1. Introduction
Leaching of sulfide minerals [
1] necessitates the employment of a sufficient oxidizing agent, which is commonly ferric iron [
2]. High ferric iron concentrations generate various forms of precipitates on the surface of ore particles, reducing the accessibility of the leaching agent and bacteria cells to the surfaces. Chemical and biological interactions oxidize ferrous iron in acidic situations; biological oxidation of iron is much faster than chemical oxidation. In the bio-oxidation process, ferrous iron is often oxidized, resulting in precipitates containing schwertmannite (Fe
8O
8(OH)
6SO
4)·nH
2O, ferrihydrite (5Fe
2O
3·9H
2O), and several forms of jarosite [
3]. The quantities of ferric iron, sulfate, and alkali cations, as well as temperature, contact time, and pH, influence the specific phases formed and their properties. Jarosite-group minerals are typically precipitated from sulfate-rich media with pH values ranging from 1 to 3, but schwertmannite precipitation [
4] is more feasible at pH values of 3–4. In cases when alkali cation concentrations were insufficient for the development of more ordered phases, schwertmannite synthesis at lower pH was observed in some cases [
5]. Jarosite (KFe
3(SO
4)
2(OH)
6) formation in the bio-leaching process requires cations, which can be obtained from salts utilized as nutrition in bacteria’s growth medium or through mineral dissolution. The type of jarosite generated is determined by the concentrations of the cations NH
4+, K
+, and Na
+. A low concentration of potassium is needed for the potassium jarosite [
6] formation, whereas a high concentration of sodium is required for the sodium jarosite [
7]. Previous findings indicate that jarosite is formed by a single nucleation event followed by two independent stages of development, each with a different Avrami exponent [
6].
If an appropriate cation is available to occupy the “A” site in the jarosite structure, schwertmannite can be a precursor to jarosite mineral formation in acidic, sulfate-rich settings. Laboratory systems as well as sediments damaged by acidic mine water flow have shown the transition from schwertmannite to jarosite [
5]. The dominant monovalent cations in bioleaching solutions and liquid media for iron-oxidizing microorganisms are typically K
+, NH
4+, and Na
+, in addition to the ubiquitous H
3O
+, but other monovalent and divalent cations (e.g., Ag
+, Pb
2+, Cu
2+, Ni
2+, and Hg
2+) may also be available and can embed into the jarosite matrix [
5].
To precipitate dissolved iron from hot acid leach solutions prior to further zinc recovery, the jarosite process is the most extensively utilized technology in the zinc business [
7]. In the present research, sulfuric acid is needed to produce high purity rare earth oxides (REO) from coal refuse sources using innovative technologies that will reduce costs and improve environmental outcomes compared to traditional rare-earth processing technologies [
8]. In this case, some iron removal is desirable prior to acid-mediated leaching to avoid contamination and the formation of other unwanted products in leaching circuits. Thus, adequate measures are needed to remove iron.
One iron removal approach is precipitation or jarosite formation. This approach has several advantages, including the ability to produce filterable iron precipitates, minimal losses of Zn, Cd, and Cu in jarosite precipitates, and simultaneous control of sulfate and alkali ions. An alkali source (typically NH
3, Na
2CO
3, or Na
2SO
4) and a neutralizing agent are added to an iron-rich, hot acid leach solution in the jarosite formation process [
7].
The overall biochemical reaction of ferrous oxidation can be written as [
9]:
Note that this ferric ion can be hydrolyzed to its hydroxide
As a result of the hydrogen ion consumption during ferric ion generation, the pH of the system initially rises. The hydrolysis of ferric iron, on the other hand, counteracts this rise in pH.
The hydroxide can combine with sulfate to form iron sulfate hydroxide as can be seen:
This is a process that accompanies the hydrolysis reaction, yielding basic ferric hydroxysulfates as a result. In presence of the required cation, hydroxysulfate precipitates or jarosite formation takes place as can be seen here:
It is very clear that at low pH, ferric ions remain dissolved, and precipitation starts at elevated pH. In most cases, the required cations for the formation of jarosite are M = K+, Na+, NH4+, Ag+, or H3O+. It is also helpful to note that as precipitation continues high ferric ion concentration will be less effective in very low pH solutions. In the case of high sulfate concentration, more iron hydroxide will be consumed and it will promote the forward reaction, and hence reduction of pH can be observed. Note that such reduction of pH may be counteracted by consumption to dissolve some gangue minerals.
In general, exopolysaccharides are thought to promote Fe (III) deposition on bacterial cell surfaces, with bacterial cell surfaces functioning as nuclei for initial crystal development. Exopolysaccharides are important in the production of biofilms in many bacteria; however, mutants were unable to generate exopolysaccharides and so became impaired or unable to form mature biofilms [
10]. Although cell surface-assisted nucleation and crystal formation were established for weakly crystalline phases such as schwertmannite, they have yet to be confirmed for biogenic jarosites.
Sodium jarosite, which is also known as natro jarosite (stoichiometric formula NaFe
3(SO
4)
2(OH)
6), is a rare mineral that has the same structure as alunite (KAl
3(SO
4)
2(OH)6). The formation of sodium–jarosite is also likely to be crucial in leaching processes that use high-salinity fluids with high sodium contents. Experiments on the production of sodium–jarosite at 70 °C revealed that at pH 1.72, sodium–jarosite forms a mixture with goethite [
11]. Only solutions with a pH of 1.42 yield a pure crystalline natro-jarosite phase. Sodium jarosite formation occurs according to the following typical reaction:
However, the formation of sodium–jarosite competes with the formation of goethite, which occurs as a result of the following reaction:
Goethite or sodium–jarosite can predominately occur depending on the solution composition and temperature. The ionic equilibrium of bisulphate and sulphate complexes (FeSO4+, FeHSO42+ FeH(SO4)2(aq), and species such as H+ and HSO4−) determines the solubility of these minerals.
2. Experiments
To study the possible approach of removing Fe in the form of jarosite from the bioleaching solution, a series of tests were conducted to study the effect of various chemicals and their concentration on the removal. These tests were conducted using a bioleaching solution from a bioreactor. The bioreactor was filled with 300 mL of stored bioleaching solution containing bacterial culture along with 1 L 9 K medium that was already prepared to make the total volume to 1.3 L in the bioleaching reactor. Overhead stirrer speed was set up at 680 rpm, and gas flow was set up at 0.5 L/min, which was changed to 0.2 L/min later. The precipitation test procedures included: (1) Sedimentation and decantation of 1 L of bioleaching solution; (2) Evaluation of the effect of oxidation and iron addition. To accomplish thisa solution sample (~300 mL) was taken, oxidized to about 670 mV, then 10 g/L of ferrous sulfate was added, and additional oxidation was performed to raise the Eh to 670 mV. Because no precipitate formed, 10 g/L of additional ferrous sulfate was introduced and the solution was oxidized to achieve an Eh of 670 mV. The process was repeated until some jarosite formation was observed. (3) Evaluation of sodium and ammonium salt additions: After observation of precipitate formation from part 2, the solution was decanted and 40 mL of solution was placed in a series of test tubes to which different amounts of sodium sulfate (2 g/40 mL, 4 g/40 mL, 6 g/40 mL) and ammonium hydroxide (1 mL/40 mL, 2 mL/40 mL, 4 mL/40 mL, 6 mL/40 mL) were added.
In order to examine jarosite formation during continuous bio-oxidation, 200 mL of the filtered bioleaching solution was also collected from the bioreactor. Based on the balance of feeding and effluent, the Fe concentration in the bioleaching solution should be around 0.5 M. This Fe concentration was adjusted to around 1.0 M by adding 27.8 g FeSO4·7H2O and 200 mL bioleaching solution to an Erlenmeyer flask. In order to provide favorable conditions, the solution was continuously stirred with a magnetic stirrer and uninterrupted airflow was maintained into the solution. The Eh change of the solution and the precipitation formation was tracked during the test.
The microstructures of the solid samples were studied using a field emission gun and a NovaNano scanning electron microscopy (SEM) (FEI, Hillsboro, USA). The voltage for our series of measurements was 30 kV. The elemental spectra and maps of the test samples were gathered using an energy dispersive X-beam spectroscopy (EDS) detector (EDAX, NJ, USA) to visualize the elemental distribution consistency and precipitation, and the EDAX Genesis programming could then be used to process the EDS findings.
A Rigaku-Miniflex benchtop X-ray diffractometer (Rigaku, Wilmington, MA, USA) using Cu K radiation was used to examine the phases and crystal structure of our precipitate set.
3. Results and Discussion
The alkaline ions required for precipitation are provided by ammonium sulfate and sodium sulfate, which are typical additions. Sodium sulfate was utilized in zinc extraction because it is readily available and inexpensive [
12]. Jarosite precipitation was investigated in the presence of various concentrations of the precipitating chemical Na
2SO
4. The rate of jarosite precipitation is clearly influenced by the Na
2SO
4 concentration. The amount of precipitated jarosite rose as the concentration of Na
2SO
4 in the solution increased, and this tendency continued with rising Na
2SO
4 concentrations to roughly 2 g/L [
12]. In the present scenario, which is different from a Zn
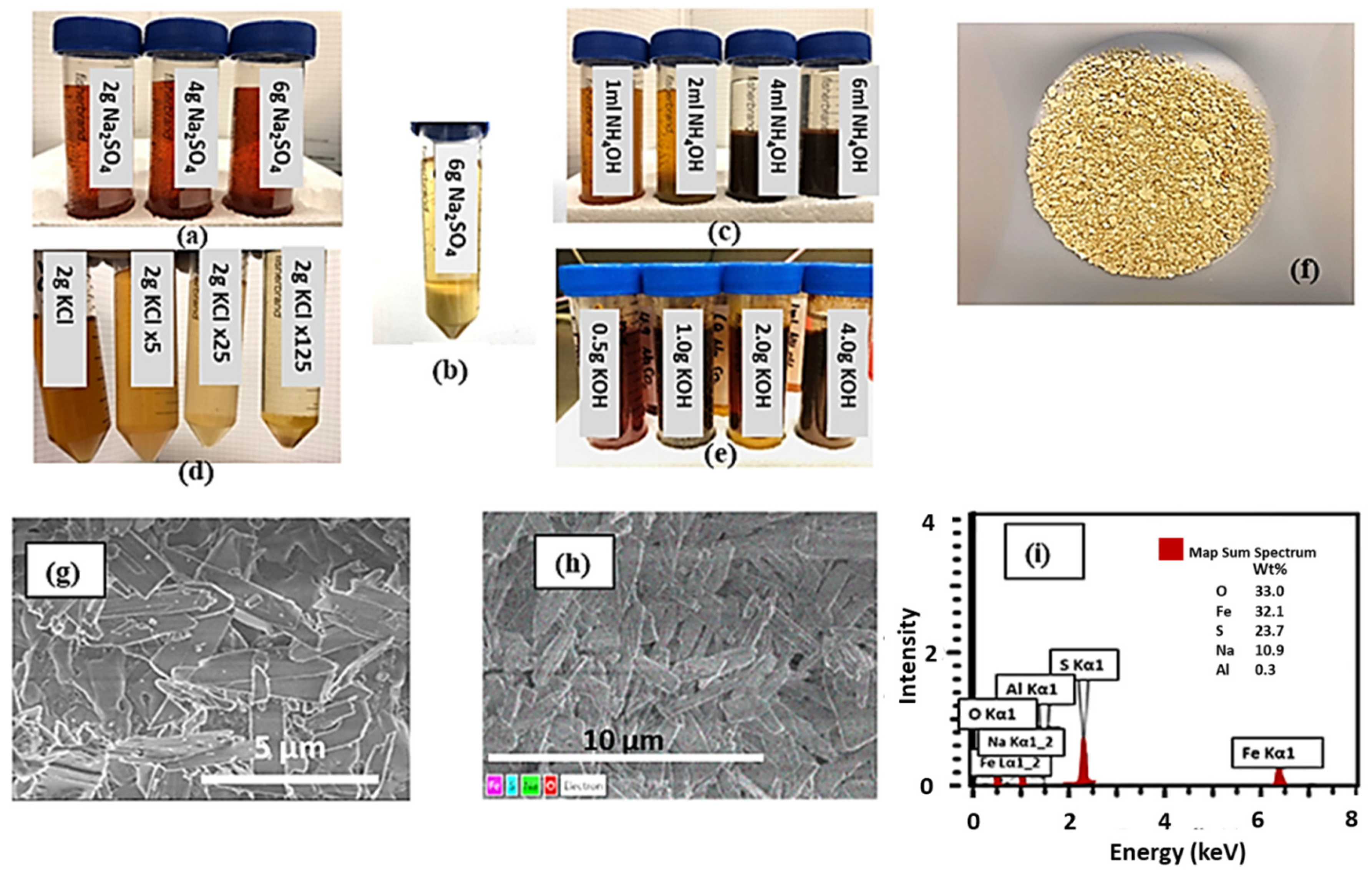
2+ based environment, no precipitate was formed after 24 h even when 6 g of sodium sulfate were added (see
Figure 1a). However, when these samples were left with 6 g of Na
2SO
4 addition after 40 days, a large amount of precipitate with an amber yellow color formed, and clearly (see
Figure 1b) the color of the solution became lighter compared with the sample after 24 h, indicating a huge Fe concentration reduction in the sample. Therefore, the addition of sodium sulfate can be an effective way to remove the iron in the bioleaching solution. After 28 days of aging in solutions with varying starting concentrations, a combination of XRD, SEM, and EDS data revealed that schwertmannite and natrojarosite-like compounds precipitated at strongly acidic and ambient conditions [
13]. The present case showed a similar outcome. However, the reaction rate for this approach is not fully understood and additional tests need to be carried out to determine the best condition for Fe removal from the solution. In general, the addition of Na, which has a large ionic radius (r = 102 pm), may eventually distort and destabilize the metastable phase’s structure, resulting in a reduction in particle size [
13]. As size decreases, the surface-to-volume ratio of the smallest particles of schwertmannite will be higher than that of bigger natrojarosite particles, resulting in an increase in total surface free energy and, as a result, an increase in solubility. During the dissolution of schwertmannite, Ostwald ripening mechanisms cause the larger natrojarosite particles to expand at the expense of the smaller ones [
13].
3.1. Effect of Different Cations
Pictures of the test solutions and the Eh/pH values after sedimentation are shown in
Figure 1 and
Table 1, respectively. It shows that the samples with 2 mL or more of ammonium hydroxide addition show the precipitate with possible jarosite formation (
Figure 1c). Since the 9 K medium used as a nutrient in bioleaching contains a high concentration of ammonium ions, jarosite formation is often favorable. Similar tests were conducted with potassium chloride (1 g/40 mL, 2 g/40 mL, 4 g/40 mL, 6 g/40 mL) and no precipitates were observed for this set of tests. Pictures of the test solutions are shown in
Figure 1d (left 4 tubes). Another set of tests were conducted with potassium hydroxide (0.5 g/40 mL, 1 g/40 mL, 2 g/40 mL, 4 g/40 mL). Precipitates formed in all the tests and the pictures of the test solutions as well as the pH are shown in
Figure 1e (right 4 tubes). All four tests show precipitates but the tests with 1 g/40 mL and 2 g/40 mL addition show possible jarosite formation. The pH and Eh values after adding these reagents are shown in
Table 1. Considering the formation and the color of precipitate as well as the pH range of 2–3 that favors jarosite formation, a preliminary judgment can be made on the effect of these reagents on jarosite formation. Precipitates from these jarosite formation tests were filtered and dried and examined with XRD to determine the composition.
By adding sodium salt to the bioleaching solution, Na–jarosite might form in the bioleaching solution. The precipitate formed in the test in which 6 g Na
2SO
4 was added was examined using SEM and EDS.
Figure 1f, g shows the picture of the precipitate and the SEM image of the precipitate. Compared with the jarosite precipitate formed in the bio-oxidation test, the precipitate form in this test is different. It looks like it consists of small flakes rather than small particles. This can be confirmed with the SEM image. The EDS analysis is shown in
Figure 1i. As it shows, the main elements in the precipitate are Fe, O, S, and Na, indicating that the precipitate is Na–jarosite.
Iron is mainly found as an elemental material in low-potential environments. At pH 0 to 6, the iron was gradually oxidized into Fe (II) ions as the potential increased. Alkaline circumstances cause the precipitation of ferrous iron. With the increasing potential value of the solution, the Fe (II) ion was gradually oxidized to Fe (III). Under high acid conditions with pH values ranging from 0 to 1.9, Fe (III) remained stable. According to the thermodynamics analysis, a Fe (III) precipitate was usually generated at pH levels > 1.9. The precipitation of ferric iron hydroxide occurred as the pH of the solution increased.
In order to form ammonium jarosite, ammonium salt or ammonia solution can be utilized as the ammonium ion source. Both of these can control the pH of the solution as well. In the case of the ferric ion-containing solution when the ammonia-based solution is introduced in it, there is a formation of ferric hydroxide as local pH turns higher [
14]. Such formation of ferric hydroxide is not problematic because ferric hydroxide is converted to jarosite under the condition of sufficiently long retention time and the presence of other requisite ions. Calculated distribution of Fe (III) species in Fe (III)–S–H
2O system suggests that amount of Fe (III)hydroxide in solution increases with temperature increment. This is favorable in the formation of solid jarosite. It was observed that at any given temperature (~25–95 °C) when pH increases the concentrations of all other species except ferric hydroxide decrease [
14]. When the temperature increases greater than 80 °C, the equilibrium FeSO
4+ amount depletes significantly with a very small increment in pH. When solid ferric hydroxide is present in the solution, the aqueous ferric hydroxide concentration is a function of temperature. A significant amount of ferric hydroxide is in form of a solid phase, which increases with pH increment.
In the formation of jarosite FeSO
4+ is one of the important species. It is believed that jarosite formation occurs via basic ferric sulfate precursor [Fe(OH)
2]
2SO
4 and Fe(OH)SO
4. These precursors’ formation occurs via the reaction of water with FeSO
4+. Another way to form these basic precursors is by colloidal or precipitated ferric hydroxide as can be seen:
These basic ferric sulfates turn polynuclear in the presence of ammonium ions and sulfate. The final reaction is as follows, where formation starts with ferric hydroxide:
Despite the fact that K
+ and NH
4+ have moreover identical hydrated ionic radii, K
+ considerably aided the creation of a jarosite phase [
5] as compared to NH
4+.
3.2. Jarosite Formation Modeling with Visual Minteq
To have a better understanding of the effects of parameters on the jarosite formation, equilibrium modeling was conducted to specifically study the effects of pH and K, and pH and Na on jarosite precipitation with 20 and 40 g/L dissolved Fe
3+. Similar to the experimental tests, the model studied the effects of pH and KOH addition, as well as the pH and NaOH addition. Plots were made to show the Fe
3+ concentration and jarosite saturation index change with the composition change of the solution at equilibrium. The saturation index is defined as the difference between log IAP (Ion activity product) and log Ks (solubility constant of the mineral at the given temperature). When undersaturation prevails, the saturation indexes are negative and given in blue. The modeling results are shown in
Figure 2. Here it can be observed that dissolved iron concentration starts decreasing rapidly above a pH of 2.4. A similar trend is observed for high and low ferric ion concentrations. The equilibrium of the following related processes leads to a rise in ferric iron content with decreasing pH values:
In previously reported cases when pH was greater than 1.72, a mixture of Na–jarosite and geothite were formed. Conditions for the precipitation of pure Na–jarosite, on the other hand, were only attained in tests where the final equilibrium pH was equal to or less than 1.42.
One of the most crucial factors in the creation of jarosite-type compounds is pH. The leftover acidity from the hot acid leach stage, as well as the acid produced during the jarosite precipitation reaction, must be controlled. In the case of Na, iron depletion starts at a relatively lower pH. Enhanced addition of sodium or potassium salt causes achievement of a high jarosite saturation index even at relatively low pH. This trend is true, both for Na or K jarosite. For the comparison point of view, the K jarosite saturation index is extended to higher values for the range of pH for which simulations were performed. For K jarosite formation [
5] (from ferric and sulfate ions), the log K
sp value is ~12.5, whereas for Na jarosite formation the log K
sp value is ~8.5. Hence, higher values for both the H and K jarosite saturation index make sense. They indicate solution can be unsaturated for extended ionic concentration, and more of the ionic solid, if available, will dissolve.
3.3. Jarosite Formation Assessment with Bio-Oxidation
Pictures of the bioleaching solution and the Eh value (mV) are shown in
Figure 3. As can be seen from the pictures, in the first 11 days, the color and Eh of the solution did not change very much, and the solution was generally clear. After that, the solution became turbid with time, indicating the generation of precipitates in the solution. Besides, the Eh of the solution increased with time faster and reached a very high value of 665 mV after 21 days, indicating that most of the ferrous ions were oxidized to ferric ions, which might facilitate the generation of precipitate as can be seen. The solution was filtered to collect both the precipitate and the solution. The weight of the precipitate after drying is 3.66 g. The picture of the dried precipitate is shown in
Figure 4a. As can be seen, the color of the precipitate is amber yellow, which is the typical color of the jarosite. This is confirmed with SEM, EDS and XRD analysis, as shown in
Figure 4b,c. From the EDS analysis, it can be seen that the main elements in the precipitates are O, Fe, and S. This is consistent with the chemical composition of the H jarosite. XRD analysis further confirmed the composition of the precipitate to be mainly jarosite because all the peaks of the sample match well with the peaks of the jarosite.
Knowing that
A. ferrooxidans is an aerobic bacterium [
15,
16,
17], it was assumed that a constant supply of O
2 (e.g., DO > 5.0 mg/L) is required for jarosite production, resulting in significant costs [
18]. Interestingly, recent research discovered that when a FeSO
4 solution containing
A. ferrooxidans LX5 was statically incubated for 40 h, a large number of yellowish hydroxysulfate precipitates were produced, showing that oxygen may not be required during the jarosite biosynthesis experiment [
18]. This assumption is not validated in the present case. Earlier, the correlations between dissolved oxygen concentrations, Fe
3+, Fe
2+, and Fe
2+ oxidation rate and the generation of Fe-precipitates were studied using the Pearson linear regression model. The analysis suggested that the generation of Fe-precipitates was significantly connected with the concentrations of generated Fe
3+ (R
2 = 0.952 and Root-MSE = 16.72) and was poorly correlated with DO concentration [R
2 = 0.158 and Root-MSE = 41.42]. Further experiments suggested that the supply of oxygen played a significant role in the oxidation of Fe
2+ to Fe
3+ by
A. ferrooxidans; however, its role in the forming of Fe-precipitates could be overlooked, presumably because the energy released during the oxidation of Fe
2+ can be used for the succeeding hydrolysis reaction of Fe
3+ into jarosite [
18]. Considering these facts, more focus was given to evaluating the effect of other parameters with a constant supply of oxygen.
Since there is not too much K, Na, or NH
4+ in the solution, it is believed that most of the jarosite is hydronium jarosite ((H
3O) Fe
3(SO
4)
2(OH)
6). Based on the mass balance, the amount of the Fe in the precipitate is calculated as:
In the initial solution, the amount of Fe is about 0.2 mol; therefore, about 11.5% of the iron was precipitated in the form of jarosite during the bio-oxidation process. As stated before, the solution was filtered, and the precipitate was collected. While the liquid was put back to the flask to continue the test. A total of 5 g of FeSO
4·7H
2O and 100 mL 9 K medium was added to the solution. After 20 days, the precipitate formed again in the solution. After filtration and drying, the weight of the precipitate is 2.02 g. The amount of Fe precipitated out in this test is:
If ferrous ions are the predominant form of iron, FeSO
4(aq) or Fe
2+ will be soluble below a pH of 9. Therefore, 6.5% of the Fe was precipitated in the form of jarosite during the second stage of continued bio-oxidation. Note that, if ferrous iron is not entirely oxidized to Fe
3+, it will precipitate with rare earth elements in the case of leaching of REE-based minerals [
19]. The substantial differences in speciation and precipitation of iron and rare earth elements allow for facile removal of iron from solution by precipitation without considerable losses of rare earth elements [
19] if the dissolved iron is oxidized to Fe
3+. Though, one species was mostly investigated. As a result of precipitation, it is feasible that one jarosite species can efficiently nucleate the precipitation of another [
19]. This is a key component of interest, but it is also becoming increasingly feasible as companies reprocess jarosite leftovers, change the jarosite species generated for environmental reasons, or vary the jarosite species precipitated. The temperature was shown to have a substantial impact on jarosite precipitation amount and pace [
12]. The effect of temperature as a critical parameter on jarosite precipitation reveals that the rate of jarosite production and iron precipitation enhanced as the temperature increased [
12]. The optimum reaction temperature for jarosite precipitation, according to the literature, is 90–100 °C. However, in our case, most of the experiments were performed at a relatively lower temperature.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}