

The Effects of SARS-CoV-2 on the Angiopoietin/Tie Axis and the Vascular Endothelium

{kind=link}
{kind=link}
Abstract
:1. Introduction to Coronaviruses
1.1. Virion Composition of Coronaviruses
1.2. SARS-CoV-2 Infection
2. The Endothelium
2.1. Angiopoietin Family
2.2. The Function of Angiopoietin 1
2.3. The Function of Angiopoietin 2
2.4. Tie Receptors
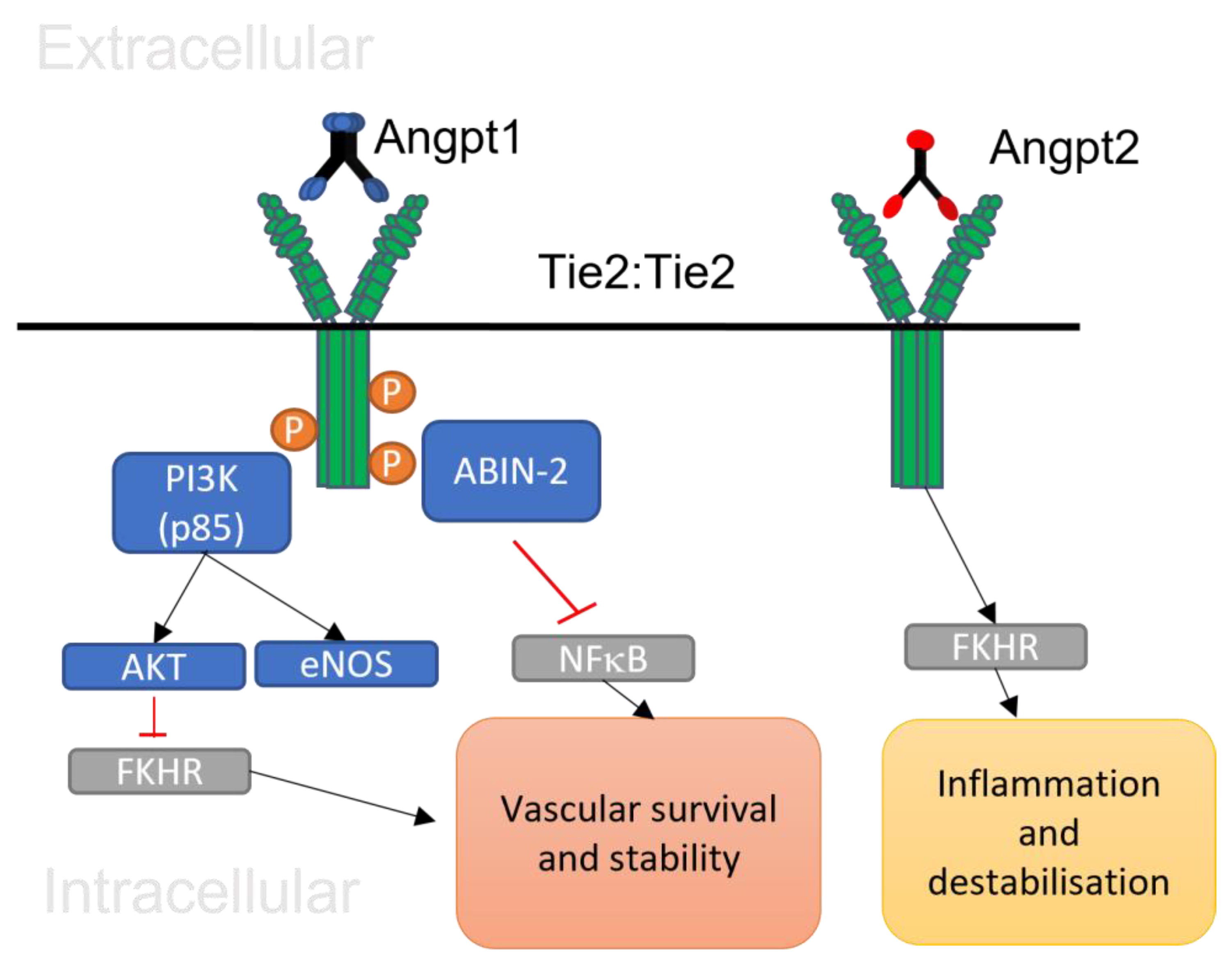
2.5. Angiopoietin 1/Tie 2 Signalling
3. The Impact of SARS-CoV-2 on Endothelial Cells
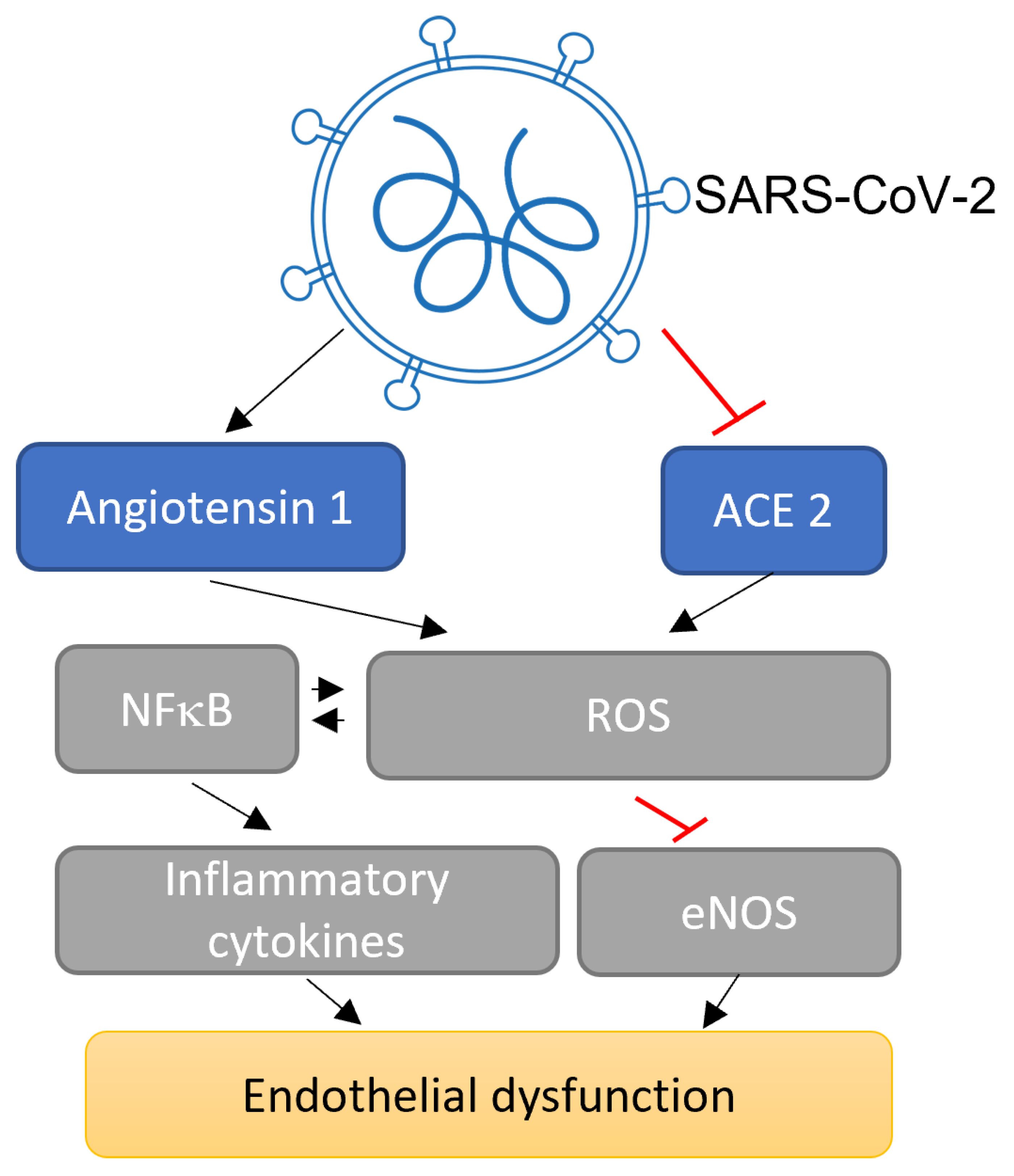
3.1. Potential Impact of SARS-CoV-2 on Angpt/Tie Signalling
3.2. Potential Clinical Implication of Angpt/Tie Signalling in COVID-19 Patients
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Kulcsar, K.; Misra, V.; Frieman, M.; Mossman, K. Bats and Coronaviruses. Viruses 2019, 11, 41. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2020, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Gaunt, E.R.; Hardie, A.; Claas, E.C.; Simmonds, P.; Templeton, K.E. Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real-time PCR method. J. Clin. Microbiol. 2010, 48, 2940–2947. [Google Scholar] [CrossRef]
- Naskalska, A.; Dabrowska, A.; Szczepanski, A.; Milewska, A.; Jasik, K.P.; Pyrc, K. Membrane Protein of Human Coronavirus NL63 Is Responsible for Interaction with the Adhesion Receptor. J. Virol. 2019, 93, e00355-19. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar]
- Lalchhandama, K. The chronicles of coronaviruses: The bronchitis, the hepatitis and the common cold. Sci. Vis. 2020, 20, 43–53. [Google Scholar] [CrossRef]
- Gierer, S.; Bertram, S.; Kaup, F.; Wrensch, F.; Heurich, A.; Kramer-Kuhl, A.; Welsch, K.; Winkler, M.; Meyer, B.; Drosten, C.; et al. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J. Virol. 2013, 87, 5502–5511. [Google Scholar] [CrossRef]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Wang, W.; Ye, L.; Ye, L.; Li, B.; Gao, B.; Zeng, Y.; Kong, L.; Fang, X.; Zheng, H.; Wu, Z.; et al. Up-regulation of IL-6 and TNF-α induced by SARS-coronavirus spike protein in murine macrophages via NF-κB pathway. Virus Res. 2007, 128, 1–8. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Riva, L.; Pu, Y.; Martin-Sancho, L.; Kanamune, J.; Yamamoto, Y.; Sakai, K.; Gotoh, S.; Miorin, L.; Paul, D.; et al. MDA5 Governs the Innate Immune Response to SARS-CoV-2 in Lung Epithelial Cells. Cell Rep. 2021, 34, 108628. [Google Scholar] [CrossRef]
- Lokugamage, K.G.; Hage, A.; de Vries, M.; Valero-Jimenez, A.M.; Schindewolf, C.; Dittmann, M.; Rajsbaum, R.; Menachery, V.D. Type I Interferon Susceptibility Distinguishes SARS-CoV-2 from SARS-CoV. J. Virol. 2020, 94, e01410-20. [Google Scholar] [CrossRef]
- Reuschl, A.-K.; Thorne, L.G.; Whelan, M.V.X.; Ragazzini, R.; Furnon, W.; Cowton, V.M.; De Lorenzo, G.; Mesner, D.; Turner, J.L.E.; Dowgier, G.; et al. Evolution of enhanced innate immune suppression by SARS-CoV-2 Omicron subvariants. Nat. Microbiol. 2024, 9, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Shin, E. Type I and III interferon responses in SARS-CoV-2 infection. Exp. Mol. Med. 2021, 53, 750–760. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. ChemRxiv: Prepr. Serv. Chem. 2020, 21, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.A.; Colley, L.; Agbaedeng, T.A.; Ellison-Hughes, G.M.; Ross, M.D. Vascular Manifestations of COVID-19—Thromboembolism and Microvascular Dysfunction. Front. Cardiovasc. Med. 2020, 7, 598400. [Google Scholar] [CrossRef]
- Pugsley, M.K.; Tabrizchi, R. The vascular system. An overview of structure and function. J. Pharmacol. Toxicol. Methods 2000, 44, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelium and haemostasis. Hamostaseologie 2015, 35, 11–16. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef]
- Gullo, A.L.; Aragona, C.O.; Scuruchi, M.; Versace, A.G.; Saitta, A.; Imbalzano, E.; Loddo, S.; Campo, G.M.; Mandraffino, G. Endothelial progenitor cells and rheumatic disease modifying therapy. Vasc. Pharmacol. 2018, 108, 8. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Pearson, J.D. Normal endothelial cell function. Lupus 2000, 9, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Buhimschi, C.S.; Bhandari, V.; Dulay, A.T.; Thung, S.; Razeq, S.S.A.; Rosenberg, V.; Han, C.S.; Ali, U.A.; Zambrano, E.; Zhao, G.; et al. Amniotic fluid angiopoietin-1, angiopoietin-2, and soluble receptor tunica interna endothelial cell kinase-2 levels and regulation in normal pregnancy and intraamniotic inflammation-induced preterm birth. J. Clin. Endocrinol. Metab. 2010, 95, 3428–3436. [Google Scholar] [CrossRef]
- Daly, C.; Eichten, A.; Castanaro, C.; Pasnikowski, E.; Adler, A.; Lalani, A.S.; Papadopoulos, N.; Kyle, A.H.; Minchinton, A.I.; Yancopoulos, G.D.; et al. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res. 2013, 73, 108–118. [Google Scholar] [CrossRef]
- Bilimoria, J.; Singh, H. The Angiopoietin ligands and Tie receptors: Potential diagnostic biomarkers of vascular disease. J. Recept. Signal Transduct. Res. 2019, 39, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Witzenbichler, B.; Maisonpierre, P.C.; Jones, P.; Yancopoulos, G.D.; Isner, J.M. Chemotactic properties of angiopoietin-1 and -2, ligands for the endothelial-specific receptor tyrosine kinase Tie2. J. Biol. Chem. 1998, 273, 18514–18521. [Google Scholar] [CrossRef] [PubMed]
- Suri, C.; Jones, P.F.; Patan, S.; Bartunkova, S.; Maisonpierre, P.C.; Davis, S.; Sato, T.N.; Yancopoulos, G.D. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996, 87, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.K.; Kim, I.; Lim, S.T.; Chung, M.J.; Kim, W.H.; Kim, H.G.; Ko, J.K.; Koh, G.Y. Coadministration of angiopoietin-1 and vascular endothelial growth factor enhances collateral vascularization. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2573–2578. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, P.; Eklund, L.; Miettinen, J.; Wirkkala, R.; Anisimov, A.; Winderlich, M.; Nottebaum, A.; Vestweber, D.; Deutsch, U.; Koh, G.Y.; et al. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat. Cell Biol. 2008, 10, 527–537. [Google Scholar] [CrossRef]
- Kim, M.; Allen, B.; Korhonen, E.; Nitschke, M.; Yang, H.; Baluk, P.; Saharinen, P.; Alitalo, K.; Daly, C.; Thurston, G.; et al. Opposing actions of angiopoietin-2 on Tie2 signalling and FOXO1 activation. J. Clin. Investig. 2016, 126, 3511–3525. [Google Scholar] [CrossRef]
- Cai, J.; Kehoe, O.; Smith, G.M.; Hykin, P.; Boulton, M.E. The angiopoietin/Tie-2 system regulates pericyte survival and recruitment in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2000, 49, 2163–2171. [Google Scholar] [CrossRef]
- Stratmann, A.; Risau, W.; Plate, K.H. Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am. J. Pathol. 1998, 153, 1459–1466. [Google Scholar] [CrossRef]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef]
- Lobov, I.B.; Brooks, P.C.; Lang, R.A. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11205–11210. [Google Scholar] [CrossRef]
- Dumont, D.J.; Gradwohl, G.; Fong, G.H.; Puri, M.C.; Gertsenstein, M.; Auerbach, A.; Breitman, M.L. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 1994, 8, 1897–1909. [Google Scholar] [CrossRef]
- Felcht, M.; Luck, R.; Schering, A.; Seidel, P.; Srivastava, K.; Hu, J.; Bartol, A.; Kienast, Y.; Vettel, C.; Loos, E.K.; et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J. Clin. Investig. 2012, 122, 1991–2005. [Google Scholar] [CrossRef]
- Fiedler, U.; Scharpfenecker, M.; Koidl, S.; Hegen, A.; Grunow, V.; Schmidt, J.M.; Kriz, W.; Thurston, G.; Augustin, H.G. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood 2004, 103, 4150–4156. [Google Scholar] [CrossRef] [PubMed]
- Scholz, A.; Plate, K.H.; Reiss, Y. Angiopoietin-2: A multifaceted cytokine that functions in both angiogenesis and inflammation. Ann. N. Y. Acad. Sci. 2015, 1347, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kontos, C.D.; Cha, E.H.; York, J.D.; Peters, K.G. The endothelial receptor tyrosine kinase Tie1 activates phosphatidylinositol 3-kinase and Akt to inhibit apoptosis. Mol. Cell. Biol. 2002, 22, 1704–1713. [Google Scholar] [CrossRef]
- Fiedler, U.; Krissl, T.; Koidl, S.; Weiss, C.; Koblizek, T.; Deutsch, U.; Martiny-Baron, G.; Marme, D.; Augustin, H.G. Angiopoietin-1 and angiopoietin-2 share the same binding domains in the Tie-2 receptor involving the first Ig-like loop and the epidermal growth factor-like repeats. J. Biol. Chem. 2003, 278, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Koblizek, T.I.; Weiss, C.; Yancopoulos, G.D.; Deutsch, U.; Risau, W. Angiopoietin-1 induces sprouting angiogenesis in vitro. Curr. Biol. CB 1998, 8, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Savant, S.; Porta, S.; Budnik, A.; Busch, K.; Hu, J.; Tisch, N.; Korn, C.; Valls, A.; Benest, A.; Terhardt, D.; et al. The orphan receptor Tie1 controls Angiogenesis and vascular remodeling by differentially regulating Tie2 in Tip and Stalk cells. Cell Rep. 2015, 12, 1761–1773. [Google Scholar] [CrossRef]
- Procopio, W.N.; Pelavin, P.I.; Lee, W.M.; Yeilding, N.M. Angiopoietin-1 and -2 coiled coil domains mediate distinct homo-oligomerization patterns, but fibrinogen-like domains mediate ligand activity. J. Biol. Chem. 1999, 274, 30196–30201. [Google Scholar] [CrossRef]
- Leppanen, V.; Saharinen, P.; Alitalo, K. Structural basis of Tie2 activation and Tie2/Tie1 heterodimerization. Proc. Natl. Acad. Sci. USA 2017, 114, 4376–4381. [Google Scholar] [CrossRef] [PubMed]
- Jeansson, M.; Gawlik, A.; Anderson, G.; Li, C.; Kerjaschki, D.; Henkelman, M.; Quaggin, S. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J. Clin. Investig. 2011, 121, 2278–2289. [Google Scholar] [CrossRef]
- Kwak, H.J.; So, J.; Lee, S.J.; Kim, I.; Koh, G.Y. Angiopoietin-1 is an apoptosis survival factor for endothelial cells. FEBS Lett. 1999, 448, 249–253. [Google Scholar] [CrossRef]
- Kwak, H.J.; Lee, S.J.; Lee, Y.; Ryu, C.H.; Koh, K.N.; Choi, H.Y.; Koh, J.Y. Angiopoietin-1 inhibits irradiation and mannitol-induced apoptosis in endothelial cells. Circulation 2000, 101, 2317. [Google Scholar] [CrossRef]
- Kim, I.; Kim, H.G.; So, J.; Kwak, H.J.; Koh, J.Y. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Circ. Res. 2000, 86, 24–29. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Fulton, D.; Mahboubi, K.; Kalb, R.; O’connor, D.; Li, F.; Altieri, D.; Sessa, W. Angiopoietin-1 inhibits endothelial cell apoptosis via the akt/survivin pathway. J. Biol. Chem. 2000, 275, 9102–9105. [Google Scholar] [CrossRef] [PubMed]
- Kontos, C.D.; Stauffer, T.P.; Yang, W.; York, J.; Huang, L.; Blanar, M.; Meyer, T.; Peters, K. Tyrosine 1101 of Tie2 is the major site of association of p85 and is required for activation of phosphatidylinositol 3-kinase and Akt. Mol. Cell Biol. 1998, 18, 4131–4140. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Master, Z.; Jones, J.; Bouchard, D.; Gunji, Y.; Sasaki, H.; Daly, R.; Alitalo, K.; Dumont, D. Identification of tek/Tie2 binding partners. Binding to a multifunctional docking site mediates cell survival and migration. J. Biol. Chem. 1999, 274, 30896–30905. [Google Scholar] [CrossRef]
- Jones, N.; Chen, S.H.; Sturk, C.; Master, Z.; Tran, J.; Kerbel, R.; Dumont, D. A unique autophosphorylation site on Tie2/tek mediates dok-R phosphotyrosine binding domain binding and function. Mol. Cell Biol. 2003, 23, 2658–2668. [Google Scholar] [CrossRef] [PubMed]
- Harfouche, R.; Gratton, J.; Yancopoulos, G.D.; Noseda, M.; Karsan, A.; Hussain, S. Angiopoietin-1 activates both anti- and proapoptotic mitogen-activated protein kinases. FASEB J. 2003, 17, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Kichina, J.V.; Goc, A.; Al-Husein, B.; Somanath, P.; Kandel, E. PAK1 as a therapeutic target. Expert Opin. Ther. Targets 2010, 14, 703–725. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Ghosh, C.; Mukherjee, A.; Parikh, S. Angiopoietin-1 requires IQGAP1 to activate Rac1 and promote endothelial barrier defense. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Tadros, A.; Hughes, D.P.; Dunmore, B.J. ABIN-2 protects endothelial cells from death and has a role in the antiapoptotic effect of Angiopoietin-1. Blood 2003, 102, 4407–4409. [Google Scholar] [CrossRef]
- Van Huffel, S.; Delaei, F.; Heyninck, K.; Valck, D.; Beyaert, R. Identification of a novel A20-binding inhibitor of nuclear factor-kappaB activation termed ABIN-2. J. Biol. Chem. 2001, 276, 30216–30223. [Google Scholar] [CrossRef]
- Korhonen, E.A.; Lampinen, A.; Giri, H.; Anisimov, A.; Kim, M.; Allen, B.; Fang, S.; D’Amico, G.; Sipila, T.; Lohela, M.; et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J. Clin. Investig. 2016, 126, 3495–3510. [Google Scholar] [CrossRef]
- Inoue, M.; Itoh, H.; Ueda, M.; Naruko, T.; Kojima, A.; Komatsu, R.; Doi, K.; Ogawa, Y.; Tamura, N.; Takaya, K.; et al. Vascular endothelial growth factor (VEGF) expression in human coronary atherosclerotic lesions: Possible pathophysiological significance of VEGF in progression of atherosclerosis. Circulation 1998, 98, 2108–2116. [Google Scholar] [CrossRef]
- Singh, H.; Hansen, T.M.; Patel, N.; Brindle, N. The molecular balance between receptor tyrosine kinases Tie1 and Tie2 is dynamically controlled by VEGF and TNFa and regulates angiopoietin signalling. PLoS ONE 2012, 7, 29319. [Google Scholar]
- Marron, M.B.; Singh, H.; Tahir, T.A.; Kavumkal, J.; Kim, H.; Koh, J.; Brindle, N. Regulated proteolytic processing of Tie1 modulates ligand responsiveness of the receptortyrosine kinase Tie2. J. Biol. Chem. 2007, 282, 30509–30517. [Google Scholar] [CrossRef]
- Findley, C.M.; Cudmore, M.J.; Ahmed, A.; Kontos, C. VEGF induces Tie2 shedding via a phosphoinositide 3-kinase/Akt-dependent pathway to modulate Tie2 signaling. Arter. Thromb. Vasc. Biol. 2007, 27, 2619–2626. [Google Scholar] [CrossRef]
- Seegar, T.C.; Eller, B.; Tzvetkova-Robev, D.; Kolev, M.; Henderson, C.; Nikilov, D.; Barton, W. Tie1-Tie2 interactions mediate functional differences between angiopoietin ligands. Mol. Cell 2010, 37, 643–655. [Google Scholar] [CrossRef]
- Schlosser, K.; Taha, M.; Deng, Y.; McIntyre, L.; Mei, S.; Stewart, D. High circulating Angiopoietin 2 levels exacerbate pulmonary inflammation but not vascular leak or mortality in endotoxin-induced lung injury in mice. Thorax 2018, 73, 248–261. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Zhang, L.; Croll, S.D.; Chopp, M. Angiopoietin-1 reduces cerebral blood vessel leakage and ischemic lesion volume after focal cerebral embolic ischemia in mice. Neuroscience 2002, 113, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Weber, C. Inflammatory mediators in atherosclerotic vascular disease. Basic Res. Cardiol. 2005, 100, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.S.; Yeo, T.W.; Piera, K.A.; Woodberry, T.; Celermajer, D.; Stephens, D.; Anstey, N. Angiopoietin-2 is increased in sepsis and inversely associated with nitric oxide-dependent microvascular reactivity. Crit. Care 2010, 14, R89. [Google Scholar] [CrossRef]
- Ghosh, C.C.; Thamm, K.; Berghelli, A.V.; Schrimpf, C.; Maski, M.; Abid, T.; Milam, K.; Rajakimar, A.; Santel, A.; Kielstein, J. Drug repurposing screen identifies Foxo1-dependent Angiopoietin-2 regulation in sepsis. Crit. Care Med. 2015, 43, 230–240. [Google Scholar] [CrossRef]
- Leow, C.C.; Coffman, K.; Inigo, I.; Breen, S.; Czapiga, M.; Soukharev, S.; Gingles, N.; Peterson, N.; Fazenbaker, C.; Woods, R.; et al. MEDI3617, a human anti angiopoietin 2 monoclonal antibody, inhibits angiogenesis and tumor growth in human tumor xenograft models. Int. J. Oncol. 2012, 40, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Shan, S.; Rusconi, C.P.; Shetty, G.; Dewhirst, M.; Kontos, C.; Sullenger, B. Inhibition of rat corneal angiogenesis by a nuclease-resistant RNA aptamer specific for Angiopoietin-2. Proc. Natl. Acad. Sci. USA 2003, 100, 5028–5033. [Google Scholar] [CrossRef]
- Yang, P.; Chen, N.; Yang, D.; Crane, J.; Yang, S.; Wang, H.; Dong, R.; Yi, X.; Xie, L.; Jing, G.; et al. The ratio of serum Angiopoietin-1 to Angiopoietin-2 in patients with cervical cancer is a valuable diagnostic and prognostic biomarker. PeerJ 2017, 5, 3387. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.; Burns, A.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Smadja, D.M.; Mentzer, S.J.; Fontenay, M.; Laffan, M.; Ackermann, M.; Helms, J.; Jonigk, D.; Chocron, R.; Pier, G.; Gendron, N.; et al. COVID-19 is a systemic vascular hemopathy: Insight for mechanistic and clinical aspects. Angiogenesis 2021, 24, 755–788. [Google Scholar] [CrossRef]
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Schimmel, L.; Chew, K.Y.; Stocks, C.J.; Yordanov, T.E.; Essebier, P.; Kulasinghe, A.; Monkman, J.; Dos Santos Miggiolaro, A.F.R.; Cooper, C.; de Noronha, L.; et al. Endothelial cells are not productively infected by SARS-CoV-2. Clin. Transl. Immunol. 2021, 10, e1350. [Google Scholar] [CrossRef]
- Garcia-Ponce, A.; Chanez Paredes, S.; Castro Ochoa, K.F.; Schnoor, M. Regulation of endothelial and epithelial barrier functions by peptide hormones of the adrenomedullin family. Tissue Barriers 2016, 4, e1228439. [Google Scholar] [CrossRef]
- Millar, F.R.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 2016, 71, 462–473. [Google Scholar] [CrossRef]
- Noris, M.; Benigni, A.; Remuzzi, G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int. 2020, 98, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, C.; Tinello, C.; Vatrella, A.; De Sarro, G.; Pelaia, G. Lung under attack by COVID-19-induced cytokine storm: Pathogenic mechanisms and therapeutic implications. Ther. Adv. Respir. Dis. 2020, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- Bernard, I.; Limonta, D.; Mahal, L.K.; Hobman, T.C. Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses 2020, 13, 29. [Google Scholar] [CrossRef]
- Grobler, C.; Maphumulo, S.C.; Grobbelaar, L.M.; Bredenkamp, J.C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. Covid-19: The Rollercoaster of Fibrin (Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes. Int. J. Mol. Sci. 2020, 21, 5168. [Google Scholar] [CrossRef] [PubMed]
- Nicin, L.; Abplanalp, W.T.; Mellentin, H.; Kattih, B.; Tombor, L.; John, D.; Schmitto, J.; Heineke, J.; Emrich, F.; Arsalan, M.; et al. Cell type specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 2020, 41, 1804–1806. [Google Scholar] [CrossRef] [PubMed]
- Sluimer, J.C.; Gasc, J.M.; Hamming, I.; Goor, H.; Michaud, A.; Van den Akker, L.H.; Jutten, B.; Cleutjens, J.; Bijnens, A.; Corvol, P.; et al. Angiotensin converting enzyme 2 (ACE2) expression and activity in human carotid atherosclerotic lesions. J. Pathol. 2008, 215, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, 9610. [Google Scholar] [CrossRef] [PubMed]
- Delorey, T.M.; Ziegler, C.G.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Stephen, J.; Ayshwarya Subramanian, F.; Montoro, D.T. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Wang, P.; Luo, R.; Zhang, M.; Wang, Y.; Song, T.; Tao, T.; Li, Z.; Jin, L.; Zheng, H.; Chen, W.; et al. A cross-talk between epithelium and endothelium mediates human alveolar-capillary injury during SARS-CoV-2 infection. Cell Death Dis. 2020, 11, 1042. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef]
- Andersson, M.I.; Arancibia-Carcamo, C.V.; Auckland, K.; Baillie, J.K.; Barnes, E.; Beneke, T.; Bibi, S.; Brooks, T.; Carroll, M.; Crook, D.; et al. SARS-CoV-2 RNA detected in blood products from patients with COVID-19 is not associated with infectious virus. Wellcome Open Res. 2020, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Ranucci, M.; Ballotta, A.; Di Dedda, U.; Baryshnikova, E.; Dei Poli, M.; Resta, M.; Falco, M.; Albano, G.; Menicanti, L. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J. Thromb. Haemost. JTH 2020, 18, 1747–1751. [Google Scholar] [CrossRef] [PubMed]
- Fodor, A.; Tiperciuc, B.; Login, C.; Orasan, O.H.; Lazar, A.L.; Buchman, C.; Hanghicel, P.; Sitar-Taut, A.; Suharoschi, R.; Vulturar, R.; et al. Endothelial Dysfunction, Inflammation, and Oxidative Stress in COVID-19-Mechanisms and Therapeutic Targets. Oxidative Med. Cell. Longev. 2021, 8671713. [Google Scholar] [CrossRef] [PubMed]
- Nagele, M.; Haubner, B.; Tanner, F.; Ruschitzka, F.; Flammer, A. Endothelial dysfunction in Covid-19: Current findings and therapeutic implications. Atherosclerosis 2020, 314, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Miesbach, W.; Makris, M. COVID-19: Coagulopathy, Risk of Thrombosis, and the Rationale for Anticoagulation. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620938149. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef]
- Iba, T.; Connors, J.M.; Levy, J.H. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm. Res. 2020, 69, 1181–1189. [Google Scholar] [CrossRef]
- Henry, B.M.; de Oliveira, M.H.S.; Cheruiyot, I.; Benoit, J.L.; Cooper, D.S.; Lippi, G.; Le Cras, T.D.; Benoit, S.W. Circulating level of Angiopoietin-2 is associated with acute kidney injury in coronavirus disease 2019 (COVID-19). Angiogenesis 2021, 24, 403–406. [Google Scholar] [CrossRef]
- Lu, R.X.Z.; Lai, B.F.L.; Rafatian, N.; Gustafson, D.; Campbell, S.B.; Banerjee, A.; Kozak, R.; Mossman, K.; Mubareka, S.; Howe, K.L.; et al. Vasculature-on-a-chip platform with innate immunity enables identification of angiopoietin-1 derived peptide as a therapeutic for SARS-CoV-2 induced inflammation. Lab Chip 2022, 22, 1171–1186. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Rendeiro, A.F.; Ravichandran, H.; Bram, Y.; Chandar, V.; Kim, J.; Meydan, C.; Park, J.; Foox, J.; Hether, T.; Warren, S.; et al. The spatial landscape of lung pathology during COVID-19 progression. Nature 2021, 593, 564–569. [Google Scholar] [CrossRef]
- Ramaiah, M.J. mTOR inhibition and p53 activation, microRNAs: The possible therapy against pandemic COVID-19. Gene Rep. 2020, 20, 100765. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Shi, F.; Basu, D.; Huq, A.; Routhier, S.; Day, R.; Jin, W. Proteolytic processing of angiopoietin-like protein 4 by proprotein convertases modulates its inhibitory effects on lipoprotein lipase activity. J. Biol. Chem. 2011, 286, 15747–15756. [Google Scholar] [CrossRef]
- Bhatraju, P.K.; Morrell, E.D.; Stanaway, I.B.; Sathe, N.A.; Srivastava, A.; Postelnicu, R.; Green, R.; Andrews, A.; Gonzalez, M.; Kratochvil, C.J.; et al. Angiopoietin-Like4 Is a Novel Marker of COVID-19 Severity. Crit. Care Explor. 2022, 5, e0827. [Google Scholar] [CrossRef]
- Reyes, A.; Corrales, N.; Galvez, N.M.S.; Bueno, S.M.; Kalergis, A.M.; Gonzalez, P.A. Contribution of hypoxia inducible factor-1 during viral infections. Virulence 2020, 11, 1482–1500. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Keskinidou, C.; Jahaj, E.; Gallos, P.; Dimopoulou, I.; Kotanidou, A.; Orfanos, S.E. ICU Admission Levels of Endothelial Biomarkers as Predictors of Mortality in Critically Ill COVID-19 Patients. Cells 2021, 10, 186. [Google Scholar] [CrossRef]
- Arab, O.A.; Bennis, Y.; Gauthier, P.; Dupont, H.; Kamel, S.; Mahjoub, Y. Association between inflammation, angiopoietins, and disease severity in critically ill COVID-19 patients: A prospective study. Br. J. Anaesth. 2021, 126, 127–130. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Weng, J. Endothelial dysfunction in COVID-19: An overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 2023, 44, 695–709. [Google Scholar] [CrossRef]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Salazar, B.; Holwerda, M.; Stüdle, C.; Piragyte, I.; Mercader, N.; Engelhardt, B.; Rieben, R.; Döring, Y. COVID-19 and the Vasculature: Current Aspects and LongTerm Consequences. Front. Cell Dev. Biol. 2022, 10, 824851. [Google Scholar] [CrossRef] [PubMed]
- Schmaier, A.A.; Pajares Hurtado, G.M.; Manickas-Hill, Z.J.; Sack, K.D.; Chen, S.M.; Bhambhani, V.; Quadir, J.; Nath, A.K.; Collier, A.Y.; Ngo, D.; et al. Tie2 activation protects against prothrombotic endothelial dysfunction in COVID-19. JCI Insight 2021, 6, e151527. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janchivlamdan, D.; Shivkumar, M.; Singh, H. The Effects of SARS-CoV-2 on the Angiopoietin/Tie Axis and the Vascular Endothelium. Encyclopedia 2024, 4, 544-557. https://doi.org/10.3390/encyclopedia4010035
Janchivlamdan D, Shivkumar M, Singh H. The Effects of SARS-CoV-2 on the Angiopoietin/Tie Axis and the Vascular Endothelium. Encyclopedia. 2024; 4(1):544-557. https://doi.org/10.3390/encyclopedia4010035
Chicago/Turabian StyleJanchivlamdan, Dolgormaa, Maitreyi Shivkumar, and Harprit Singh. 2024. "The Effects of SARS-CoV-2 on the Angiopoietin/Tie Axis and the Vascular Endothelium" Encyclopedia 4, no. 1: 544-557. https://doi.org/10.3390/encyclopedia4010035
APA StyleJanchivlamdan, D., Shivkumar, M., & Singh, H. (2024). The Effects of SARS-CoV-2 on the Angiopoietin/Tie Axis and the Vascular Endothelium. Encyclopedia, 4(1), 544-557. https://doi.org/10.3390/encyclopedia4010035