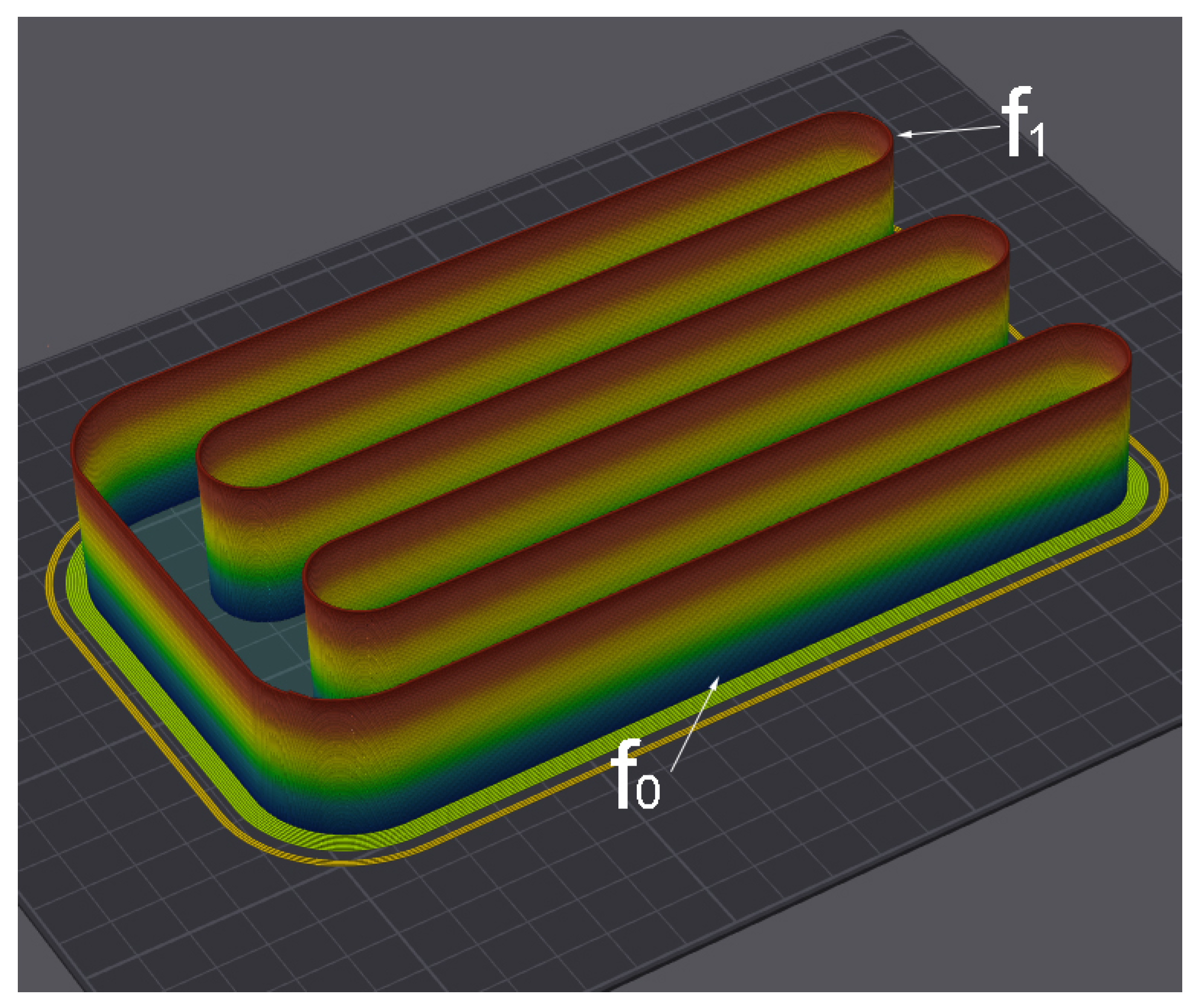
3.1. Parameters of Additive Manufacturing
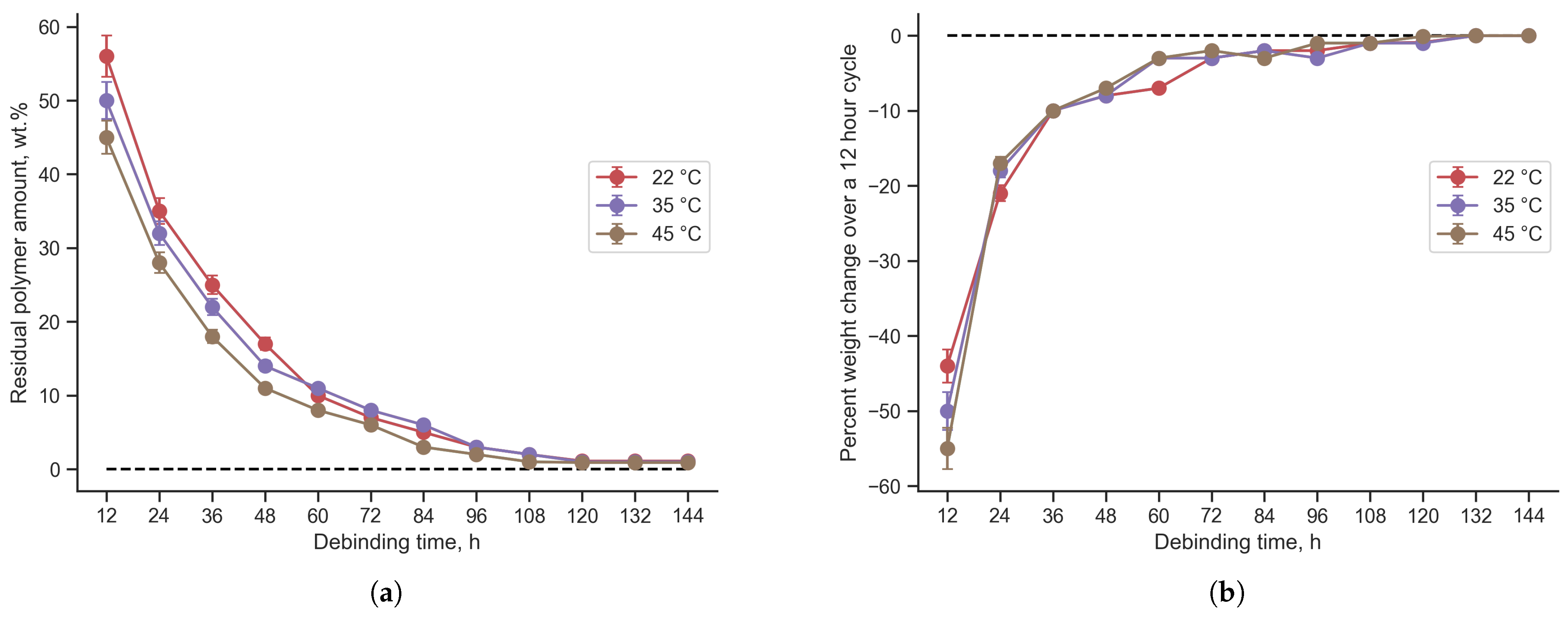
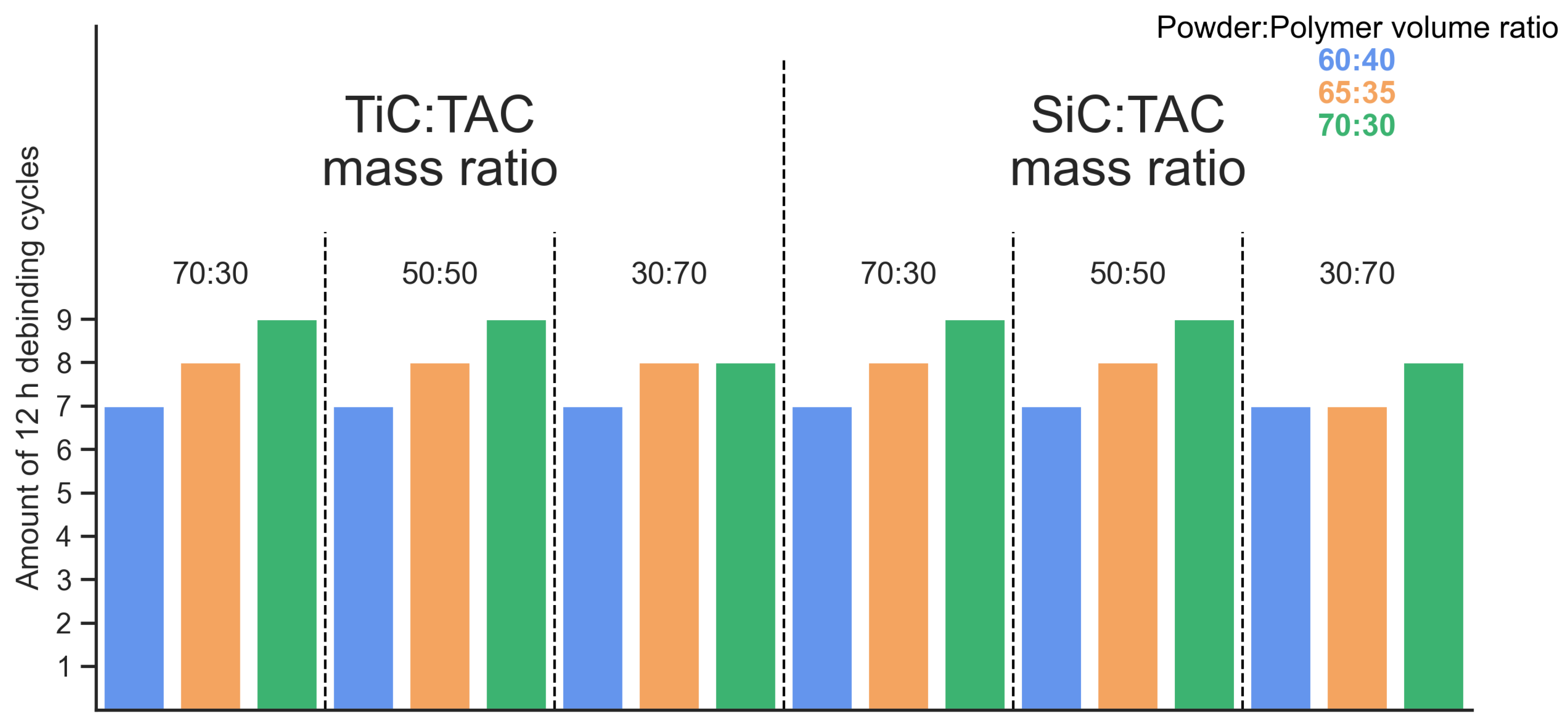
3.2. Debinding
3.3. Structure, Phase Composition, and Mechanical Properties
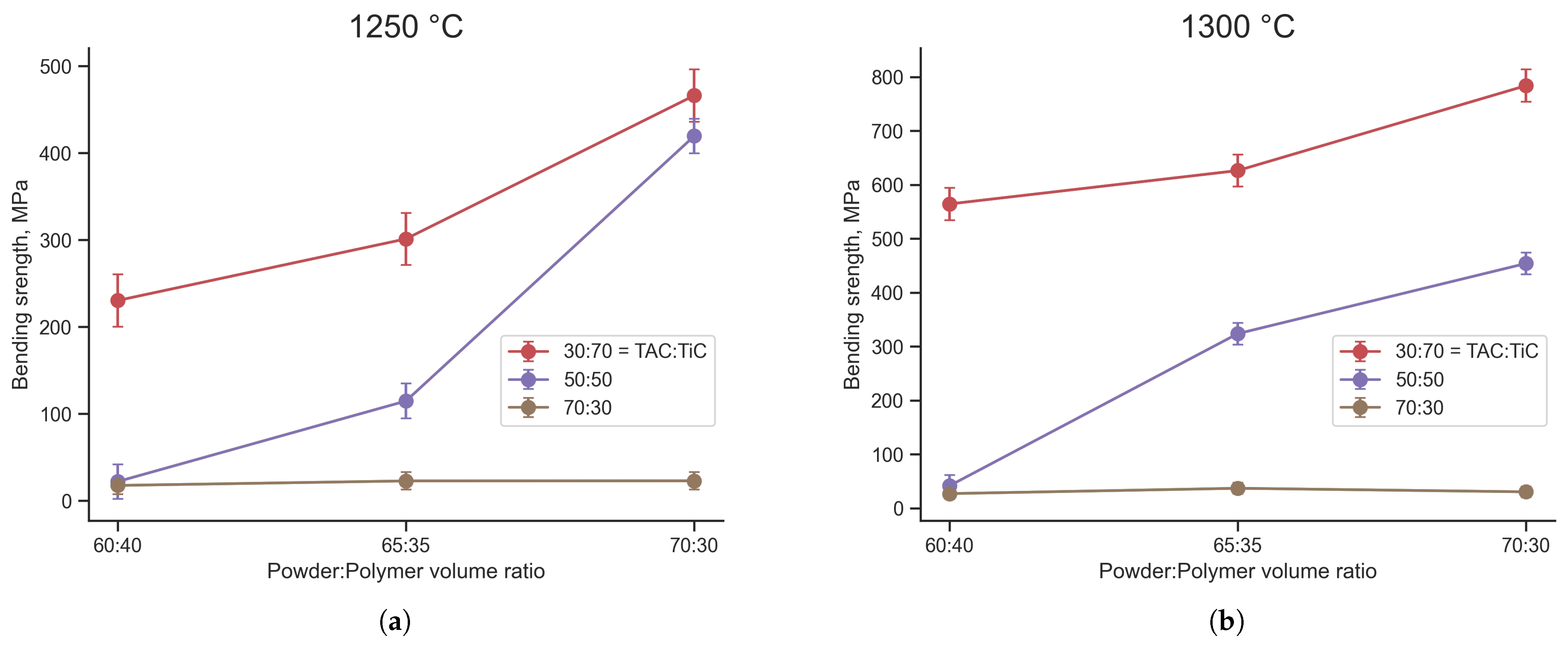
3.3.1. TiC-Containing Composites
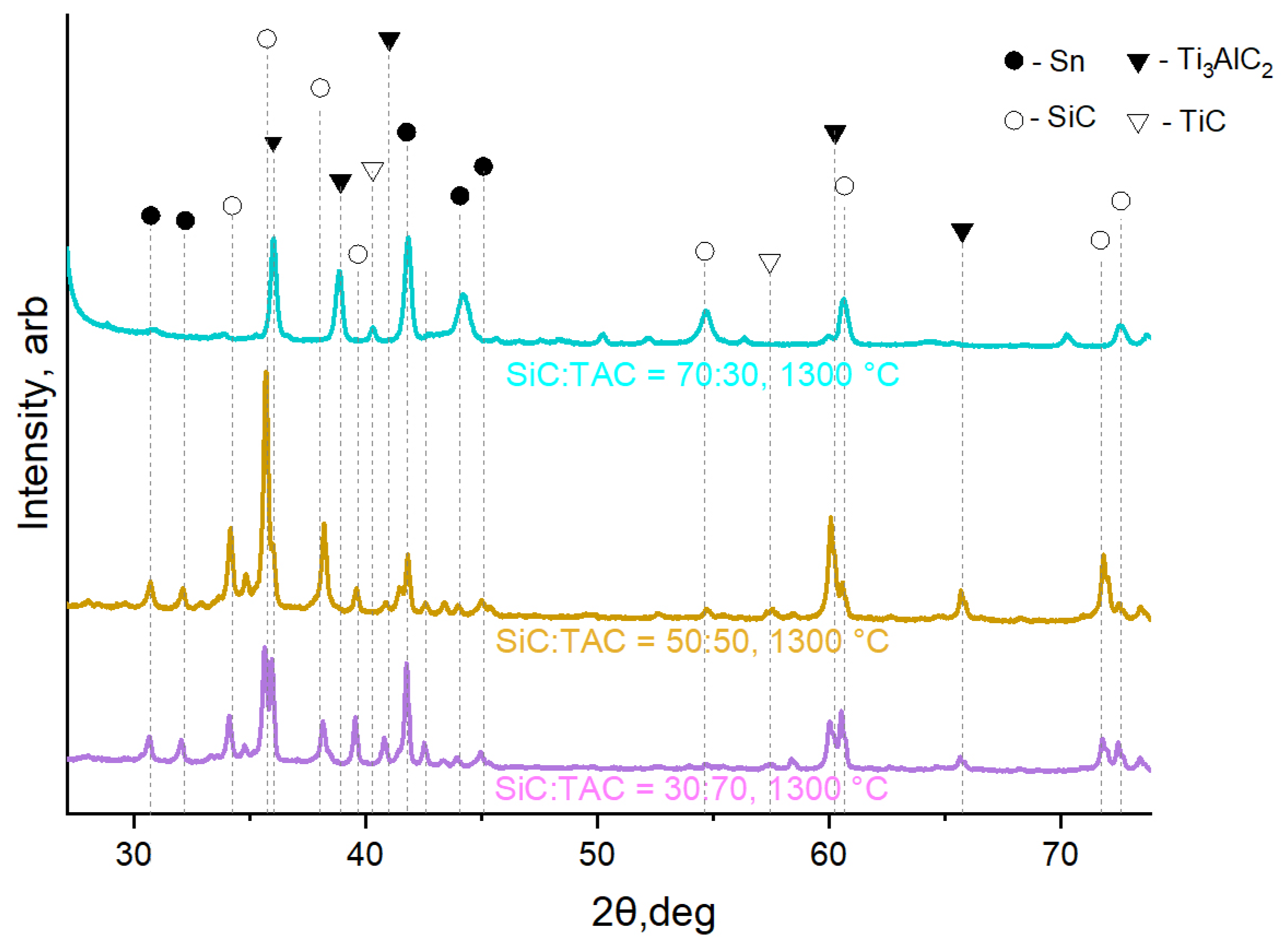
3.3.2. SiC-Containing Composites
4. Conclusions
- -
- FGF samples of TiC:TAC and SiC:TAC composites were successfully printed without any macroscopic printing defects. Debinding and sintering parameters have been optimized. Minimum debinding time for samples with a mm cross-section that is enough to remove 99% of polymer is 84 h. The optimal sintering temperature to form the best mechanical properties for both TiC:TAC and SiC:TAC composites is 1300 °C.
- -
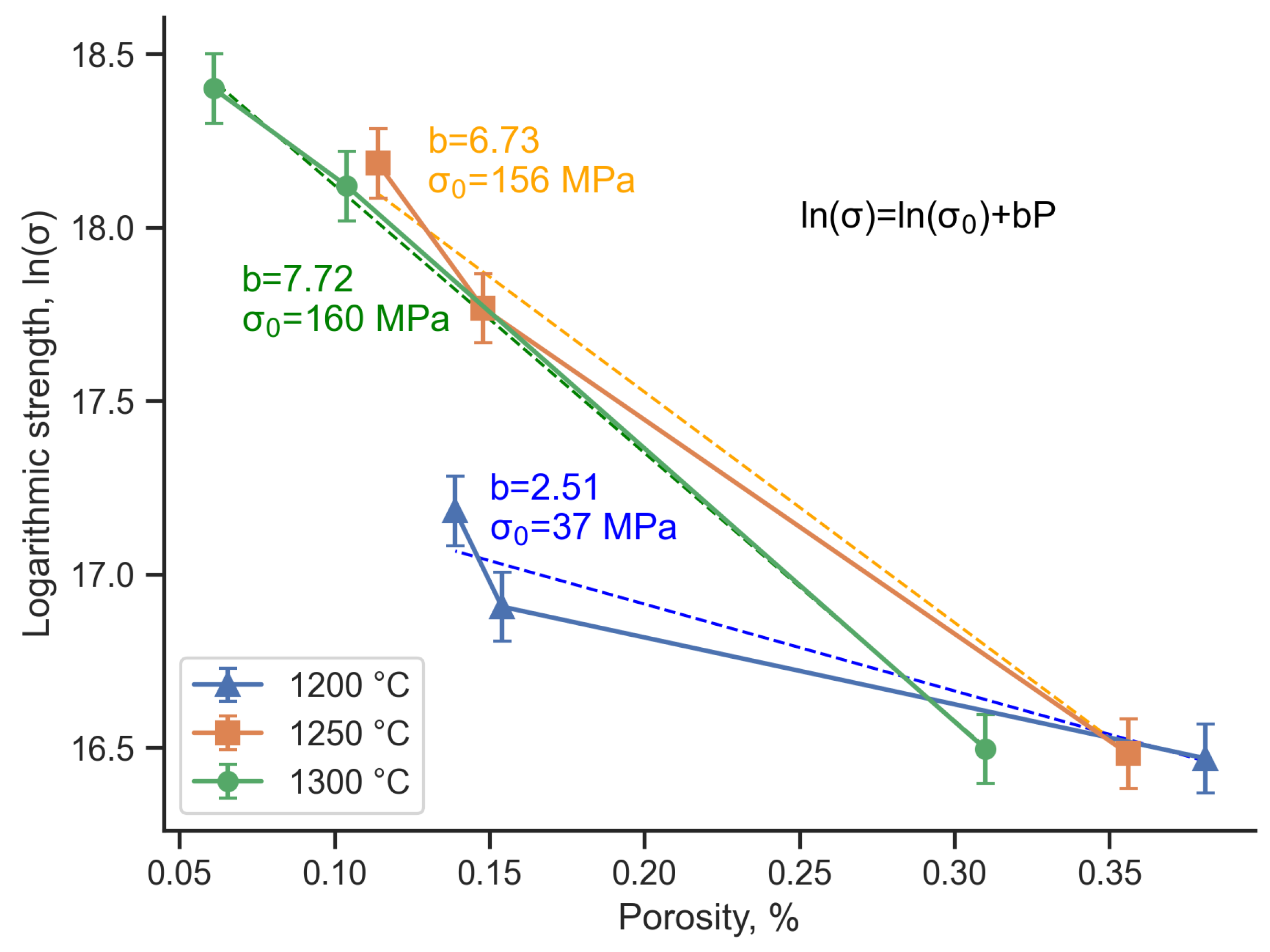
- Both SiC:TAC and TiC:TAC samples are porous, and the more TAC that is in the sample, the more porous it is. Thus, TAC content, sintering temperature, and polymer content in the feedstock are the main parameters that regulate the porosity of FGF samples.
- -
- Mechanical properties of the samples depend on composition, porosity of the samples, and sintering temperature. The highest bending strength is 784 ± 9 MPa for TiC:TAC composition and 95 ± 7 MPa for SiC:TAC composition after sintering at 1300 °C. Calculated bending strength for nonporous SiC:TAC sample is 156 MPa, which is comparable with the literature data.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ni, D.; Cheng, Y.; Zhang, J.; Liu, J.X.; Zou, J.; Chen, B.; Wu, H.; Li, H.; Dong, S.; Han, J.; et al. Advances in ultra-high temperature ceramics, composites, and coatings. J. Adv. Ceram. 2022, 11, 1–56. [Google Scholar] [CrossRef]
- Sun, J.; Ye, D.; Zou, J.; Chen, X.; Wang, Y.; Yuan, J.; Liang, H.; Qu, H.; Binner, J.; Bai, J. A review on additive manufacturing of ceramic matrix composites. J. Mater. Sci. Technol. 2023, 138, 1–16. [Google Scholar] [CrossRef]
- An, Q.; Chen, J.; Ming, W.; Chen, M. Machining of SiC ceramic matrix composites: A review. Chin. J. Aeronaut. 2021, 34, 540–567. [Google Scholar] [CrossRef]
- Wei, S.; Xu, B.Q.; Bin, Y.; Sun, H.Y.; Song, J.X.; Wan, H.L.; Dai, Y.N. Preparation of TiC powders by carbothermal reduction method in vacuum. Trans. Nonferrous Met. Soc. China 2011, 21, 185–190. [Google Scholar]
- Zhang, H.; Li, F.; Jia, Q.; Ye, G. Preparation of titanium carbide powders by sol–gel and microwave carbothermal reduction methods at low temperature. J. Sol-Gel Sci. Technol. 2008, 46, 217–222. [Google Scholar] [CrossRef]
- Nguyen, V.H.; Delbari, S.A.; Asl, M.S.; Van Le, Q.; Shokouhimehr, M.; Namini, A.S.; Mohammadi, M. Microstructure–property correlation in nano-diamond and TiN added TiC-based ceramics. Ceram. Int. 2021, 47, 449–460. [Google Scholar] [CrossRef]
- Ishizaki, K.; Komarneni, S.; Nanko, M. Porous Materials: Process Technology and Applications; Springer Science & Business Media: New York, NY, USA, 2013; Volume 4. [Google Scholar]
- Choyke, W.; Pensl, G. Physical properties of SiC. Mrs Bull. 1997, 22, 25–29. [Google Scholar] [CrossRef]
- Snead, L.L.; Nozawa, T.; Katoh, Y.; Byun, T.S.; Kondo, S.; Petti, D.A. Handbook of SiC properties for fuel performance modeling. J. Nucl. Mater. 2007, 371, 329–377. [Google Scholar] [CrossRef]
- Taslicukur, Z.; Balaban, C.; Kuskonmaz, N. Production of ceramic foam filters for molten metal filtration using expanded polystyrene. J. Eur. Ceram. Soc. 2007, 27, 637–640. [Google Scholar] [CrossRef]
- Sun, B.; Fan, T.; Zhang, D. Porous TiC ceramics derived from wood template. J. Porous Mater. 2002, 9, 275–277. [Google Scholar] [CrossRef]
- Gringoz, A.; Glandut, N.; Valette, S. Electrochemical hydrogen storage in TiC0.6, not in TiC0.9. Electrochem. Commun. 2009, 11, 2044–2047. [Google Scholar] [CrossRef]
- Sang, K.; Lü, Z.; Jin, Z. A study of the SiC-L composite ceramics for self-lubrication. Wear 2002, 253, 1188–1193. [Google Scholar] [CrossRef]
- Gao, F.; Lu, J.; Liu, W. Tribological behavior of carbide-derived carbon coating on SiC polycrystal against SAE52100 steel in moderately humid air. Tribol. Lett. 2007, 27, 339–345. [Google Scholar] [CrossRef]
- Barsoum, M.W. The MN+ 1AXN phases: A new class of solids: Thermodynamically stable nanolaminates. Prog. Solid State Chem. 2000, 28, 201–281. [Google Scholar] [CrossRef]
- Li, S.B.; Zhai, H.X.; Bei, G.P.; Zhou, Y.; Zhang, Z.L. Synthesis and microstructure of Ti3AlC2 by mechanically activated sintering of elemental powders. Ceram. Int. 2007, 33, 169–173. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y. Stability and selective oxidation of aluminum in nano-laminate Ti3AlC2 upon heating in argon. Chem. Mater. 2003, 15, 3716–3720. [Google Scholar] [CrossRef]
- Pang, W.K.; Low, I.M.; Sun, Z.M. In situ high-temperature diffraction study of the thermal dissociation of Ti3AlC2 in vacuum. J. Am. Ceram. Soc. 2010, 93, 2871–2876. [Google Scholar] [CrossRef]
- Shuck, C.E.; Han, M.; Maleski, K.; Hantanasirisakul, K.; Kim, S.J.; Choi, J.; Reil, W.E.; Gogotsi, Y. Effect of Ti3AlC2 MAX phase on structure and properties of resultant Ti3C2T x MXene. ACS Appl. Nano Mater. 2019, 2, 3368–3376. [Google Scholar] [CrossRef]
- Ruan, M.M.; Feng, X.M.; Ai, T.T.; Yu, N.; Hua, K. Microstructure and mechanical properties of TiC/Ti3AlC2 in situ composites prepared by hot pressing method. Mater. Sci. Forum. Trans. Tech. Publ. 2015, 816, 200–204. [Google Scholar] [CrossRef]
- Chen, W.; Chen, J.; Zhu, M.; Zheng, J.; Ma, N.n.; Liu, X.; Chen, Z.; Huang, Z. Fabrication of SiC ceramics with invariable value resistivity in the range of 20–400 °C using MAX phase-Ti3AlC2 additives. J. Eur. Ceram. Soc. 2021, 41, 6248–6254. [Google Scholar] [CrossRef]
- ASTM D1238-23; Standard Test Method for Melt Flow Rates of Thermoplastics by Extrusion Plastometer. ASTM International: West Conshohocken, PA, USA, 2023.
- ASTM D3835; Standard Test Method for Determination of Properties of Polymeric Materials by Means of a Capillary Rheometer. ASTM International: West Conshohocken, PA, USA, 1996.
- ASTM E290; Standard Test Methods for Bend Testing of Material for Ductility. ASTM International: West Conshohocken, PA, USA, 1997.
- ASTM E384; Standard Test Method for Microindentation Hardness of Materials. ASTM International: West Conshohocken, PA, USA, 1999.
- Lin, X.; Gao, J.; Wang, J.; Wang, R.; Gong, M.; Zhang, L.; Lu, Y.; Wang, D.; Zhang, L. Desktop printing of 3D thermoplastic polyurethane parts with enhanced mechanical performance using filaments with varying stiffness. Addit. Manuf. 2021, 47, 102267. [Google Scholar] [CrossRef]
- Krinitcyn, M.; Sharafeev, S.; Afanasyev, A. Structure and Properties of Ti3AlC2-SiC and Ti3AlC2-TiC Materials Obtained by Powder Injection Molding Technology. Coatings 2023, 13, 1013. [Google Scholar] [CrossRef]
- Ai, M.; Zhai, H.; Zhou, Y.; Tang, Z.; Huang, Z.; Zhang, Z.; Li, S. Synthesis of Ti3AlC2 powders using Sn as an additive. J. Am. Ceram. Soc. 2006, 89, 1114–1117. [Google Scholar]
- Vasanthakumar, K.; Bakshi, S.R. Effect of C/Ti ratio on densification, microstructure and mechanical properties of TiCx prepared by reactive spark plasma sintering. Ceram. Int. 2018, 44, 484–494. [Google Scholar] [CrossRef]
- Panigrahi, B.B.; Gracio, J.J.; Chu, M.C.; Cho, S.J.; Reddy, N.S. Powder Synthesis, Sintering Kinetics, and Nickel-Activated Pressureless Sintering of Ti3AlC2. Int. J. Appl. Ceram. Technol. 2010, 7, 752–759. [Google Scholar] [CrossRef]
- Omori, M.; Takei, H. Pressureless sintering of SiC. J. Am. Ceram. Soc. 1982, 65, c92–c92. [Google Scholar] [CrossRef]
- Raju, K.; Yoon, D.H. Sintering additives for SiC based on the reactivity: A review. Ceram. Int. 2016, 42, 17947–17962. [Google Scholar] [CrossRef]
- TANAKA, H. Sintering of silicon carbide and theory of sintering. J. Ceram. Soc. Jpn. 2002, 110, 877–883. [Google Scholar] [CrossRef]
- Chi, W.; Jiang, D.; Huang, Z.; Tan, S. Sintering behavior of porous SiC ceramics. Ceram. Int. 2004, 30, 869–874. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, Z.; Wang, X.; Chen, S. Ab initio geometry optimization and ground state properties of layered ternary carbides Ti3MC2 (M = Al, Si and Ge). J. Phys. Condens. Matter 2001, 13, 10001. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, S. First-principles study of Ti3AC2 (A = Si, Al)(0 0 1) surfaces. Acta Mater. 2007, 55, 4645–4655. [Google Scholar] [CrossRef]
- Kleykamp, H. Gibbs energy of formation of SiC: A contribution to the thermodynamic stability of the modifications. Berichte Bunsenges. Phys. Chem. 1998, 102, 1231–1234. [Google Scholar] [CrossRef]
- Zhu, C.C.; Zhu, J.; Wu, H.; Lin, H. Synthesis of Ti3AlC2by SHS and thermodynamic calculation based on first principles. Rare Met. 2015, 34, 107–110. [Google Scholar] [CrossRef]
- Rice, R. Evaluation and extension of physical property-porosity models based on minimum solid area. J. Mater. Sci. 1996, 31, 102–118. [Google Scholar] [CrossRef]
- Rice, R. Comparison of physical property-porosity behaviour with minimum solid area models. J. Mater. Sci. 1996, 31, 1509–1528. [Google Scholar] [CrossRef]
- Li, J.; Lin, H.; Li, J. Factors that influence the flexural strength of SiC-based porous ceramics used for hot gas filter support. J. Eur. Ceram. Soc. 2011, 31, 825–831. [Google Scholar] [CrossRef]
- Salvo, M.; Rizzo, S.; Casalegno, V.; Handrick, K.; Ferraris, M. Shear and bending strength of SiC/SiC joined by a modified commercial adhesive. Int. J. Appl. Ceram. Technol. 2012, 9, 778–785. [Google Scholar] [CrossRef]
- Vargas-Gonzalez, L.; Speyer, R.F.; Campbell, J. Flexural strength, fracture toughness, and hardness of silicon carbide and boron carbide armor ceramics. Int. J. Appl. Ceram. Technol. 2010, 7, 643–651. [Google Scholar] [CrossRef]
- Gulden, T. Mechanical Properties of Polycrystalline β-SiC. J. Am. Ceram. Soc. 1969, 52, 585–590. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krinitcyn, M.; Kopytov, G.; Ryumin, E. Additive Manufacturing of Ti3AlC2/TiC and Ti3AlC2/SiC Ceramics Using the Fused Granules Fabrication Technique. J. Manuf. Mater. Process. 2024, 8, 123. https://doi.org/10.3390/jmmp8030123
Krinitcyn M, Kopytov G, Ryumin E. Additive Manufacturing of Ti3AlC2/TiC and Ti3AlC2/SiC Ceramics Using the Fused Granules Fabrication Technique. Journal of Manufacturing and Materials Processing. 2024; 8(3):123. https://doi.org/10.3390/jmmp8030123
Chicago/Turabian StyleKrinitcyn, Maksim, Georgy Kopytov, and Egor Ryumin. 2024. "Additive Manufacturing of Ti3AlC2/TiC and Ti3AlC2/SiC Ceramics Using the Fused Granules Fabrication Technique" Journal of Manufacturing and Materials Processing 8, no. 3: 123. https://doi.org/10.3390/jmmp8030123