Abstract
Familial hemiplegic migraine type 2 is a premonitory subtype of migraine caused by an ATP1A2 gene mutation. It is an autosomal dominant genetic disease. Here, we report a 51-year-old woman who had a migraine attack due to a pathogenic ATP1A2 gene mutation. With frequent attacks, the patient developed complete left hemiplegia, a confusion of consciousness and partial seizures. Magnetic resonance imaging showed extensive angiogenic edema in the right cerebral hemisphere. In this article, we review the latest literature and try to explain the above symptoms in our patient with cortical spreading depression (CSD) and ATP1A2 gene mutations.
1. Introduction
Familial hemiplegic migraine type 2 is a premonitory subtype of migraine caused by ATP1A2 gene mutations. It is characterized by an aura of reversible motor weakness and is associated with the involvement of at least one first- or second-degree relative. Some other neurological phenomena that patients have include coma and epilepsy []. We report a 51-year-old woman who had a migraine attack due to a pathogenic ATP1A2 gene mutation. With frequent attacks, the patient developed complete left hemiplegia, a confusion of consciousness and partial seizures. Magnetic resonance imaging showed extensive angiogenic edema in the right cerebral hemisphere.
2. Case Presentation
The patient is a 51-year-old woman; at the age of 30, she experienced her first attack. Before each attack, she experienced the following: fatigue before each attack; general weakness that was sometimes slightly obvious on one side, but she was able to walk; gradual to complete difficulty in orientation and understanding; unable to find the location of familiar objects; unable to remember the names of acquaintances; decreased attention; slow response; memory loss; speech disorder (slurred language, incomprehensible speech); and no flashes and scotomas before the attack. There are hallucinations during the attack, and a headache occurs within less than 1 h. Each attack starts from the right front area of the head, followed by full head pain and throbbing pain. Relief is experienced after 1–2 h, and the headache recurs. The attacks become more frequent. The severity gradually increases, and the pattern gradually becomes regular. The attacks occur 1–2 times a month, and the attack normally occurs at 3–5 p.m. Her aunt and father had a similar form of migraine attack.
At the age of 51, she had a migraine attack with weakness in the left limb. The headache became increasingly serious, so she was admitted to a hospital six days after the attack. Upon admission, she had severe headaches and left limb weakness, and the neurological examination revealed left hemiparalysis with muscle strength at 3/5 by manual muscles testing (MMT). A brain MRI (magnetic resonance imaging) on day two of admission revealed cortical swelling in the right cerebral hemisphere (Figure 1A), and on the day three of admission, the magnetic resonance angiography (MRA) showed a slight decrease in the branches of the left middle cerebral artery (MCA). The main trunk of the left posterior cerebral artery (PCA) was slender contralateral, and its branches were significantly reduced contralaterally (Figure 1B). Symptoms persisted. On day five of admission, the patient had a focal epileptic seizure, which was followed by two more seizures. Subsequently, the patient’s left limb weakness gradually worsened, the neurologic examination revealed left hemiparalysis with muscle strength at 0/5 by MMT, and she gradually developed unconsciousness to light coma. Brain CT scans during coma revealed swelling of the right cerebral cortex (Figure 1C). Contrast-enhanced brain MRI showed swelling and diffuse enhancements in the cortex of the right cerebral hemisphere (Figure 1D). Perfusion-weighted imaging (PWI) showed right hemisphere hyperfusion (Figure 1E). Based on her medical history and presentation at admission, a lumbar puncture was performed, and the cerebrospinal fluid pressure (CSF) was 204 mm H2O. The biochemical and cytologic examination of the CSF revealed normal chloride, glucose and cell counts and classifications, in addition to normal protein levels. The CSF tested negative for bacteria, viruses, fungi and autoimmune encephalitis. Whole-exome sequencing showed a missense mutation (c.2473G > A, p.Glu825Lys) in exon 18 of ATP1A2. Based on a series of tests, the patient was diagnosed with FHM2 caused by a pathogenic ATP1A2 mutation. During hospitalization, we dehydrated the patient, gave the patient paracetamol for symptomatic treatment and an intramuscular injection of phenobarbital to control seizures. On the 15th day after admission, consciousness and hemiplegia improved, and the headache was relieved; the patient was discharged after 22 days. She was back to normal, and at later follow-up, the patient’s brain MRI (Figure 2A), contrast-enhanced brain MRI (Figure 2B), PWI (Figure 2C) and computed tomography angiography (CTA) all returned to normal (Figure 2D). The time course diagram of the disease in our patient is presented in Figure 3.
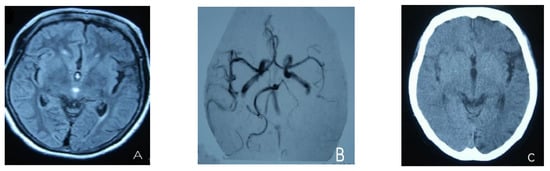
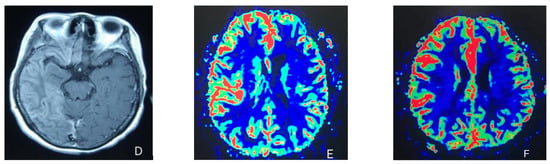
Figure 1.
(A) Cortical swelling in the right cerebral cortex (the 2nd day); (B) the MCA and PCA branches decreased (the 3rd day); (C) swelling of right cerebral cortex (the 7th day); (D) swelling and diffuse enhancement in the cortex of the right cerebral hemisphere; (E,F) right hemisphere hyperfusion.
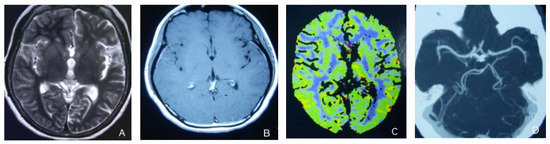
Figure 2.
(A,B) Previously demonstrated the swelling of cerebral cortex has resolved; (C) hyperfusion in the right hemisphere has returned to normal. (D) Bilateral MCA and PCA were relatively symmetrical.
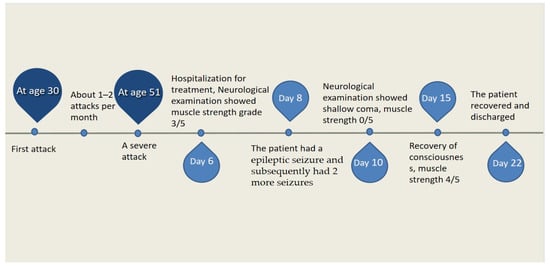
Figure 3.
Time course of the disease. Diagram illustrating time course of disease in our patient.
3. Discussion
Over the past few decades, neurophysiological and neuroimaging studies have provided consistent evidence supporting CSD as a neurophysiological process in migraines. Cortical spreading depression is a depolarizing wave of neurons and glial cells that propagates in adjacent gray matter in a relatively concentric manner. Indeed, during SD, any action potential generation and synaptic transmission are blocked due to the loss of membrane potential; thus, despite depolarization, brain function is inhibited, and the recovery of synaptic activity is required after SD of 10 min or more, with brief bursts of neuronal excitation before propagation []. The ATP1A2 gene encodes the α2 subunit of Na, K-ATPase, which are mainly located in astrocytes and are key players in potassium clearance from the synaptic cleft. The ATP1A2 mutations lead to reduced expression and a loss of function of Na, K-ATPase, which in turn lead to the accumulation of K+ and glutamate in the synaptic cleft []. Heterozygotes of ATP1A2-deficient mice mimicking FHM2 exhibited lower thresholds for the induction of CSD, faster spread and slower recovery []. Growing evidence supports an important role for cortical spreading inhibition in the pathogenesis of migraines. Here, we attempted to use mutations in the CSD and ATP1A2 genes to explain a variety of symptoms in our patients, including aura, headache, seizures, radiographic vasogenic edema and reversible motor weakness.
3.1. CSD and Aura
The transmission of CSD is limited by the sulci and aorta; therefore, the patient’s somatosensory, motor and language auras are difficult to interpret. A new idea that CSD involves the cerebral cortex and/or thalamic nucleus in causing precursors has been proposed. The SD waves propagating to the thalamic reticular nucleus or higher order thalamic nucleus affect multiple cortical regions of different cortical functional networks via the thalamo–corticothalamic connection []. This, in turn, may interfere with large-scale functional networks in the brain [], and it even affects the contralateral cortex, which explains the complex sensory aura and even the bilateral aura []. Studies using resting-state fMRIs (RS-fMRIs) showed that high resting state functional connectivity (RS-Fc) in the interictal lingual gyrus (“aura generator”) may promote CSD initiation, while increased RS-FC in the insula (sensorimotor network) is more conducive to CSD propagation, leading to a complex precursor phenotype []. In conclusion, visual precursors are a result of CSD, and subsequent complex precursors depend on a complex network of brain functional connections.
3.2. CSD and Migraine
Although the mechanisms of migraine onset are not fully understood, the activation and sensitization of pain fibers that govern the dura matter and associated large blood vessels (i.e., meningeal nociceptors) are believed to play a key role in migraine onset []. Direct experimental data confirm that the firing rate of the neurons continued to increase, beginning during CSD, immediately after or after a delay of about 25 min after CSD and lasting 30–70 min []. While the dura is dominated by pain fibers, it is also densely packed with immune cells, especially mast cells, which are closely packed with nociceptive fibers. Aseptic meningeal inflammation is considered to be a key endogenous factor in the sensitization and subsequent activation of meningeal nociceptors. Studies have shown that j-activated meninges nociceptive receptors release neuropeptides, such as substance P, calcitonin gene-related peptide (CGRP) and pituitary adenylate cyclase-activating peptide (PACAP), which cause vascular dilation, plasma protein extravasation and mast cell degranulation []. Mast cells release prostaglandins, serotonin [] and trypsin-like tumor necrosis factor −α []. These inflammatory factors in turn act on meningeal nociceptors. It has been proved that mast cell degranulation can trigger the prolonged excitation of meningeal nociceptors and promote the activation of nociceptive trigeminovascular brainstem neurons []. Therefore, the prolonged activation and sensitization of the trigeminal cervical nerve complex with primary and central nociceptors in vivo due to aseptic meningitis are considered to be the basis of throbbing headaches and allodynia during migraine. It is believed that the increases in K+ and glutamate concentrations in the extracellular interstitium are caused by CSD, which then trigger the migraine (such as sleep deprivation and acute psychological stress). In addition, increased cell swelling and intracellular Ca2+ concentrations caused by CSD can open the Panx1 channel in neurons and trigger a cascade of inflammatory signals. This results in the enhanced pro-inflammatory transcription activity of astrocytes and the release of NO, prostaglandin and cytokines, and these mediators reach the meningeal notoriety receptors through the meningeal–astrocyte network and activate them. This mechanism explains the occurrence of headaches in migraine patients with and without auras [].
3.3. CSD, ATP1A2 and Neurovascular Coupling
TakahiroIizuka et al. monitored cerebral blood flow in three patients with FHM2 during 11 episodes of aura lasting more than 24 h and found that around 18–19 h after the onset of aura, blood flow in the affected cerebral hemispheres would change from hypoperfusion to hyperperfusion, followed by sustained hyperperfusion []. The supply of oxygen and nutrients and the removal of metabolic wastes help maintain the integrity of brain function, which depends on the blood supply to the brain. Neurovascular coupling refers to the increase in regional cerebral blood flow caused by neuronal excitation. Neurovascular coupling promotes the balance between neuronal activity and local blood supplies. The cerebral blood flow is regulated by the interaction of intrinsic myogenic and exogenous vasoactive factors []. Sufficient data show that SD leads to neurovascular uncoupling and releases many potential vasoactive substances during SD, many of which are related to shaping hemodynamic responses []. It was found that the expression of Na and K-ATPase in the cerebral arteries of mice carrying the ATP1A2 gene mutation decreased, while the decrease in Na and K-ATPase allowed the automatic phosphorylation of cSrc kinases []. The α2 isomer-dependent regulation of cSrc kinase can increase the sensitivity of cerebral artery smooth muscle cells to calcium ions. It also means that the change in blood perfusion may be independent of changed neuronal activities at least to some extent but originated from the vascular wall. Moreover, the increased contractility may be confined to the cerebral arteries []. It was previously believed that the mechanism of neurovascular coupling is the dilation of parenchymal arterioles (SM) caused by active neurons in excited neurons and vasoactive substances released by adjacent astrocytes. SM is considered to be the target of action, but SM cannot form direct synaptic connections with most neurons in the brain. On the contrary, each neuron is closely connected with cerebral capillary endothelial cells (CECs). Therefore, the existing view is that neurons excite, release K+ to the extracellular interstitium and activate inward rectifier Kir2.1 channels []. The Kir2.1 channels of smooth muscle cells are open, and hyperpolarization inactivates the voltage-gated calcium channels on the membrane, resulting in a decrease in intracellular Ca2+ concentrations and vascular relaxation []. The hyperpolarization of endothelial cells activates calcium influx, activates calcium-dependent enzymes and causes the release of NO, prostaglandin E2 and epoxide eicosatrienoic acid, which are considered to be effector molecules of neurovascular coupling []. In turn, vascular smooth muscle cells and blood vessels are relaxed []. Animal experiments have found that the basis of cerebral blood flow hyperperfusion in mice carrying the mutant ATP1A2 gene is the increased expression of the endothelial Kir2.1 channel, which leads to the increased sensitivity of cerebral capillaries and to the increase in extracellular stroma K+ concentrations, resulting in abnormally high cerebral vascular hyperemia [].
3.4. CSD, Seizure and Vasogenic Edema
A retrospective study of patients with FHM2 found that, on the 7th–10th day of the attack, MRIs showed swelling and/or cortical hyperintensity on T2/FLAIR-weighted images of the affected hemispheres’ CSD and seizures. This seems to be related to the prolongation of the duration of the attack and suggests angiogenic edema, which is caused by the destruction of the blood–brain barrier []. CSD has been shown to alter the permeability of the blood–brain barrier by activating matrix metalloprotein-9 (MMP-9) []. Additionally, seizures can trigger a pathway involved in glutamate signaling through cytosolic phospholipase A2 (cPLA2), which increases MMP-2 and MMP-9 levels []. All of the above suggest that the swelling and/or cortical hyperintensity on T2/FLAIR-weighted images are due to vasogenic edema caused by blood–brain barrier disruption. Barrier leakage can also trigger seizures [], so cPLA2 may be a potential pharmaceutical target for repairing barrier dysfunction and improve epilepsy treatments []. The MMP inhibitor IPR-179 also showed good antiseizure and antiepileptogenic effects in rodent models of epilepsy and alleviated the cognitive decline caused by epilepsy []. Most of these radiographic changes occurred in the late stage of the attack (day 7–9) and may be related to the prolonged duration of the attack [].
3.5. CSD, ATP1A2 and Seizure
Although the pathophysiological mechanism of FHM and epilepsy comorbidities has not been fully elucidated, functional data and clinical evidence support the supposition that migraine and epilepsy are parts of the common pathogenic mechanism associated with genetically determined modifications in the ion channel and pump alterations []. A mouse model for FHM2 showed increased susceptibility to both epilepsy and CSD. In addition, epileptiform activities were superimposed on CSDs []. SD and seizures are not separate physiological events in the brain; they persist along a dynamical continuum of our membrane of neurons, and in vitro experiments showed that the extracellular K+ concentration causing seizures was lower than that causing SD []. Epileptic seizures can be converted to SD when glial K+ buffer capacities decrease, accompanied by increased cell size and decreased extracellular clearance []. SD has been observed to be triggered by highly focal seizures under experimental and clinical conditions, which was reinforced by a human migraine mutation []. This seems to explain why the prevalence of epilepsy is higher in migraines with auras than in the general population and why ATP1A2 mutations were found in the largest number of patients with epilepsy and FHM (the proportion of patients with epilepsy was 7.7%, 57.7%, 16.7% and 17.9% in FHM1, FHM2, FHM3 and FHM4, respectively) []. According to Isra Tamim et al., microseizures may trigger the migraine aura (SD), which, once triggered, immediately inhibits epileptic seizures by its electrical silencing effect and prevents its generalization; thus, in terms of survival advantage, it is better for the body to experience migraine aura than seizures []. This idea is better supported by the fact that people with epilepsy are more likely to have in-attack or post-attack migraines than those with migraine-induced epilepsy []. The study also found that the increase in power density in the δ frequency range predicts the occurrence of SD []. Meanwhile, previous studies found that the EEG of patients with HM shows a consistent expression of delta range slowing throughout the affected hemisphere []. This seems to explain why epilepsy and HM do not occur together in most cases in patients with hemiplegic migraines.
3.6. ATP1A2, CSD and Transient Motor Impairment
Optogenetically triggered CSD results in the transient and unilateral inhibition of motor function and prolonged reduction in spontaneously active behavior in freely moving mice []. Recent animal experiments found that mice with conditional α2-Na, K ATPase, knockout display episodic paralysis, which is related to spontaneous CSD. The experiment also found that the loss of α2-Na, K ATPase, led to an increase in serine and glycine in the brain, and a diet free of serine and glycine can rescue the transient motor impairment []. Since D-serine is a synaptic NMDA receptor co-agonist, the increase in serine and its related molecules may lead to cortical overexcitation via increased glutamatergic transmissions [], which may explain this possibility.
4. Conclusions
In conclusion, we attempted to use CSD and ATP1A2 gene mutations to explain the symptoms of our patients, including complex auras, headaches, hemiplegia, epilepsy, etc. Interestingly, contrary to the previous view that CSD can trigger epileptic seizures, CSD is now considered to be an anti-epileptic mechanism; in particular, CSD is more likely to occur when the power density in the δ frequency range increases. Moreover, a previous literature review found that the EEG of HM patients mostly showed diffuse slow wave activity on the contralateral side of hemiplegia, and this seems to explain why HM and seizures are mostly independent. In the future, attempts should be made to metabolically explain why increases in serine and glycine in the brain can trigger the transient motor impairment and to explain how CSD plays a role in anti-seizure behavior.
Author Contributions
C.L. analyzed the clinical data and wrote the manuscript. W.Y. contributed to the editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Tianjin Natural Science Foundation (Grant Number 19JCZDJC36500 to W.Y.).
Institutional Review Board Statement
Ethical review and approval were waived for this study due to no identifiable information about the patient was published.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
No new data were created in this study.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Di Stefano, V.; Rispoli, M.G.; Pellegrino, N.; Graziosi, A.; Rotondo, E.; Napoli, C.; Pietrobon, D.; Brighina, F.; Parisi, P. Diagnostic and therapeutic aspects of hemiplegic migraine. J. Neurol. Neurosurg. Psychiatry 2020, 91, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Ayata, C. Cortical Spreading Depression Triggers Migraine Attack: Pro. Headache J. Head Face Pain 2010, 50, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, T.; Tavraz, N.N.; Junghans, C. ATP1A2 Mutations in Migraine: Seeing through the Facets of an Ion Pump onto the Neurobiology of Disease. Front. Physiol. 2016, 7, 239. [Google Scholar] [CrossRef]
- Unekawa, M.; Ikeda, K.; Tomita, Y.; Kawakami, K.; Suzuki, N. Enhanced susceptibility to cortical spreading depression in two types of Na+, K+-ATPase α2 subunit-deficient mice as a model of familial hemiplegic migraine 2. Cephalalgia 2018, 38, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.; Bertolero, M.A.; Liu, W.B.; D’Esposito, M. The Human Thalamus Is an Integrative Hub for Functional Brain Networks. J. Neurosci. 2017, 37, 5594–5607. [Google Scholar] [CrossRef]
- Sepulcre, J.; Sabuncu, M.R.; Yeo, T.B.; Liu, H.; Johnson, K.A. Stepwise Connectivity of the Modal Cortex Reveals the Multimodal Organization of the Human Brain. J. Neurosci. 2012, 32, 10649–10661. [Google Scholar] [CrossRef]
- Vuralli, D.; Karatas, H.; Yemisci, M.; Bolay, H. Updated review on the link between cortical spreading depression and headache disorders. Expert Rev. Neurother. 2021, 21, 1069–1084. [Google Scholar] [CrossRef]
- Silvestro, M.; Tessitore, A.; Di Nardo, F.; di Clemente, F.S.; Trojsi, F.; Cirillo, M.; Esposito, F.; Tedeschi, G.; Russo, A. Functional connectivity changes in complex migraine aura: Beyond the visual network. Eur. J. Neurol. 2022, 29, 295–304. [Google Scholar] [CrossRef]
- Levy, D.; Burstein, R.; Kainz, V.; Jakubowski, M.; Strassman, A.M. Mast cell degranulation activates a pain pathway underlying migraine headache. Pain 2007, 130, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Levy, D. Endogenous Mechanisms Underlying the Activation and Sensitization of Meningeal Nociceptors: The Role of Immuno-Vascular Interactions and Cortical Spreading Depression. Curr. Pain Headache Rep. 2012, 16, 270–277. [Google Scholar] [CrossRef]
- Waeber, C.; Moskowitz, M.A. Migraine as an inflammatory disorder. Neurology 2005, 64, S9–S15. [Google Scholar] [CrossRef]
- Zhang, X.-C.; Strassman, A.M.; Burstein, R.; Levy, D. Sensitization and Activation of Intracranial Meningeal Nociceptors by Mast Cell Mediators. J. Pharmacol. Exp. Ther. 2007, 322, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Erdener, Ş.E.; Kaya, Z.; Dalkara, T. Parenchymal neuroinflammatory signaling and dural neurogenic inflammation in migraine. J. Headache Pain 2021, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Tominaga, N.; Kaneko, J.; Sato, M.; Akutsu, T.; Hamada, J.; Sakai, F.; Nishiyama, K. Biphasic neurovascular changes in prolonged migraine aura in familial hemiplegic migraine type 2. J. Neurol. Neurosurg. Psychiatry 2015, 86, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Colinas, O.; Moreno-Domínguez, A.; Zhu, H.-L.; Walsh, E.J.; Pérez-García, M.T.; Walsh, M.P.; Cole, W.C. α5-Integrin-mediated cellular signaling contributes to the myogenic response of cerebral resistance arteries. Biochem. Pharmacol. 2015, 97, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Ayata, C.; Lauritzen, M. Spreading Depression, Spreading Depolarizations, and the Cerebral Vasculature. Physiol. Rev. 2015, 95, 953–993. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Lai, F.; Banerjee, M.; Duan, Q.; Li, Z.; Si, S.; Xie, Z. Expression of Mutant α1 Na/K-ATPase Defective in Conformational Transition Attenuates Src-mediated Signal Transduction*. J. Biol. Chem. 2013, 288, 5803–5814. [Google Scholar] [CrossRef]
- Staehr, C.; Hangaard, L.; Bouzinova, E.V.; Kim, S.; Rajanathan, R.; Jessen, P.B.; Luque, N.; Xie, Z.; Lykke-Hartmann, K.; Sandow, S.L.; et al. Smooth muscle Ca2+ sensitization causes hypercontractility of middle cerebral arteries in mice bearing the familial hemiplegic migraine type 2 associated mutation. J. Cereb. Blood Flow Metab. 2019, 39, 1570–1587. [Google Scholar] [CrossRef]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykocki, N.R.; Brayden, J.E.; Hill-Eubanks, D.; Nelson, M.T. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef]
- Longden, T.A.; Nelson, M.T. Vascular Inward Rectifier K+ Channels as External K+ Sensors in the Control of Cerebral Blood Flow. Microcirculation 2015, 22, 183–196. [Google Scholar] [CrossRef]
- Hosford, P.S.; Gourine, A.V. What is the key mediator of the neurovascular coupling response? Neurosci. Biobehav. Rev. 2019, 96, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef]
- Staehr, C.; Rajanathan, R.; Postnov, D.D.; Hangaard, L.; Bouzinova, E.V.; Lykke-Hartmann, K.; Bach, F.W.; Sandow, S.L.; Aalkjaer, C.; Matchkov, V.V. Abnormal neurovascular coupling as a cause of excess cerebral vasodilation in familial migraine. Cardiovasc. Res. 2020, 116, 2009–2020. [Google Scholar] [CrossRef]
- Roth, C.; Ferbert, A.; Huegens-Penzel, M.; Siekmann, R.; Freilinger, T. Multimodal imaging findings during severe attacks of familial hemiplegic migraine type 2. J. Neurol. Sci. 2018, 392, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Gürsoy-Ozdemir, Y.; Qiu, J.; Matsuoka, N.; Bolay, H.; Bermpohl, D.; Jin, H.; Wang, X.; Rosenberg, G.A.; Lo, E.H.; Moskowitz, M.A. Cortical spreading depression activates and upregulates MMP-9. J. Clin. Investig. 2004, 113, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Rempe, R.G.; Hartz, A.M.; Soldner, E.L.; Sokola, B.S.; Alluri, S.R.; Abner, E.L.; Kryscio, R.J.; Pekcec, A.; Schlichtiger, J.; Bauer, B. Matrix Metalloproteinase-Mediated Blood-Brain Barrier Dysfunction in Epilepsy. J. Neurosci. 2018, 38, 4301–4315. [Google Scholar] [CrossRef]
- Seiffert, E. Lasting Blood-Brain Barrier Disruption Induces Epileptic Focus in the Rat Somatosensory Cortex. J. Neurosci. 2004, 24, 7829–7836. [Google Scholar] [CrossRef]
- Broekaart, D.W.; Bertran, A.; Jia, S.; Korotkov, A.; Senkov, O.; Bongaarts, A.; Mills, J.D.; Anink, J.J.; Seco-Moral, J.; Baayen, J.C.; et al. The matrix metalloproteinase inhibitor IPR-179 has antiseizure and antiepileptogenic effects. J. Clin. Investig. 2021, 131, e138332. [Google Scholar] [CrossRef]
- Pisano, T.; Spiller, S.; Mei, D.; Guerrini, R.; Cianchetti, C.; Friedrich, T.; Pruna, D. Functional characterization of a novel C-terminal ATP1A2 mutation causing hemiplegic migraine and epilepsy. Cephalalgia 2013, 33, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Kros, L.; Lykke-Hartmann, K.; Khodakhah, K. Increased susceptibility to cortical spreading depression and epileptiform activity in a mouse model for FHM2. Sci. Rep. 2018, 8, 16959. [Google Scholar] [CrossRef]
- Wei, Y.; Ullah, G.; Schiff, S.J. Unification of Neuronal Spikes, Seizures, and Spreading Depression. J. Neurosci. 2014, 34, 11733–11743. [Google Scholar] [CrossRef] [PubMed]
- Ullah, G.; Wei, Y.; Dahlem, M.A.; Wechselberger, M.; Schiff, S.J. The Role of Cell Volume in the Dynamics of Seizure, Spreading Depression, and Anoxic Depolarization. PLoS Comput. Biol. 2015, 11, e1004414. [Google Scholar] [CrossRef]
- Tamim, I.; Chung, D.Y.; de Morais, A.L.; Loonen, I.C.M.; Qin, T.; Misra, A.; Schlunk, F.; Endres, M.; Schiff, S.J.; Ayata, C. Spreading depression as an innate antiseizure mechanism. Nat. Commun. 2021, 12, 2206. [Google Scholar] [CrossRef]
- Hasırcı Bayır, B.R.; Tutkavul, K.; Eser, M.; Baykan, B. Epilepsy in patients with familial hemiplegic migraine. Seizure 2021, 88, 87–94. [Google Scholar] [CrossRef]
- Mantegazza, M.; Cestèle, S. Pathophysiological mechanisms of migraine and epilepsy: Similarities and differences. Neurosci. Lett. 2018, 667, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Houben, T.; Loonen, I.; Baca, S.M.; Schenke, M.; Meijer, J.H.; Ferrari, M.D.; Terwindt, G.M.; Voskuyl, R.A.; Charles, A.; Maagdenberg, A.M.V.D.; et al. Optogenetic induction of cortical spreading depression in anesthetized and freely behaving mice. J. Cereb. Blood Flow Metab. 2017, 37, 1641–1655. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Chen, X.; Brier, L.M.; Bumstead, J.R.; Rensing, N.R.; Ringel, A.E.; Shin, H.; Oldenborg, A.; Crowley, J.R.; Bice, A.R.; et al. Astrocyte deletion of α2-Na/K ATPase triggers episodic motor paralysis in mice via a metabolic pathway. Nat. Commun. 2020, 11, 6164. [Google Scholar] [CrossRef]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.-P.; Oliet, S.H. Synaptic and Extrasynaptic NMDA Receptors Are Gated by Different Endogenous Coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).