Abstract
Classical (MD) and ab initio (AIMD) molecular dynamics simulations were conducted to investigate the fundamental properties of solid and liquid MgO. AIMD was performed by DFT using the Strongly Conditioned and Appropriately Normed (SCAN) exchange correlation functional. The obtained pair-correlation functions of liquid MgO were used as reference data for the optimization of parameters of classical MD. For the latter, a Born–Mayer–Huggins (BMH) potential was applied, and parameters were adjusted until a best fit of both structural properties was obtained by AIMD and physical properties by experimental data. Different structural, dynamic and thermodynamic properties of solid and liquid MgO were then calculated by classical MD and compared with the literature data. Good agreement was found for the Mg-O bond length, self-diffusion coefficients, density of liquid MgO and for heat content and density of crystalline MgO. Using a void-melting approach, the melting temperature of MgO was found as 3295 ± 30 K, which is in good agreement with the recent experimental work by Ronchi et al. (3250 ± 20 K). The optimized parameters of BMH potential describe well the structural, dynamic and thermodynamic properties of solid and liquid MgO and may be combined with our previous results of a CaO-Al2O3-TiO2 system to calculate the properties of a quaternary CaO-MgO-Al2O3-TiO2 system.
1. Introduction
Magnesium oxide is one of the most important components in various scientific and technical fields: from the applied cement and glass industries to fundamental Earth and planetology science. Despite the fact that there are a large number of experimental data for the pure solid oxide available, reliable physico-chemical information about molten MgO is still limited. This is due to the very high melting temperature, which makes experimental investigations very challenging. The only available experimental data in the literature for molten MgO are on the melting temperature at ambient pressure [,,,,,,,,], and some authors have also measured the melting point at high pressure (up to 45 GPa) [,,,]. However, due to the experimental difficulties of measuring the melting temperature of MgO, the values vary in the range of 200 K from 3040 ± 100 [] to 3250 ± 20 K []. It should be noted that in the most recent experimental work by Ronchi et al. [], the authors discussed in detail why the earlier measurements are underestimated and why the real melting temperature is higher than the recommended one of 3070 K.
In general, in case of experimental difficulties, numerical simulations may be a good alternative to obtain the desired data. There are different calculation approaches available, from theory [] to classical (MD) [,,,,] and ab initio (AIMD) molecular dynamics [,,,,,,,] simulations. The results obtained by ab initio calculations are considered as being the most precise data, but even first principles data depend on the number of atoms in a simulation box, the calculation time, the chosen form of exchange correlation, the value of cutoff energy, etc. The majority of ab initio results for MgO were derived using the local density approximation (LDA) [,,] or the generalized gradient approximation (GGA) [,,] exchange correlation. However, Rang and Kresse [] in the most recent work used the Strongly Constrained and Appropriately Normed (SCAN) functional which was considered to give results with the highest precision at that time.
Due to the fact that MgO plays a key role in Earth science, a large number of related works were devoted to the investigation of the melting slope at different pressures [,,,]. In addition, other properties of liquid MgO were calculated from first principles: structural [,,,,], dynamic [,], physical [,,,] and thermodynamic [,,,] properties. The main drawback of ab initio calculations is that it requires significant computing time to obtain high quality results.
In contrast, classical molecular dynamics simulation needs significantly less computing resources. Thus, combining the advantages of both methods, an efficient approach could be to conduct selected ab initio molecular dynamics simulations, then to adjust the interaction parameters of classical MD using AIMD as a reference data and further to calculate properties at all required conditions by classical MD in large enough cells and for longer equilibration times. One of the available methods to achieve this goal is described in the work by Carre et al. []. The basis of this method is to calculate a difference function between partial pair-correlated functions obtained by ab initio and classical molecular dynamics simulations. And minimizing this objective function, the optimal parameters of classical molecular dynamics potential are determined.
The overall aim of our work is to construct reliable classical MD models for pure end-member oxide components (including CaO, Al2O3, MgO, TiO2, SiO2, Fe2O3, etc), which could be used in many scientific fields and technological applications. Recently, we published our results on the investigation of pure CaO by classical and ab initio molecular dynamics simulations including a full comparison with experimental data from the reference []. In a subsequent step, the three-component system CaO-Al2O3-TiO2 was studied for both pure liquid oxides and their mixtures []. In both works, the Born–Mayer–Huggins (BMH) potential was used for the description of the interatomic interaction of different species. Following these works, the aim of the present study is to investigate pure MgO in both the liquid and the solid state. Despite the fact that it is possible to use published BMH potential parameters from the literature [,,], the goal here is to adjust and homogenize the results of this work with our previous data in the CaO-Al2O3-TiO2 system []. We therefore need to re-optimize parameters with two constraints: we should keep the same charge for the O atom and all parameters for the O-O interaction as published in the work by []. Thus, the present work consists of three parts: performing ab initio molecular dynamics calculations to extract the atomic structure of molten MgO using the SCAN exchange functional, then optimizing interaction parameters for classical molecular dynamics simulations using the BMH potential and the ab initio data as a reference and finally carrying out calculations to derive the different structural, dynamic, thermodynamic and physical properties of solid and liquid MgO by classical MD. The results will be compared to the available literature data to benchmark our new potential.
2. Simulations
2.1. Ab Initio Molecular Dynamics Simulations
Density functional theory (DFT) [,] was used to carry out ab initio molecular dynamics simulations of solid and liquid MgO at finite temperatures. The Vienna ab initio simulation package (VASP, v.5.4.4) [,] was used. The projected augmented-wave (PAW) method was used to describe the electron–ion interactions [,]. Exchange–correlation effects were taken into account within the Strongly Conditioned and Appropriately Normed (SCAN) many-body exchange correlation functional []. The 2s2p3s orbitals for Mg and the 2s2p orbitals for O were considered as valence states with a cutoff energy set to 600 eV (beyond the recommended value) to obtain accurate calculations of the pressure. For the solid phases at K, the -centered grid of k-points in the irreducible part of the Brillouin zone was set to 2 × 2 × 2, following the Monkhorst–Pack scheme []. For the liquid states and the high temperature solid states, only the -point was kept. The lattice parameters at 0 K were fully relaxed using the linear tetrahedron method with Blöchl corrections [] to calculate the electronic density of states (DOS). The relaxations were performed with a convergence criterion of eV/Å for the total energy.
In addition to the static calculations described above, ab initio molecular dynamics (AIMD) simulations were performed using the VASP code in the same conditions. The dynamics were performed in the canonical ensemble at a constant number of atoms, volume and temperature (NVT). Newton’s equations of motion were integrated using the Verlet algorithm in the velocity form with a time step of 1 fs. A total of 216 atoms (108 atoms of Mg and 108 atoms of O) were used in all AIMD simulations. The solid state simulations were conducted at 300 K and 3000 K, with the primitive lattice of MgO (B1 structure) as cell geometry. The liquid state simulations were conducted at 3000 K, 3250 K, 3500 K and 4000 K using cubic simulation supercells with periodic boundary conditions in all three directions of space. The volume of the simulation box was adjusted to reach ambient pressure for each temperature for all AIMD simulations. The process included three stages for the solid state. The first one was progressively heating the system up from 0 K (the initial relaxed crystalline configuration was obtained by static calculations describe above) to the target temperature. The second stage was an equilibration at finite temperature within 20 ps. During this time, the volume of the simulation box was adjusted to reach the ambient pressure. And the third stage was the calculation of the solid state properties within 60 ps. For the liquid state, an additional step was included in the procedure to correctly melt the system. At the beginning, the system was progressively heated up to 4000 K where complete melting was observed. After that, the system was cooled down to the target temperature within 20 ps. Then, the same procedure as in the solid state was followed for the equilibration and calculation of the liquid state properties.
From the ab initio simulations, different structural, dynamic and thermodynamic properties were calculated. From the structural point of view, the partial pair-correlation function ((r)) was determined from the following equation:
where (r) represents the mean number of particles j in a spherical shell of radius r and thickness Δr centered on particle i (i, j—Mg and O species).
From the dynamic point of view, the mean-square displacement of each atom type ((t)) (i = Mg, O) was calculated (Equation (2)):
where (t) denotes the position of atom l at time t, and is the number of particles of species i.
The self-diffusion coefficients of Mg and O were defined as a slope of the mean-square displacement which becomes
The angular bracket corresponds to an average over various time origins .
2.2. Classical Molecular Dynamics Simulations and Fitting Parameters Optimization
The first step for the classical molecular dynamics simulation method is to choose the form of interaction potential between particles of the studied substance. One of the least complex interaction potentials, but which works well in oxide systems, is a Born–Mayer–Huggins potential (BMH) [] written in the following form:
where i, j represent different species (Mg and O in the present work), and —interatomic distance between two ions of species i and j. The first term in Equation (4) corresponds to a long-range Coulomb potential. The second term represents a repulsion term, while the two last terms are the contribution of the dipolar expansion: we took only the van der Waals term (C/r6) into account, while the term (D/r8) has a negligible influence, as was shown in our previous works for the aluminosilicate (AS) binary [] and calcium aluminosilicate (CAS) ternary [] systems.
The overall aim of the work is to simulate the pure MgO system and to have an opportunity to combine the obtained results with results of our previous work on the calcium titanoaluminate (CAT) system [] to construct a model for the full quaternary system. To achieve this, additional constraints must be met: the electrical charge of O () and the set of parameters for O-O interaction (, , and ) in the present work must be the same as in []. In addition, to take into account the charge neutrality ( = −qO), we have 8 variable parameters (4 pairs of parameters for Mg-Mg and Mg-O interaction).
The parameter optimization in Equation (4) was made by the method proposed by Carre et al. []. The basis of the method is to identify a set of parameters that gives a good agreement with the structural properties of liquid MgO between the ones obtained by classical MD with the ab initio results, which are considered as reference values. For that, the following objective function () was minimized (Equation (5)):
where and are the partial pair-correlation functions obtained by ab initio and classical molecular dynamics simulations, respectively. In Equation (5), L represents the size of the AIMD simulation box and n an integer. Taking into account that a larger value of n corresponds to putting more weight on larger distances of and that AIMD simulation boxes are rather small and induce possibly finite size effects in the vicinity of L/2, n = 0 was chosen in this work.
The empirical potential optimization of MgO was made at 3500 K and in the NVT ensemble. The temperature was selected in such a way as to ensure that the simulation was carried out in a fully liquid state. To speed up the iterative fitting procedure, only a small system was used, namely 512 atoms. All the equilibration and production stages were conducted within 20 ps.
The first stage of optimization was to generate an initial configuration of the parameters. It was taken from the literature []. Guillot et al. [] used a similar potential as in the present work, excepting the parameter in Equation (4). However, using parameters (Mg-Mg and Mg-O) from [] and fixed parameters (O-O and qO = −qMg) from [], good approximate results for the structure of liquid MgO (gij) were found. Nevertheless, combining parameters and the numerical form of the potentials from different articles led to the value for pressure being out of range with respect to the experimental conditions. Nevertheless, it can still be considered as a good starting point for the optimization process.
The second stage of optimization was the minimization of the objective function () (see Equation (5)), using the Nelder–Mead numerical method. The initial set of parameters was taken from the previous step. For each combination of the parameters, a classical MD simulation (see next section) was performed using a NVT ensemble to determine the partial pair-correlation functions from which a value of was calculated. This method allows us to obtain the best structure matching between ab initio and classical MD simulations. However, this method does not take into account the thermodynamic parameter such as pressure. It should be noted that the ultimate goal of this description of interatomic potentials is to be able to compute the thermodynamic properties for a better understanding and, therefore, more efficient use of these material properties in applications. It is difficult to integrate pressure into our method. Therefore, it is necessary to establish a compromise between the structural properties and the physical pressure. By implementing the minimization method, it has been noted that there are several sets of potential parameters that give very close values of the objective function (Equation (5)). On the other hand, it should be noted that these different sets of parameters do not give the same values of pressure. Thus, we chose a set of parameters that corresponds to the expected structure, obtained by ab initio MD and, at the same time, has the physically correct pressure (close to ambient). Therefore, a parameter CMgo was adjusted to obtain a balance between a good description of both structural and physical properties at NVT conditions and physical properties at NPT conditions. Finally, the optimized parameter set from Equation (5) is presented in Table 1.

Table 1.
Optimized parameters of MgO of BMH potential used in this work (see Equation (4)).
The LAMMPS code [] was used to perform all classical molecular dynamics simulations. The total number of atoms in the simulation box was set to 5832 (2916 Mg atoms and 2916 O atoms). All simulations were performed at NPT ensemble and ambient pressure, except the simulations, which were compared with the ab initio results (they were performed at NVT ensemble). Equations of motion were solved numerically within the Verlet algorithm in the velocity form using a time step of 1 fs, as in the case of ab initio calculations. For the solid state, the starting point was crystalline MgO (B1 structure) at 300 K. A simulation of this state was performed in the NPT ensemble during 100 ps of equilibration stage and 200 ps of production stage. After that a stepwise heating with a temperature step of 500 K was applied to the system, followed by the same equilibration/production stages at each step. The heating continued up to 3500 K (temperature just above the melting temperature). For the liquid state, the system was firstly heated up from crystalline B1 structure at 300 K to 4000 K. After a full melt was observed, the system was cooled down in NPT ensemble stepwise with the temperature step of 250 K with the same equilibration/production stages at each step as for the solid state. The cooling was stopped when a particular crystallization was observed (2500 K).
During the production stage of the simulation, different properties of MgO were calculated. From the structural point of view, the partial pair-correlation function (gij(r)) was determined (see Equation (1)). Concerning the dynamics, the self-diffusion coefficients of Mg and O (D) were defined as a slope of the mean-square displacement (see Equation (3). In addition, the density of solid and liquid MgO was obtained by classical molecular dynamics simulations. From the thermodynamic point of view, the enthalpy was directly calculated during the simulation from the positions and velocities of all atoms as follows:
where is the pressure imposed to the simulation box, and the volume of the box V is a dynamical variable. The heat capacity at constant pressure Cp was determined as a numerical derivative of the enthalpy vs. temperature curve at constant pressure (Equation (7)):
One additional property that we calculated from classical MD simulations was the melting temperature of MgO. It is well known that heating a perfect crystal and observing melting leads to a considerably overestimated value for the melting temperature due to finite size effects. From the many years of inferring the melting temperature, two reliable techniques have been identified, which allow us to estimate the melting temperatures: the direct simulation of the solid–liquid interface [] and void-nucleated melting techniques [,]. As in our previous works [,,], the void-nucleated melting techniques were chosen here. At the beginning, a perfectly crystalline MgO at 300 K containing 5832 atoms was stepwise heated with an average heating rate of 1012 K/s. In the second step, a single spherical void of n atoms in the center of the simulation box was created by removing atoms in a relaxed crystalline configuration of MgO. The same procedure was carried out with this system as for a perfect crystal (n = 0). Different properties of the system such as enthalpy, volume and self-diffusion coefficients of Mg and O atoms were monitored during the simulation, and the melting was observed from a sharp change in these properties. This procedure was applied to different values of n from 0 (indicating a perfect crystal) to a maximum value, when the system ceased to be stable at low temperatures. All MD simulations were performed in the NPT ensemble.
3. Results and Discussion
3.1. Structure and Diffusion
The pair-correlation functions of liquid MgO at 3500 K are presented in Figure 1. The results for the other temperatures (3000 K, 3250 K, 4000 K) are presented in the Supplementary Information file. The first intense peak in the Mg-O partial (orange curve in Figure 1) corresponds to the Mg-O bond length. The simulated value is 1.95 Å by ab initio MD and 2.01 Å by classical MD without any significant shift with temperature (from 3000 K to 4000 K). The value obtained by AIMD is in good agreement with the literature data [,]. Haskins et al. [] found 2.0 Å at 3000 K, which is close to other results within the calculation uncertainty. However, in a deeper analysis, it can be noted that the Mg-O curve obtained by classical MD is shifted to a higher distance, despite the fact that the parameters of the BMH potential were first optimized by using only ab initio rdf data at 3500 K. This seems to be a consequence of the above mentioned adjustment of the Mg-O interaction parameter to adjust the obtained results to the available experimental physical and thermodynamic data. For the symmetric Mg-Mg and O-O pair-correlation functions, a good agreement is observed between ab initio and classical MD for both peak positions and their intensities.
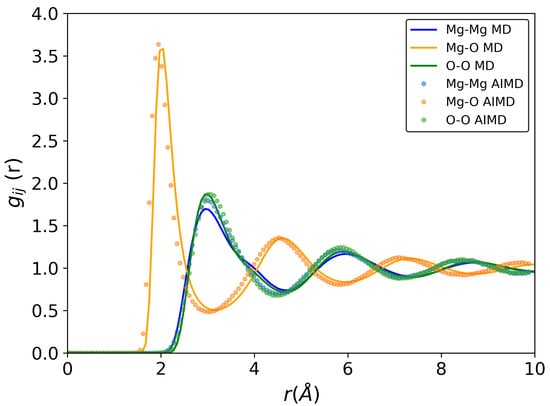
Figure 1.
Pair-correlation functions of liquid MgO at 3500 K. Solid lines—classical molecular dynamics simulation, circles—ab initio MD simulation. All calculations were conducted in NVT ensemble.
For crystalline MgO at 300 K (Figure 2) and 3000 K (Supplementary Information), a larger shift was observed between classical and ab initio MD for all pair-correlation functions (Mg-O, Mg-Mg and O-O) as compared to the results for liquid MgO. However, this result was expected because the parameter optimization was carried out for MgO in the liquid state.
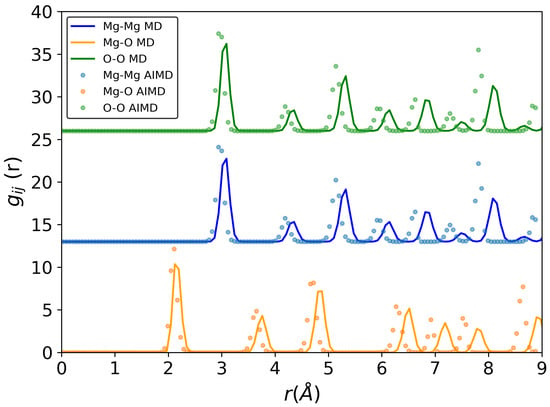
Figure 2.
Pair-correlation functions of crystalline MgO at 300 K. Solid lines—classical molecular dynamics simulation, circles—ab initio MD simulation. All calculations were conducted in NVT ensemble.
Summarizing all this information, we can confirm that from the structural point of view, we obtained reliable data for both ab initio and classical molecular dynamics simulations in the absence of experimental data.
For dynamic properties, the mean-square displacement of Mg and O species was calculated for liquid MgO at different temperatures (3000 K, 3250 K, 3500 K and 4000 K). The results at 3500 K are presented in Figure 3. Other plots at 3000 K, 3250 K and 4000 K are given in Supplementary Information file. As can be seen in Figure 3, for both ab initio and classical MD simulations, a linear behavior was observed for both Mg and O atoms, as expected in the liquid state. Using Equation (3), self-diffusion coefficients of Mg and O were calculated and are listed in Table 2.
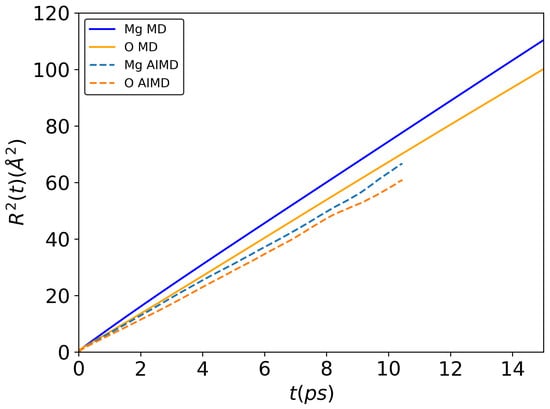
Figure 3.
Mean-square displacement of Mg and O atoms at 3500 K. A total of 216 and 5832 atoms for ab initio and classical MD simulations, respectively. All calculations were conducted in NVT ensemble.
Table 2.
Self-diffusion coefficients of Mg and O at different temperatures. Values are given in .
The self-diffusion coefficients of both Mg and O obtained by classical molecular dynamics simulation (see Table 2) are greater than by ab initio ones, and the difference increases with temperature. This result may be explained by a different number of atoms during the simulation (216 atoms for ab initio MD and 5832 atoms for classical MD). To confirm this, we carried out additional classical MD simulations for 216 atoms (see Figure 4) at 3500 K. As can be seen, the slopes in the mean-square displacement dependencies show a similar behavior for ab initio and classical MD of both Mg and O atoms. This indicates that the self-diffusion coefficients are also the same. Comparing our simulation results with the literature data, an excellent agreement of self-diffusion coefficients between results obtained by classical MD in this research and a work of Haskins et al. [] is observed. In [], the authors used ab initio MD with a box of 300 atoms. They investigated the size effect (with 300, 600 and 1200 atoms) by calculating the density of oxide. Thus, there is no guarantee that for dynamic properties, the size effect also was not observed. Karki et al. [] presented only the total diffusion coefficients. However, again, the results obtained by classical MD simulations in this work are in good agreement with the data found in []. Thus, we may conclude that the parameters of BMH potential optimized in the present work are in agreement with the published results and can be used to derive the dynamic properties of molten MgO.
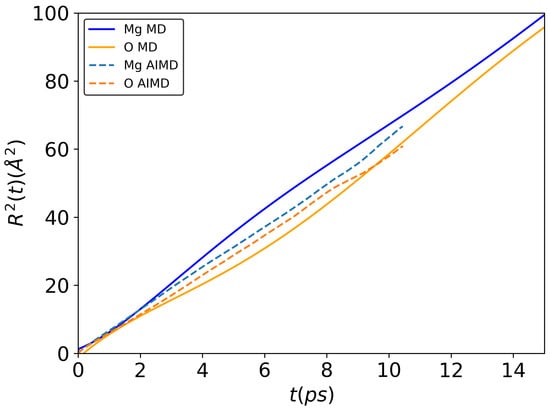
Figure 4.
Mean-square displacement of Mg and O atoms at 3500 K with 216 atoms by ab initio and classical MD simulations. All calculations were conducted in NVT ensemble.
3.2. Thermodynamic Properties and Melting Temperature
The enthalpies of solid and liquid MgO were directly derived from the classical molecular dynamics simulations (see Equation (6)). The results for the heat content (H(T)-H(273)) are presented in Figure 5. The calculated heat content by classical MD for solid MgO was found to be in good quantitative agreement with the available experimental data [,,,,,] up to 2500 K (the absolute difference does not exceed 5 kJ/mol from room temperature to 1500 K and reached 5% at 2500 K []). It should be noted that the literature data for the heat content [,,,] were corrected from H(298) to H(273) using the fitted data from Gurvich [] for the heat capacity of solid MgO from 273 K to 298 K. At higher temperatures (from 2500 K to 3000 K), the compiled results from Gurvich, which are based on the extrapolation of the fitted low-temperature data [], differ about 7% as compared to our results. In addition, an ab initio MD simulation was performed at 3000 K, and as a result, the derived heat content differs 8% with classical MD and 3% with Gurvich’s estimation [], which is considered satisfactory.
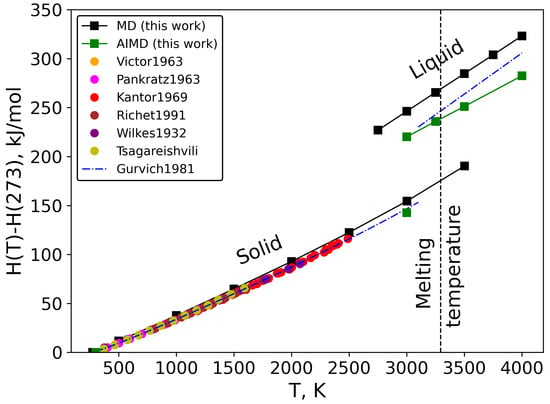
Figure 5.
Heat content of solid and liquid MgO as a function of temperature [,,,,,,].
The analysis of the heat content data of liquid MgO is more difficult, as there are no experimental data available for comparison. All calculated data increase linearly with temperature but with different slopes. For example, the difference between classical MD and AIMD is 26 kJ/mol at 3000 K and 41 kJ/mol at 4000 K. It is interesting to mention that the heat content of molten MgO from Gurvich’s estimation [] lies in between the two curves, is close to the AIMD result at 3000 K and becomes closer to classical MD at 4000 K. Nevertheless, such a big spread of the heat content of the liquid state compare to the solid state leads to a different enthalpy of melting that will be discussed below.
Another thermodynamic property that was derived from our classical MD simulation is the heat capacity at constant pressure. The result was derived from the calculated heat content (see Equation (7)) and is presented in Figure 6. A good agreement between heat capacity of solid MgO obtained by classical MD simulations, experimental data and calculated from heat content data in the temperature range from 1000 to 1800 K [,,,,,] was observed. In addition, we found excellent agreement with Kantor et al. [] data, which are the highest high-temperature experimental data (up to 2500 K) for this substance. However, even for temperatures higher than 2500 K up to melting point, the discrepancies between MD data and Gurvich’s estimation [] are less than 5%, which indicates that the values obtained by classical molecular dynamics simulations are acceptable. At temperatures lower than 1000 K, there are not enough raw data to derive a good calculation of heat capacity from the heat content data.
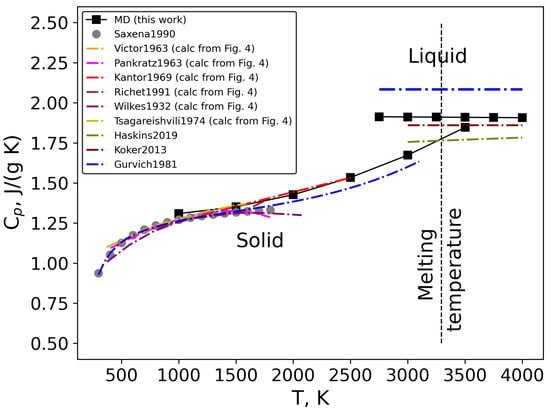
Figure 6.
Heat capacity of solid and liquid MgO as a function of temperature [,,,,,,,,,].
For the liquid state, a huge spread of data was observed. It should be noted that there is no direct experimental investigation of the heat capacity of liquid MgO. Thus, in the literature, only an estimation can be found [], and values are calculated by ab initio methods [,]. As can be seen in Figure 6, the result obtained by classical MD in this work (1.91 J/(g K)) is in good agreement with the literature data [,] and only 8% lower than the heat capacity estimated by Gurvich [] (2.08 J/(g K)), which is a good result in the absence of experimental data. In addition to estimated values from Gurvich data [] and ab initio methods [,], in the literature, two estimations for the heat capacity of molten MgO, obtained by the additive rule by fitting the direct experimental heat capacity data of multicomponent oxide glasses and liquids [,], were also found. The obtained results were 2.47 J/(g K) [] and 2.34 J/(g K) [], which are significantly higher than the value obtained in the present work by classical MD (1.91 J/(g K)). However, taking into account that it was fitting and not direct experimental data, we believe that our value for heat capacity of liquid MgO is acceptable.
Melting is a fundamental phenomenon and a first-order phase transition. Knowledge of the melting temperature of MgO makes it possible to construct a physically correct phase diagram or to estimate the enthalpy of fusion. As was noted earlier in this contribution, the melting temperature of MgO was calculated by classical molecular dynamics using a void-nucleated melting technique [,]. The obtained results are presented in Figure 7. It is well known that heating a perfect crystal leads to a considerably overestimated value for the melting temperature. As a result, 3827 K was found as a melting temperature under these conditions (n = 0). The close value for a perfect crystal was also found in the literature []. In our simulations, a plateau of the melting temperature at 3295 K was found in the 12–25% interval of the void (Figure 7). Thus, the melting temperature calculated here by classical MD was taken as 3295 ± 30 K. As mentioned in the introduction, due to experimental difficulties to measure precisely the melting temperature of MgO, a wide range of experimental data was found in the literature from 3040 ± 100 K [] to 3250 ± 20 K []. Our value (3295 ± 30 K) seems to be slightly overestimated as compared to the currently recommended melting temperature of 3070 K; however, it is in good agreement with the most recent experimental work of Ronchi et al. [], in which the authors discussed in detail why the earlier measurements are underestimated and why the recommended value should be revised.
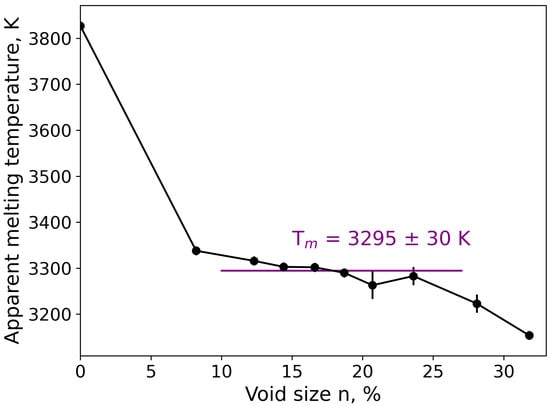
Figure 7.
Apparent melting temperature as a function of the volume of the void (in % of the simulation box volume).
Finally, the heat of fusion was calculated from the above data. By using Figure 5, the heat of fusion obtained by classical MD at 3295 K was found as 93 kJ/mol. This value is close to other data in the literature: 90 kJ/mol (at 3100 K) [] and 86 kJ/mol (at 3032 K) []. Taking into account the large difference in the heat content of molten MgO by classical and ab initio MD (see Figure 5), the value 78 kJ/mol at 3000 K was found for the heat of fusion by AIMD, which is 17% lower than by classical MD. However, the AIMD value is in good agreement with estimated heat of fusion from Gurvich [] (77 kJ/mol at 3100 K). Thus, the calculated heat of fusion by both classical and ab initio is considered to be acceptable in the absence of any experimental data.
3.3. Density
The density of solid and liquid MgO was calculated by classical MD simulations as the reverse volume of the simulation box. The results are presented in Figure 8 (black squares). As can be seen for the solid state, the calculated data have a systematic underestimation of 0.06 g/cm3 compared with experimental data [,,], and this small shift is almost constant even at high temperature close to the melting point. For the liquid state, there are no experimental data in the literature; however, our results can be compared with available calculated data [,,,,]. The majority of these values are listed at around 3000 K. As can be seen in Figure 8, the density obtained by classical MD in this work is in excellent agreement with [,,,,]. It should also be noted that in two works [,], the authors calculated the density of both solid and liquid states of MgO, and both these results agree well with our classical MD data.
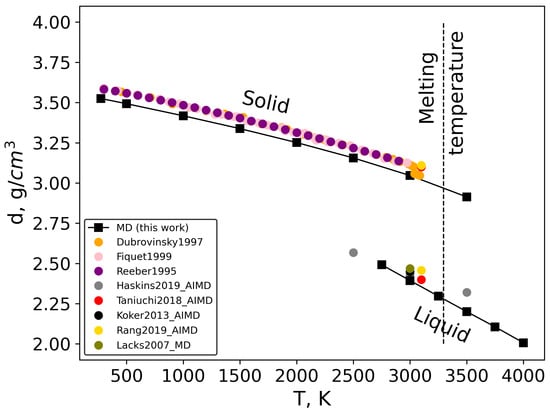
Figure 8.
Density of solid and liquid MgO as a function of temperature [,,,,,,,].
4. Summary and Conclusions
In this study, the structural, dynamic, thermodynamic and physical properties of solid and liquid MgO were investigated by classical and ab initio molecular dynamics. AIMD was performed by DFT and the Strongly Conditioned and Appropriately Normed (SCAN) many-body exchange correlation functional. A simulation box of 216 atoms and a cutoff energy set of 600 eV were used. The obtained structural properties (partial pair-correlation function) of liquid MgO were further used as reference data for the optimization of the interaction parameters in classical MD. For the latter one, a Born–Mayer–Huggins potential was applied, and the parameters were adjusted to find a best fit of both structural properties obtained by AIMD and physical properties derived from experimental data. In addition, the charge of the O atom and the parameters for the O-O interaction were considered as fixed and were taken from our previous study of CaO-Al2O3-TiO2 system to be able to combine both of these results and to calculate a quaternary CaO-MgO-Al2O3-TiO2 system.
From a structural point of view, the partial pair-correlation functions of molten MgO were calculated by classical MD and AIMD at 3000 K, 3250 K, 3500 K and 4000 K in the NVT ensemble. In the absence of experimental data, these results were compared with earlier AIMD simulations, and good agreement was found between all of these results. A bond length of 1.95 Å by ab initio MD and 2.01 Å by classical MD was found without any significant shift with increasing temperature (from 3000 K to 4000 K). For the solid state, an acceptable agreement of the calculated results between MD and AIMD at 300 K and 3000 K was observed.
The mean-square displacement of Mg and O atoms was calculated for liquid MgO at different temperatures (3000 K, 3250 K, 3500 K and 4000 K) with further calculation of the self-diffusion coefficients. A size effect was observed for dynamic properties, and it was concluded that a simulation box of 216 atoms is not enough for the calculation of diffusion coefficients of atoms. Comparing our dynamic properties of molten MgO with the literature data obtained by AIMD showed that our optimized parameters of the BMH potential gives reasonable results for the dynamic properties of liquid MgO.
The heat content of both solid and liquid MgO was calculated by classical and ab initio molecular dynamics. An excellent agreement for the solid state was found with experimental data in the literature for temperatures below 1500 K and with Gurvich’s extrapolated data up to melting point (7% difference with classical MD and 3% difference with ab initio MD). Due to the lack of experimental data for molten MgO, the simulated heat content obtained in the liquid state as obtained by classical and ab initio MD was compared with each other and with Gurvich’s estimation. A big spread of these data was observed (from 26 kJ/mol at 3000 K to 41 kJ/mol at 4000 K), which led to a difference in the heat of fusion. A heat of fusion of 93 kJ/mol was found by classical MD and 78 kJ/mol by AIMD. This must be compared to various estimations in the literature ranging from 77 to 90 kJ/mol. This indicates that the simulated heat of fusion by both classical and ab initio MD values in the present work is in the acceptable range in the absence of experimental data. Using the heat content data, the heat capacity at constant pressure of liquid and solid MgO was calculated. An acceptable agreement with experimental literature data in the temperature range 1000–1800 K for solid MgO was observed. At higher temperatures (from 2000 K up to melting), the discrepancies between classical MD data and Gurvich’s estimation were found to be less than 5%, which confirms that values obtained by classical molecular dynamics simulations data are acceptable for the solid state. Since we observed a large spread in the heat content data for molten MgO, a huge discrepancy between the heat capacity at constant pressure can be noticed. For classical MD, we found 1.91 J/(g K). These data are smaller than Gurvich’s estimation (2.08 J/(g K)). However, the result for the heat content at constant pressure obtained by classical MD in this work is in good agreement with other literature calculations and estimations. To sum up, we may conclude that the parameters of the BMH potential optimized here in this work describes in a coherent manner the thermodynamic properties of liquid and solid MgO.
Using a void-melting approach, the melting temperature of MgO obtained by classical MD and the BMH potential was found to be 3295 ± 30 K, which is in good agreement with the recent experimental work of Ronchi et al. [] (3250 ± 20 K) using a laser fusion technique. Despite the fact that, by now, the recommended value of melting temperature is 3070 K, Ronchi et al. [] discussed in detail why the earlier measurements are underestimated and why the recommended value should be revised. Last but not least, the density as a function of temperature was simulated and compared to the experimental literature data. We found a systematic but constant underestimation of 0.06 g/cm3 of the density of solid MgO compared to experimental data from room temperature up to melting by classical MD simulations. For the liquid state, our results of MD are in good agreement with other available calculated data in the literature.
To conclude, we derived a new, optimized set of parameters for the Born–Mayer–Huggins potential in the present work to simulate the structural, dynamic, thermodynamic and physical properties of solid and liquid MgO. Our results may now be combined with our previous results of the CaO-Al2O3-TiO2 system to calculate properties in a quaternary CaO-MgO-Al2O3-TiO2 system. This may be very useful for studying glasses, as it is easy to simulate quenched glass using molecular dynamics simulations. Since the parameters found in this study describe the properties of the liquid state well, they should also work reasonably well for glasses containing MgO, which may be of interest to high-refractory materials.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ceramics7030078/s1, Figure S1: Pair-correlation functions of liquid MgO at 3000 K; Figure S2: Pair-correlation functions of liquid MgO at 3250 K; Figure S3: Pair-correlation functions of liquid MgO at 4000 K; Figure S4: Pair-correlation functions of solid MgO at 3000 K; Figure S5: Mean-square displacement of Mg and O atoms at 3000 K; Figure S6: Mean-square displacement of Mg and O atoms at 3250 K; Figure S7: Mean-square displacement of Mg and O atoms at 4000 K.
Author Contributions
Conceptualization, N.J. and A.P.; methodology, A.S.A. and N.J.; software, A.S.A. and N.J.; validation, A.P. and N.J.; formal analysis, A.S.A. and N.J.; investigation, A.S.A. and N.J.; resources, N.J.; writing—original draft preparation, A.S.A.; writing—review and editing, A.P., I.A.U. and N.J.; visualization, A.S.A. and I.A.U.; supervision, A.P. and I.A.U.; funding acquisition, N.J. and I.A.U. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been partially supported by MIAI@Grenoble-Alpes (ANR-19-P3IA-0003). The investigations were partially financially supported by the Russian Ministry of Science and Education, grant No. 075-15-2021-1353.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in this article.
Acknowledgments
We acknowledge the CINES, IDRIS, and TGCC under Project No. INP2227/ 72914/gen5054, as well as CIMENT/GRICAD for computational resources. Discussions within the French collaborative network in high-temperature thermodynamics GDR CNRS3584 (TherMatHT) and in artificial intelligence in materials science GDR CNRS 2123 (IAMAT) are also acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zerr, A.; Boehler, R. Constraints on the melting temperature of the lower mantle from high-pressure experiments on MgO and magnesioüstite. Nature 1994, 371, 506–508. [Google Scholar] [CrossRef]
- Riley, B. Determination of melting points at temperatures above 2000° Celcius. Rev. Int. Hautes Temp. Refract. 1966, 3, 327–336. [Google Scholar]
- Ronchi, C.; Sheindlin, M. Melting point of MgO. J. Appl. Phys. 2001, 90, 3325–3331. [Google Scholar] [CrossRef]
- Dubrovinsky, L.S.; Saxena, S.K. Thermal expansion of periclase (MgO) and tungsten (W) to melting temperatures. Phys. Chem. Miner. 1997, 24, 547–550. [Google Scholar] [CrossRef]
- Chernyshev, A.P.; Petrov, V.A.; Titov, V.E.; Vorobyev, A.Y. Thermal radiative properties of magnesium oxide at high temperatures. Thermochim. Acta 1993, 218, 195–209. [Google Scholar] [CrossRef]
- McNally, R.N.; Peters, F.I.; Ribbe, P.H. Laboratory furnace for studies in controlled atmospheres; melting points of MgO in a N2 atmosphere and of Cr2O3 in N2 and in air atmospheres. J. Am. Ceram. Soc. 1961, 44, 491–493. [Google Scholar] [CrossRef]
- Panek, Z. Melting Temperatures of CaO and MgO. Silikaty 1979, 23, 97–102. [Google Scholar]
- Zhang, L.; Fei, Y. Melting behavior of (Mg,Fe)O solid solutions at high pressure. Geophys. Res. Lett. 2008, 35, L13302. [Google Scholar] [CrossRef]
- Kimura, T.; Ohfuji, H.; Nishi, M.; Irifune, T. Melting temperatures of MgO under high pressure by micro-texture analysis. Nat. Commun. 2017, 8, 15735. [Google Scholar] [CrossRef]
- Du, Z.; Lee, K.K. High-pressure melting of MgO from (Mg,Fe)O solid solutions. Geophys. Res. Lett. 2014, 41, 8061–8066. [Google Scholar] [CrossRef]
- Leu, A.L.; Ma, S.M.; Eyring, H. Properties of molten magnesium oxide. Proc. Natl. Acad. Sci. USA 1975, 72, 1026–1030. [Google Scholar] [CrossRef]
- Vočadlo, L.; Price, G.D. The melting of MgO—Computer calculations via molecular dynamics. Phys. Chem. Miner. 1996, 23, 42–49. [Google Scholar] [CrossRef]
- Ferneyhough, R.; Fincham, D.; Price, G.D.; Gillan, M.J. The melting of MgO studied by molecular dynamics simulation. Model. Simul. Mater. Sci. Eng. 1994, 2, 1101. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Cheng, X.-L.; Zhang, H.; Cai, L.-C. Molecular dynamics study for the melting curve of MgO at high pressure. Chin. Phys. 2004, 13, 384. [Google Scholar] [CrossRef]
- Lacks, D.J.; Rear, D.B.; Van Orman, J.A. Molecular dynamics investigation of viscosity, chemical diffusivities and partial molar volumes of liquids along the MgO–SiO2 join as functions of pressure. Geochim. Cosmochim. Acta 2007, 71, 1312–1323. [Google Scholar] [CrossRef]
- Jahn, S. Atomic structure and transport properties of MgO-Al2O3 melts: A molecular dynamics simulation study. Am. Mineral. 2008, 93, 1486–1492. [Google Scholar] [CrossRef]
- Alfè, D. Melting curve of MgO from first-principles simulations. Phys. Rev. Lett. 2005, 94, 235701. [Google Scholar] [CrossRef]
- Rang, M.; Kresse, G. First-principles study of the melting temperature of MgO. Phys. Rev. B 2019, 99, 184103. [Google Scholar] [CrossRef]
- Karki, B.B.; Bhattarai, D.; Stixrude, L. First-principles calculations of the structural, dynamical, and electronic properties of liquid MgO. Phys. Rev. B 2006, 73, 174208. [Google Scholar] [CrossRef]
- Aguado, A.; Madden, P.A. New Insights into the Melting Behavior of MgO from Molecular Dynamics Simulations: The Importance of Premelting Effects. Phys. Rev. Lett. 2005, 94, 068501. [Google Scholar] [CrossRef]
- Haskins, J.B.; Stern, E.C.; Bauschlicher, C.W.; Lawson, J.W. Thermodynamic and transport properties of meteor melt constituents from ab initio simulations: MgSiO3, SiO2, and MgO. J. Appl. Phys. 2019, 125, 235102. [Google Scholar] [CrossRef]
- Taniuchi, T.; Tsuchiya, T. The melting points of MgO up to 4 TPa predicted based on ab initio thermodynamic integration molecular dynamics. J. Phys. Condens. Matter 2018, 30, 114003. [Google Scholar] [CrossRef]
- Belonoshko, A.B.; Arapan, S.; Martonak, R.; Rosengren, A. MgO phase diagram from first principles in a wide pressure-temperature range. Phys. Rev. B 2010, 81, 054110. [Google Scholar] [CrossRef]
- de Koker, N.; Karki, B.B.; Stixrude, L. Thermodynamics of the MgO–SiO2 liquid system in Earth’s lowermost mantle from first principles. Earth Planet. Sci. Lett. 2013, 361, 58–63. [Google Scholar] [CrossRef]
- Strachan, A.; Çağin, T.; Goddard, W.A., III. Phase diagram of MgO from density-functional theory and molecular-dynamics simulations. Phys. Rev. B 1999, 60, 15084. [Google Scholar] [CrossRef]
- Carré, A.; Ispas, S.; Horbach, J.; Kob, W. Developing empirical potentials from ab initio simulations: The case of amorphous silica. Comput. Mater. Sci. 2016, 124, 323–334. [Google Scholar] [CrossRef]
- Alvares, C.; Deffrennes, G.; Pisch, A.; Jakse, N. Thermodynamics and structural properties of CaO: A molecular dynamics simulation study. J. Chem. Phys. 2020, 152, 084503. [Google Scholar] [CrossRef]
- Jakse, N.; Alvares, C.M.S.; Pisch, A. Ab initio based interionic interactions in calcium aluminotitanate oxide melts: Structure and diffusion. J. Phys. Condens. Matter 2021, 33, 285401. [Google Scholar] [CrossRef]
- Guillot, B.; Sator, N. A computer simulation study of natural silicate melts. Part I: Low pressure properties. Geochim. Cosmochim. Acta 2007, 71, 1249–1265. [Google Scholar] [CrossRef]
- Mitchell, P.J.; Fincham, D. Shell model simulations by adiabatic dynamics. J. Phys. Condens. Matter 1993, 5, 1031. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Sun, J.; Ruzsinszky, A.; Perdew, J.P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 2015, 115, 036402. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Huggins, M.L.; Mayer, J.E. Interatomic distances in crystals of the alkali halides. J. Chem. Phys. 1933, 1, 643–646. [Google Scholar] [CrossRef]
- Bouhadja, M.; Jakse, N. Structural and dynamic properties of aluminosilicate melts: A molecular dynamics study. J. Phys. Condens. Matter 2019, 32, 104002. [Google Scholar] [CrossRef] [PubMed]
- Bouhadja, M.; Jakse, N.; Pasturel, A. Structural and dynamic properties of calcium aluminosilicate melts: A molecular dynamics study. J. Chem. Phys. 2013, 138, 224510. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; In’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Morris, J.R.; Wang, C.Z.; Ho, K.M.; Chan, C.T. Melting line of aluminum from simulations of coexisting phases. Phys. Rev. B 1994, 49, 3109–3115. [Google Scholar] [CrossRef]
- Agrawal, P.M.; Rice, B.M.; Thompson, D.L. Molecular dynamics study of the melting of nitromethane. J. Chem. Phys. 2003, 119, 9617–9627. [Google Scholar] [CrossRef]
- Solca, J.; Dyson, A.J.; Steinebrunner, G.; Kirchner, B.; Huber, H. Melting curves for neon calculated from pure theory. J. Chem. Phys. 1998, 108, 4107–4111. [Google Scholar] [CrossRef]
- Jakse, N.; Pasturel, A. Molecular-dynamics study of liquid nickel above and below the melting point. J. Chem. Phys. 2005, 123, 244512. [Google Scholar] [CrossRef]
- Victor, A.C.; Douglas, T.B. Thermodynamic properties of magnesium oxide and beryllium oxide from 298 to 1200 K. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1963, 67, 325. [Google Scholar] [CrossRef]
- Pankratz, L.B.; Kelley, K.K. Thermodynamic Data for Magnesium Oxide (Periclase); U.S. Bureau of Mines Report of Investigations 6295; U.S. Bureau of Mines: Washington, DC, USA, 1963.
- Richet, P.; Fiquet, G. High-temperature heat capacity and premelting of minerals in the system MgO-CaO-Al2O3-SiO2. J. Geophys. Res. Solid Earth 1991, 96, 445–456. [Google Scholar] [CrossRef]
- Wilkes, G.B. The specific heat of magnesium and aluminum oxides at high temperatures. J. Am. Ceram. Soc. 1932, 15, 72–77. [Google Scholar] [CrossRef]
- Tsagareishvili, D.S.; Gvelisiani, G.G. Entalpiya i teploemkost okisi magniya pri visokih temperaturah. Teplofiz. Visok. Temp. 1974, 1, 208–210. [Google Scholar]
- Kantor, P.B.; Fomichev, E.N. Izmerenie entalpii i teploemkosti okisi magniya i dvuokisi tsirkoniya v intervale temperatur 1200-2500 K. Teplofiz. Svoystva Tverd. Visok. Temp. 1969, 1, 406–408. [Google Scholar]
- Glushko, V.P.; Gurvich, L.V.; Bergman, G.A.; Veyts, I.V.; Medvedev, V.A.; Khachkuruzov, G.A.; Yungman, V.S. Thermodynamic Properties of Individual Substances; Nauka: Moscow, Russia, 1981; Volume III. [Google Scholar]
- Saxena, S.K.; Zhang, J. Thermochemical and pressure-volume-temperature systematics of data on solids, examples: Tungsten and MgO. Phys. Chem. Miner. 1990, 17, 45–51. [Google Scholar] [CrossRef]
- Stebbins, J.F.; Carmichael, I.S.E.; Moret, L.K. Heat capacities and entropies of silicate liquids and glasses. Contrib. Mineral. Petrol. 1984, 86, 131–148. [Google Scholar] [CrossRef]
- Lange, R.A.; Navrotsky, A. Heat capacities of Fe2O3-bearing silicate liquids. Contrib. Mineral. Petrol. 1992, 110, 311–320. [Google Scholar] [CrossRef]
- Fiquet, G.; Richet, P.; Montagnac, G. High-temperature thermal expansion of lime, periclase, corundum and spinel. Phys. Chem. Miner. 1999, 27, 103–111. [Google Scholar] [CrossRef]
- Reeber, R.R.; Goessel, K.; Wang, K. Thermal expansion and molar volume of MgO, periclase, from 5 to 2900 K. Eur. J. Mineral. 1995, 7, 1039–1048. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).