
Influence of Copper(I) Halides on the Reactivity of Aliphatic Carbodiimides †


Abstract
:1. Introduction
2. Methods
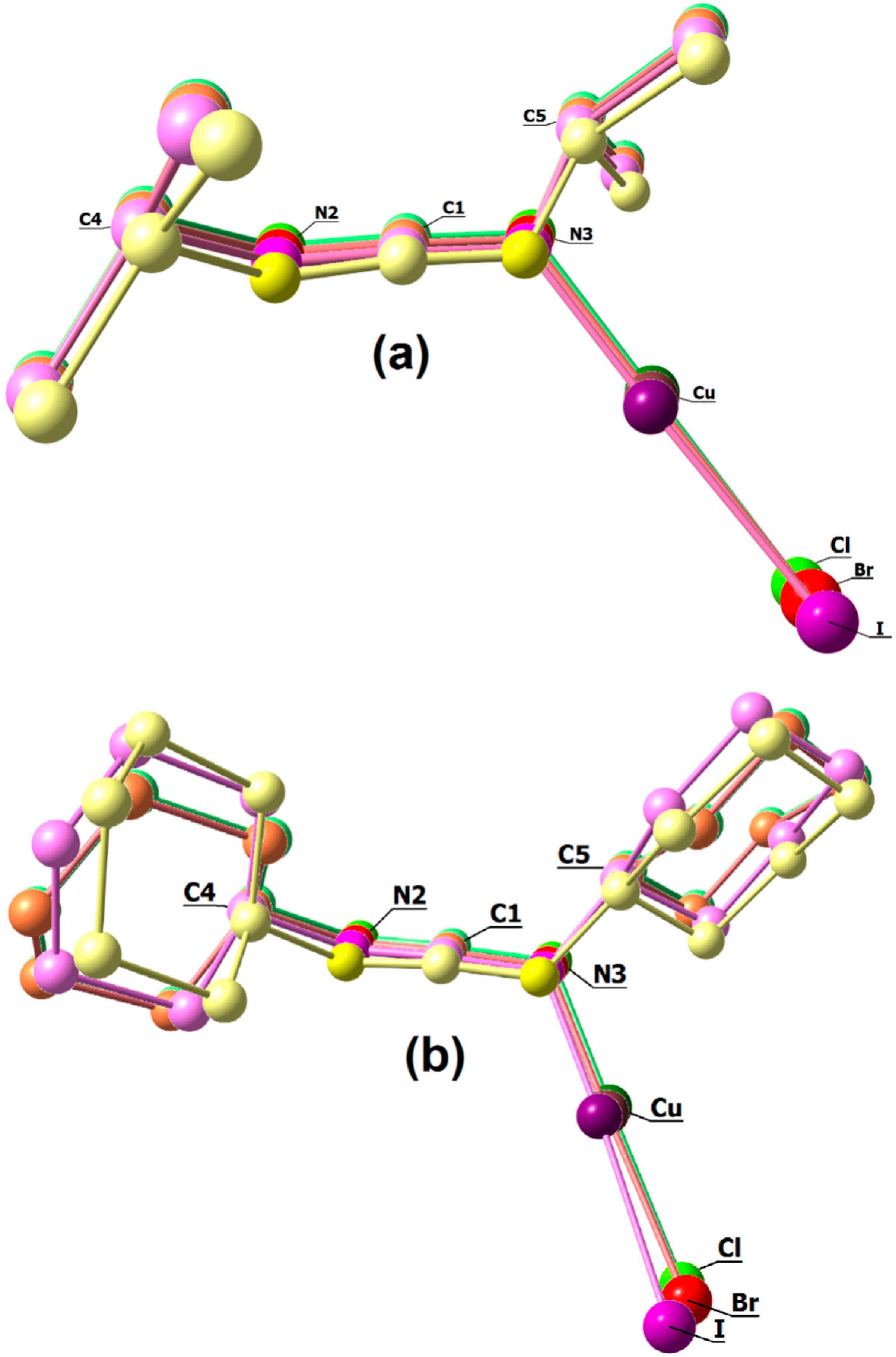
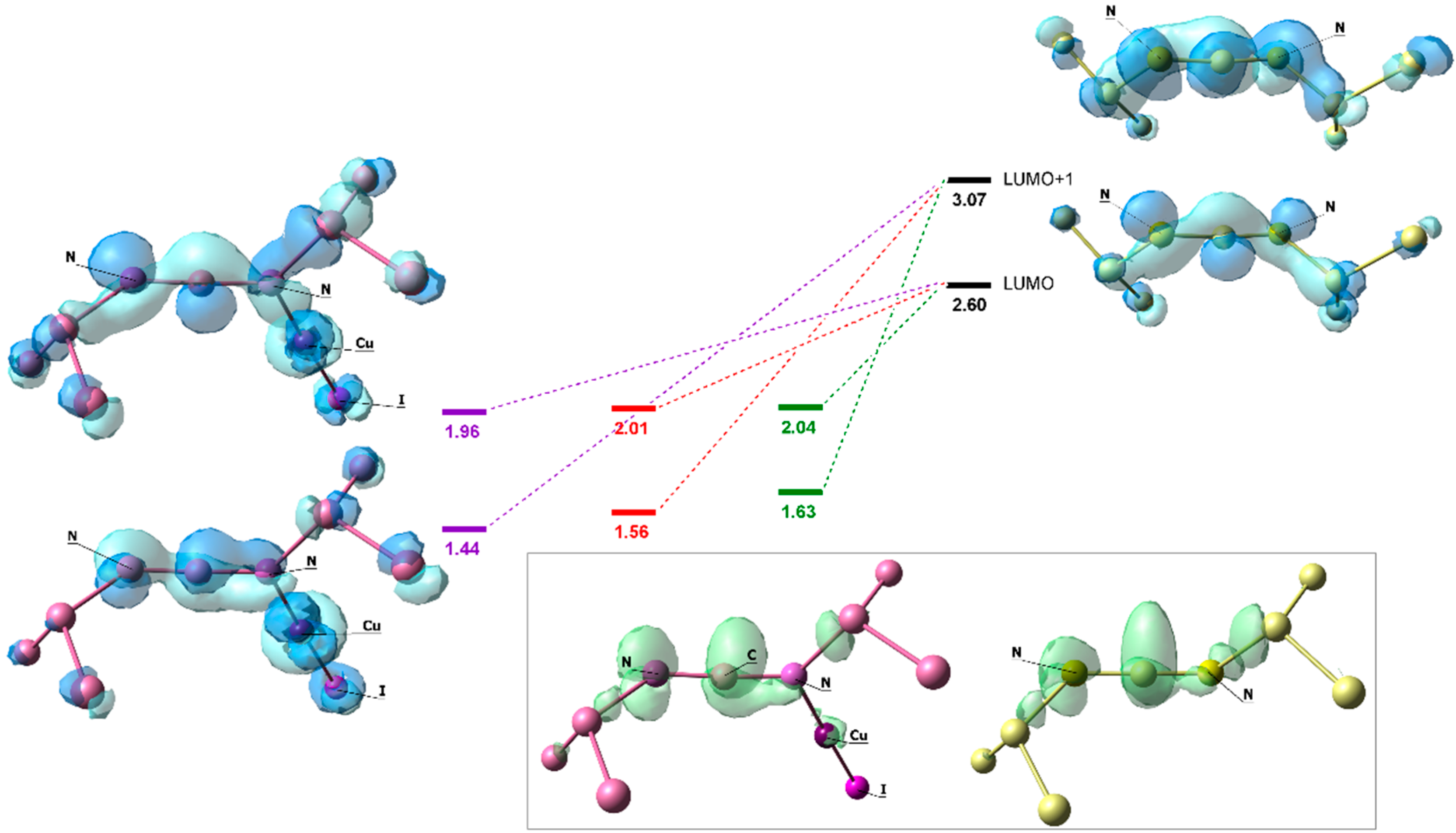
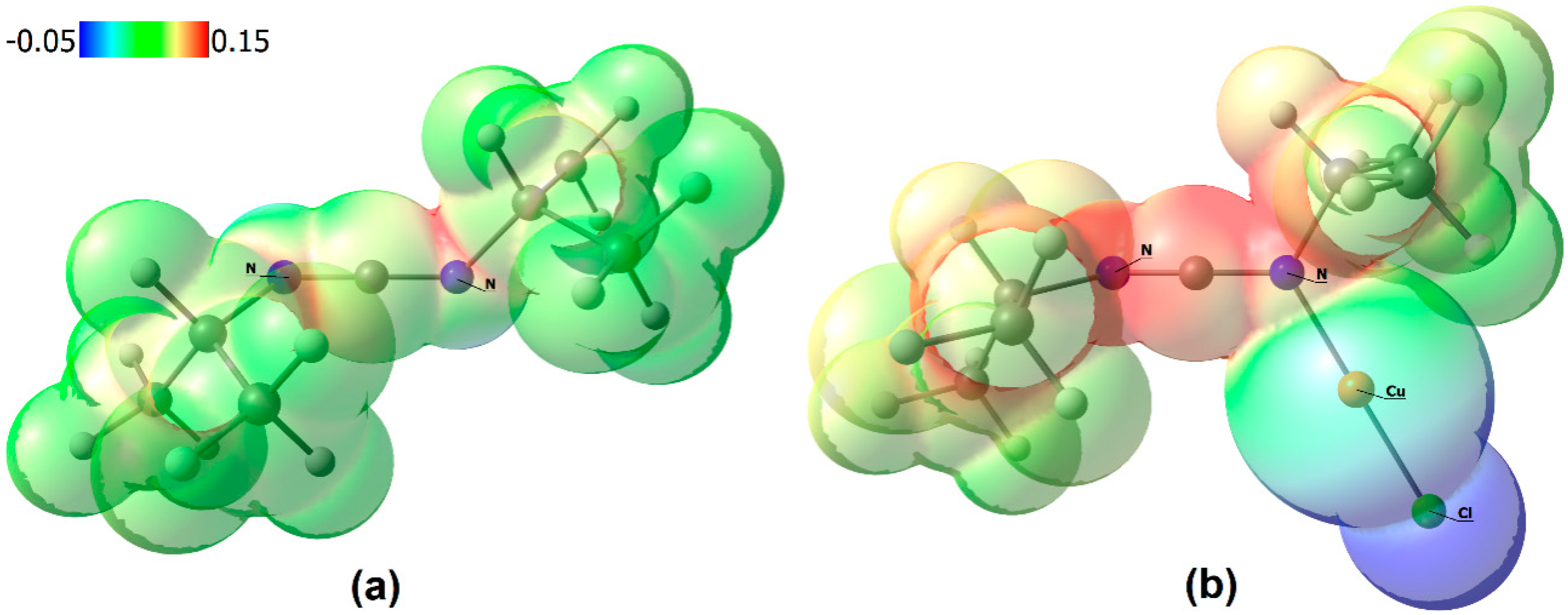
3. Results and Discussion
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
References
- Khorana, H.G. The Chemistry of Carbodiimides. Chem. Rev. 1953, 53, 145–166. [Google Scholar] [CrossRef]
- Kurzer, F.; Douraghi-Zadeh, K. Advances in the Chemistry of Carbodiimides. Chem. Rev. 1967, 67, 107–152. [Google Scholar] [CrossRef]
- Williams, A.; Ibrahim, I.T. Carbodiimide chemistry: Recent advances. Chem. Rev. 1981, 81, 589–636. [Google Scholar] [CrossRef]
- Ulrich, A. Chemistry and Technology of Carbodiimides; Wiley: Chichester, UK, 2007. [Google Scholar]
- Schmidt, E.; Moosmüller, F. Zur Kenntnis aliphatischer Carbodiimide. Liebigs Ann. Chem. 1956, 597, 235–240. [Google Scholar] [CrossRef]
- Alexandre, C.; Rouessac, F. La dicyclohexylcarbodiimide, agent de deshydratation intramoleculaire des cetols. Tetrahedron Lett. 1970, 11, 1011–1012. [Google Scholar] [CrossRef]
- Schuster, E.; Hesse, C.; Schumann, D. Dehydration of 2-Hydroxyimines with Dicyclohexylcarbodiimide: An Access to Cyclic Alkenylimines. Synlett 1991, 916–918. [Google Scholar] [CrossRef]
- Knochel, P.; Seebach, D. Dehydratisierung von Nitroaldolen mit Dicyclohexylcarbodiimid: Herstellung von Nitroolefinen unter milden Bedingungen. Synthesis 1982, 1017–1018. [Google Scholar] [CrossRef]
- Mathias, L.J. Esterification and Alkylation Reactions Employing Isoureas. Synthesis 1979, 561–576. [Google Scholar] [CrossRef]
- Gibson, F.S.; Sook Park, M.; Rapoport, H. Bis[[4-(2,2-dimethyl-1,3-dioxolyl)]methyl]- carbodiimide (BDDC) and Its Application to Residue-Free Esterifications, Peptide Couplings, and Dehydrations. J. Org. Chem. 1994, 59, 7503–7507. [Google Scholar] [CrossRef]
- Sinha, S.; Ilankumaran, P.; Chandrasekaran, S. One pot conversion of alcohols to disulfides mediated by benzyltriethylammonium tetrathiomolybdate. Tetrahedron 1999, 55, 14769–14776. [Google Scholar] [CrossRef]
- Schur, C.; Becker, N.; Bergsträßer, U.; Hartung, J.; Gottwald, T. Tertiary alkoxyl radicals from 3-alkoxythiazole-2(3H)-thiones. Tetrahedron 2011, 67, 2338–2347. [Google Scholar] [CrossRef]
- Kiełbasiński, P.; Żurawiński, R.; Orabowicz, J.; Mikołajczyk, M. Organosulphur compounds: XLVII Alkylation of sulphinic acids by o-alkylisoureas: O-versus s-reactivity and asymmetric synthesis of alkyl sulphinates. Tetrahedron 1988, 44, 6687–6692. [Google Scholar] [CrossRef]
- Bakibaev, A.A.; Shtrykova, V.V. Isoureas: Synthesis, properties, and applications. Russ. Chem. Rev. 1995, 64, 929–938. [Google Scholar] [CrossRef]
- Bortoluzzi, M.; Paolucci, G.; Sartor, F.; Bertolasi, V. Synthesis and characterization of novel pyridine–isourea complexes of Pd(II). Polyhedron 2012, 37, 66–67. [Google Scholar] [CrossRef]
- Hartke, K.; Rossbach, F. Dimerisierung von Carbodiimiden. Angew. Chem. 1968, 80, 83. [Google Scholar] [CrossRef]
- Minenkov, Y.; Singstad, Å.; Occhipinti, G.; Jensen, V.R. The accuracy of DFT-optimized geometries of functional transition metal compounds: A validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012, 41, 5526–5541. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Gerber, I.C.; Ángyán, J.G. Hybrid functional with separated range. Chem. Phys. Lett. 2005, 415, 100–105. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef]
- Li, Y.; Evans, J.N.S. The Fukui Function: A Key Concept Linking Frontier Molecular Orbital Theory and the Hard-Soft-Acid-Base Principle. J. Am. Chem. Soc. 1995, 117, 7756–7759. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Cramer, C.J. Essentials of Computational Chemistry, 2nd ed.; Wiley: Chichester, UK, 2004. [Google Scholar]
- Hirshfeld, R.F. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Mayer, I. Charge, bond order and valence in the AB initio SCF theory. Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar] [CrossRef]
- Sizova, O.V.; Skripnikov, L.V.; Sokolov, A.Y. Symmetry decomposition of quantum chemical bond orders. J. Mol. Struct. 2008, 870, 1–9. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Dapprich, S.; Frenking, G. Investigation of Donor-Acceptor Interactions: A Charge Decomposition Analysis Using Fragment Molecular Orbitals. J. Phys. Chem. 1995, 99, 9352–9362. [Google Scholar] [CrossRef]
- Gorelsky, S.I.; Ghosh, S.; Solomon, E.I. Mechanism of N2O Reduction by the μ4-S Tetranuclear CuZ Cluster of Nitrous Oxide Reductase. J. Am. Chem. Soc. 2006, 128, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Bond | DICDI | [CuCl(DICDI)] | [CuBr(DICDI)] | [CuI(DICDI)] |
|---|---|---|---|---|
| C1–N2 | 1.217 | 1.193 | 1.193 | 1.193 |
| C1–N3 | 1.215 | 1.243 | 1.243 | 1.243 |
| N2–C4 | 1.478 | 1.470 | 1.470 | 1.470 |
| N3–C5 | 1.471 | 1.494 | 1.494 | 1.494 |
| Bond | DCC | [CuCl(DCC)] | [CuBr(DCC)] | [CuI(DCC)] |
| C1–N2 | 1.216 | 1.192 | 1.192 | 1.191 |
| C1–N3 | 1.217 | 1.243 | 1.244 | 1.244 |
| N2–C4 | 1.469 | 1.465 | 1.464 | 1.464 |
| N3–C5 | 1.470 | 1.489 | 1.489 | 1.488 |
| Bond | DICDI | [CuCl(DICDI)] | [CuBr(DICDI)] | [CuI(DICDI)] |
|---|---|---|---|---|
| C1–N2 | 2.107 (2.393) | 2.217 (2.490) | 2.217 (2.490) | 2.216 (2.491) |
| C1–N3 | 2.112 (2.377) | 1.692 (2.085) | 1.693 (2.086) | 1.683 (2.088) |
| N2–C4 | 0.864 (1.196) | 0.842 (1.164) | 0.842 (1.164) | 0.842 (1.165) |
| N3–C5 | 0.804 (1.179) | 0.711 (1.096) | 0.710 (1.097) | 0.714 (1.098) |
| Bond | DCC | [CuCl(DCC)] | [CuBr(DCC)] | [CuI(DCC)] |
| C1–N2 | 2.072 (2.386) | 2.196 (2.487) | 2.198 (2.489) | 2.199 (2.493) |
| C1–N3 | 2.083 (2.379) | 1.700 (2.078) | 1.700 (2.078) | 1.682 (2.077) |
| N2–C4 | 0.859 (1.200) | 0.867 (1.171) | 0.868 (1.171) | 0.842 (1.172) |
| N3–C5 | 0.837 (1.181) | 0.629 (1.102) | 0.627 (1.102) | 0.664 (1.104) |
| Bond | DICDI | [CuCl(DICDI)] | [CuBr(DICDI)] | [CuI(DICDI)] |
|---|---|---|---|---|
| C1–N2 | 0.436 (−1.340) | 0.450 (−1.506) | 0.450 (−1.505) | 0.450 (−1.505) |
| C1–N3 | 0.435 (−1.325) | 0.418 (−1.159) | 0.419 (−1.161) | 0.419 (−1.161) |
| N2–C4 | 0.248 (−0.410) | 0.241 (−0.443) | 0.241 (−0.442) | 0.241 (−0.443) |
| N3–C5 | 0.248 (−0.404) | 0.236 (−0.374) | 0.236 (−0.374) | 0.236 (−0.375) |
| Bond | DCC | [CuCl(DCC)] | [CuBr(DCC)] | [CuI(DCC)] |
| C1–N2 | 0.435 (−1.330) | 0.449 (−1.507) | 0.450 (−1.509) | 0.451 (−1.513) |
| C1–N3 | 0.435 (−1.327) | 0.418 (−1.153) | 0.418 (−1.152) | 0.419 (−1.142) |
| N2–C4 | 0.249 (−0.414) | 0.243 (−0.459) | 0.243 (−0.461) | 0.243 (−0.463) |
| N3–C5 | 0.248 (−0.412) | 0.239 (−0.385) | 0.239 (−0.385) | 0.239 (−0.385) |
| Carbodiimide | CuCl | CuBr | CuI |
|---|---|---|---|
| DICDI | 0.237 (0.374) | 0.248 (0.391) | 0.255 (0.415) |
| DCC | 0.240 (0.379) | 0.251 (0.398) | 0.254 (0.415) |
| Atom | DICDI | [CuCl(DICDI)] | [CuBr(DICDI)] | [CuI(DICDI)] |
|---|---|---|---|---|
| C1 | 0.152 (0.219) | 0.199 (0.345) | 0.199 (0.345) | 0.199 (0.355) |
| N2 | −0.184 (−0.346) | −0.110 (−0.211) | −0.110 (−0.211) | −0.110 (−0.212) |
| N3 | −0.184 (−0.339) | −0.142 (−0.303) | −0.141 (−0.289) | −0.142 (−0.275) |
| C4 | 0.040 (0.103) | 0.054 (0.071) | 0.054 (0.071) | 0.054 (0.071) |
| C5 | 0.039 (0.105) | 0.048 (0.073) | 0.048 (0.074) | 0.048 (0.074) |
| Atom | DCC | [CuCl(DCC)] | [CuBr(DCC)] | [CuI(DCC)] |
| C1 | 0.154 (0.210) | 0.199 (0.327) | 0.200 (0.326) | 0.199 (0.325) |
| N2 | −0.189 (−0.354) | −0.108 (−0.206) | −0.107 (−0.205) | −0.107 (−0.205) |
| N3 | −0.182 (−0.329) | −0.142 (−0.325) | −0.142 (−0.311) | −0.144 (−0.287) |
| C4 | 0.032 (0.118) | 0.046 (0.041) | 0.046 (0.041) | 0.046 (0.069) |
| C5 | 0.034 (0.074) | 0.041 (0.110) | 0.040 (0.112) | 0.040 (0.098) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferraro, V.; Bortoluzzi, M. Influence of Copper(I) Halides on the Reactivity of Aliphatic Carbodiimides. Chem. Proc. 2021, 3, 20. https://doi.org/10.3390/ecsoc-24-08096
Ferraro V, Bortoluzzi M. Influence of Copper(I) Halides on the Reactivity of Aliphatic Carbodiimides. Chemistry Proceedings. 2021; 3(1):20. https://doi.org/10.3390/ecsoc-24-08096
Chicago/Turabian StyleFerraro, Valentina, and Marco Bortoluzzi. 2021. "Influence of Copper(I) Halides on the Reactivity of Aliphatic Carbodiimides" Chemistry Proceedings 3, no. 1: 20. https://doi.org/10.3390/ecsoc-24-08096
APA StyleFerraro, V., & Bortoluzzi, M. (2021). Influence of Copper(I) Halides on the Reactivity of Aliphatic Carbodiimides. Chemistry Proceedings, 3(1), 20. https://doi.org/10.3390/ecsoc-24-08096