Abstract
For most living beings, oxygen is an essential molecule for survival, being the basis of biological oxidations, which satisfy most of the energy needs of aerobic organisms. Oxygen can also behave as a toxic agent posing a threat to the existence of living beings since it can give rise to reactive oxygen species (ROS) that can oxidise biological macromolecules, among which proteins and lipids are the preferred targets. Oxidative damage can induce cell, tissue, and organ dysfunction, which leads to severe body damage and even death. The survival of the aerobic organism depends on the development of an elaborate antioxidant defence system adapted to the normal level of atmospheric oxygen. The production of ROS in the aerobic organism can occur accidentally from exposure to pollutants or radiation, but occurs constantly during normal metabolic reactions. Cells have evolved using ROS to their advantage. Indeed, ROS are used as signalling molecules in numerous physiological processes, including muscle contraction, regulation of insulin release, and adaptation to environmental changes. Therefore, supplementation with antioxidants must be used wisely. A low level of ROS is essential for adaptation processes, so an excess of antioxidants can be harmful. Conversely, in conditions where ROS production increases, antioxidants can be useful to avoid cellular dysfunction.
1. Introduction
Aerobic organisms require the presence of oxygen to complete the processes underlying the transformations necessary to obtain the chemical energy that is useful for maintaining cellular homeostasis and supporting all activities related to life, such as growth, locomotion, and reproduction [1].
Oxygen utilisation poses challenges to living organisms, as reactive oxygen species (ROS) can form from it [1]. ROS can oxidise biological macromolecules, generating damage that can alter their functionality [2]. For this reason, the evolution of aerobiosis has evolved in concert with that of antioxidant defence systems [3]. Furthermore, in aerobic organisms, mechanisms that have enabled them to use ROS as signal molecules for cellular adaptation to environmental changes have evolved [2]. The present review briefly describes how cells use oxygen, the formation of ROS, the damage they impose on the cell, and how cells evolve using ROS to their advantage.
2. The Source of Energy
Living organisms are systems characterised by the ability to convert available external energy into work necessary to maintain their homeostatic functions [4,5]. Although, in theory, it is possible to convert one form of energy into another, living organisms lack the conditions for such a conversion.
In essence, life energy comes from the constant stream of solar photons reaching our planet. When a photon interacts with an electron of a molecule on earth, the electron reaches a higher energy level, from which it returns to its initial state and releases energy [6]. Living organisms have developed mechanisms to capture and use the released energy to their advantage [6]. These systems consist of a gradual release of energy during the passage of electrons among the outer orbitals of atoms with different energy levels [6]. The dissipation of electronic energy is associated with the synthesis of ATP, which is a fundamental molecule for living organisms [6].
ATP consists of a base of adenine and a ribose sugar, with three phosphate groups linked by two high-energy bonds called phosphohydride bonds. The removal of phosphate groups by hydrolysis with the following conversion of ATP into ADP and AMP releases energy. The released energy can be transferred to other molecules to make unfavourable reactions in a cell favourable.
The Earth’s atmosphere initially consisted of CO2, N2, and H2O, with traces of H2. It is assumed that, in the prebiotic environment, oxygen did not exist in its free form. A decisive phase in the evolution of life was the appearance of photosystem II [7,8], capable of effectively capturing photons and using water to obtain the hydrogen necessary to synthesise NADPH, ATP and, therefore, for the synthesis of carbohydrates using CO2 [9] (Figure 1). While the use of water has freed bacteria from the need to use other environmental reducing agents, such as sulphur species, it has also resulted in the release of oxygen as a side effect. The latter created the fundamental conditions for the evolution of heterotrophic organisms that depend on respiration for the synthesis of ATP. Heterotrophs are unable to capture solar photons, so they take carbohydrates and other high-energy substances by eating plants or animals and get most of the energy they need through respiration, an enormously exergonic form of metabolism that was an important driver, if not essential, to the evolution of complex life forms and eukaryotes [10,11].
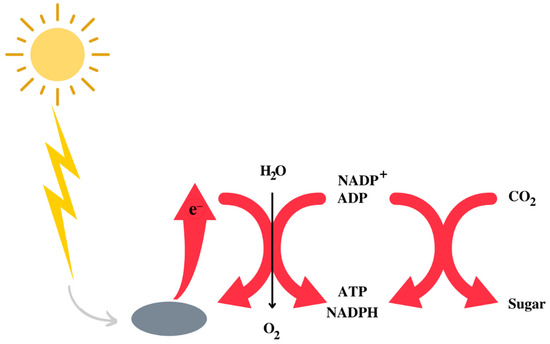
Figure 1.
Schematic representation of the utilisation of the sun energy to obtain a form of metabolically utilisable energy by living beings.
Therefore, in heterotrophic aerobic organisms, most of the energy required for life processes is obtained from respiration, in which ATP is obtained by moving electrons from the high energy level of nutrient molecules, such as glucose, to a low energy level in the molecule of water through the passage among a series of electron acceptors.
In conclusion, the only energy source that heterotrophs can use is the chemical one, taken from the outside in the form of compounds with high energy content. This ability depends on the development of biochemical and physiological systems that allow for the transformation of chemical energy into other forms of energy essential for life. Ultimately, the energy supply of living systems essentially occurs in the form of an exchange of matter.
3. Oxygen Toxicity
For most living beings, oxygen is an essential molecule for survival, being the basis of biological oxidations which satisfy most of the energy needs of aerobic organisms. Although oxygen is essential for these organisms, it can also behave as a toxic agent and prove to be a threat to their existence. This inconsistent aspect of aerobic life was defined by Davies as the “paradox of aerobic life” [12]. Indeed, the organisms can survive in the presence of oxygen because they have developed an elaborate antioxidant defence system, adapting to the Earth’s oxidising atmosphere. However, this system is only adequate for the atmospheric oxygen pressure (approximately 156 mm Hg), and there is strong evidence that exposure to oxygen pressures higher than atmospheric ones causes severe damage.
The toxicity of oxygen was already known in the 1770s when Joseph Priestley, who discovered oxygen, together with but independently from Carl Wilhelm Scheele, compared its effect on the body to candle burning, observing that a candle burns out faster in oxygen than in air and wondering if “the animal powers be too soon exhausted in this pure kind of air” [13].
The deleterious consequences of breathing oxygen at elevated partial pressures were first recognised in the late 19th century. Paul Bert was the first to describe the toxic effects of hyperbaric oxygen on the central nervous system [14] and Lorrain Smith was the first to describe the effects on the lungs [15].
Another example of oxygen toxicity is the retinopathy of the prematurity (ROP). In the preterm infant, supplemental oxygen administration has been frequently used as a life-saving treatment since the 1780s [16,17] and, in the 1940s regular supplemental oxygen use was established for the preterm infant [18]. The spread of the practice of oxygen supplementation in preterm infants led to an epidemic of severe retinopathy of prematurity and blindness between the 1940s and 1950s. In 1942, the phenomenon of retrolental fibroblastic overgrowth [19] was described, and in 1950 the role of oxygen in the development of this new sudden blindness observed in preterm infants, which was called retrolental fibroplasia [20], now known as ROP, was defined. Several studies have established the role of hyperoxia in ROP [21,22,23].
The susceptibility of the human retina to hyperoxia is due to vasculature being not fully developed until full term of gestation, and the photoreceptors developing after the small vessels. Excess oxygen leads to the destruction and arrest of the development of the neurovascular retina, followed by abnormal neovascularisation during the vulnerable retinal development stage [24,25].
Currently, a more careful control of oxygen use (continuous monitoring, with intermittent supplementation of O2, provided only to maintain blood O2 levels) has significantly decreased the severity of retinopathy [26]. Administration of the antioxidant α-tocopherol to preterm infants may reduce ROP but has been shown to have harmful effects in very preterm babies [27]. Nowadays, the use of vitamin E in the prevention of the exacerbation of ROP, and of other dysfunctions related to preterm, could be reconsidered in the light of current knowledge in the field of modern neonatal medicine [28].
High partial pressures of oxygen can also be harmful to adult humans due to their effects on the central nervous system, lungs, and eyes, even if other systems can also be affected [29]. The partial pressure of oxygen in inspired air, at the sea level, is about 160 mm Hg. This value can be increased by either breathing 100% oxygen, or by increasing the pressure of the breathing mixture [30]. These conditions can occur using hyperbaric oxygen for therapeutic purposes, or during underwater diving.
The signs of oxygen toxicity are detectable in various tissues; however, the most worrying are those affecting the central nervous system and the lungs, which can be considered real target organs.
The lungs’ sensitivity to the high partial pressure of O2, or a high percentage of oxygen in the breathing air, may be due to its greater exposure to high oxygen levels compared to other tissues in the body [29].
The pulmonary oxygen toxicity onset is directly related to the partial pressure of inspired oxygen, and usually it does not occur at oxygen concentrations lower than 50% in young healthy subjects [31]. Pulmonary toxicity occurs after 4–22 h at an oxygen percentage greater than 95% at atmospheric pressure and within 3 h at 100% oxygen at 2–3 times the atmospheric pressure [32]. The first manifestation of pulmonary toxicity is tracheobronchial irritation associated with cough and progressive dyspnoea in men exposed to 98% oxygen for 30–74 h [33]. Longer exposures to oxygen may induce diffuse alveolar damage characterised by clinical symptoms and pathological signs of alveolar damage, like those of acute respiratory distress syndrome from other causes [34]. Prolonged exposure to sublethal concentrations of oxygen may result in chronic pulmonary fibrosis and emphysema with tachypnoea as well as progressive hypoxaemia that can cause, after a few days, a lung damage so severe it may, paradoxically, cause death from anoxia [35].
Central nervous system (CNS) toxicity occurs following short-term exposure to air containing highly concentrated oxygen at a pressure above atmospheric. Exposure for minutes to hours at 160 kPa of partial pressure, approximatively eight times the atmospheric concentration, is generally associated with CNS toxicity [29]. CNS toxicity does not occur during normobaric exposure to high O2 concentrations, and conditions related to CNS toxicity can only be met in special situations such as immersion or hyperbaric oxygen treatment. The appearance time of the CNS toxicity initial symptoms (tunnel vision, tinnitus, nausea, facial twitching, dizziness, and confusion) is inversely related to the oxygen pressure and may be as short as 10 min at pressures of 4–5 atmospheres absolute [32]. Initial symptoms may be followed by tonic–clonic seizures and subsequently by unconsciousness. The onset of the initial disturbances does not follow a defined pattern before the onset of the seizures, which are the most dramatic and dangerous symptom of oxygen toxicity. However, the latter are reversible and leave no neurological damage if the partial pressure of the inspired oxygen is reduced. The onset of seizures depends on the partial pressure of oxygen and the duration of exposure [29]. Moreover, the exposure time at which symptoms begin depends on many factors and can change in the same individual day by day [30,31].
Many factors such as underwater activity, cold weather, and physical activity can reduce the time to onset of CNS symptoms [29]. Oxygen toxicity is very dangerous during diving as it may cause drowning due to an epileptic attack. Understanding that a diver exhibits symptoms of oxygen toxicity is difficult before seizure activity develops, as the first symptoms are non-specific and do not follow a typical sequence.
4. Oxygen Reactivity
The toxicity of oxygen was initially attributed to its ability to inhibit cellular enzymes [36,37]. There are several examples of oxygen-mediated enzymatic inactivation in anaerobic organisms [38]. However, most enzymes in aerobic cells are insensitive to oxygen and those that are inactivated are poorly sensitive. Furthermore, the rate at which oxygen-sensitive enzymes are inactivated is too low and does not match the rate at which the toxic effects develop [1].
Subsequently, the oxygen toxicity was attributed to the formation of oxygen radicals [39]. Despite being a free radical, O2 does not have high reactivity. The reactions in which it is involved do not take place at ordinary temperatures or in the absence of catalysts, even if its high oxidising power makes most of the substances of biological interest thermodynamically unstable in its presence [1].
This apparent contradiction is explained by the electronic configuration of diatomic oxygen. It has two unpaired electrons, each located in a different π* antibonding orbital [1]. The two electrons have the same quantum number of spins (they have parallel spins) in the ground state of oxygen. If oxygen attempts to oxidise another atom or molecule by accepting a pair of electrons from it, both electrons must have antiparallel spins to fit the empty space in the π* orbitals. However, a pair of electrons in an atomic or molecular orbital does not meet this criterion since they have opposite spins according to the Pauli principle. This represents a restriction (called a spin restriction) on the transfer of electrons to oxygen, which can accept electrons only one at a time. This explains why O2 reacts slowly with many non-radicals [39].
In aerobic organisms, the energy necessary for the vital processes derives from the oxidation reactions in which oxygen is consumed; therefore, it is evident that processes are operative in such organisms through which the spin restriction is, in some way, eliminated with a consequent increase in oxygen reactivity.
The elimination of the spin restriction could be achieved by putting enough energy in O2 to allow the inversion of the spin of one of its electrons in the π* orbital, thus passing O2 from the basal to an excited state named singlet oxygen. In the singlet oxygen, the elimination of the spin restriction makes oxygen much more reactive [40]. Moreover, the two electrons may remain in different orbitals or couple in one orbital. Therefore, two species with different reactivities are generated. Of the two singlet states of O2, the one with unpaired electrons has higher energy, truly short life in solution and decays quickly [41]. Differently, singlet oxygen with paired electrons has a much longer lifespan, so it is believed to be the only singlet oxygen species that react in solution [41].
The energy of light in the visible spectrum would be sufficient for the formation of singlet oxygen, but, fortunately, oxygen does not absorb visible light. The formation of singlet oxygen occurs when an appropriate dye, called a photosensitiser, absorbs visible light, and then collides with molecular oxygen transferring energy to it, giving rise to singlet oxygen [41]. There are many of these dyes in nature that support singlet oxygen formation. These include photosynthetic pigments, protoporphyrin IX (a heme precursor), Bengal rose, or methylene blue [41]. Singlet oxygen participates in many biochemical processes, such as photosynthesis, cell signalling, immune responses, or the degradation of polymers. However, the transformations are generally small and, therefore, cannot explain the evidence according to which many of the major changes occurring in cells consist of the O2 oxidation of various types of molecules.
Another way to avoid the spin restriction is to add electrons to oxygen one at a time, at a rate that allows for electronic rotational reversals between collision events [42]. The univalent oxygen reduction pathway requires the generation of oxygen reduction intermediates, which are reactive and can damage biological molecules [42].
These reactions are due to the intervention of substances that can transfer, in the right direction, one or more electrons from the molecule to be oxidised to O2 [1]. Among such substances, there are transition metal ions of varying valence, such as iron and copper, which have unpaired electrons with parallel spins and enzymes, such as cytochrome oxidase (Cox), which have metal ions in their active sites [1].
Cox is the terminal oxidase of cell respiration, accepts electrons from the reduced form of the cytochrome c (Cyt c), promotes the four-electrons reduction in oxygen to water, completes the electron transport of the mitochondrial respiratory chain, and can protect cells from the damage due to the formation of toxic intermediate of oxygen reduction [43]. In mammalian, Cox is a bigenomic enzyme. It contains 13 subunits, 3 catalytic subunits encoded by the mitochondrial genes and the remaining 10 encoded by the nuclear genome. The latter play a role in the regulation and/or in the assembly of the enzyme. Cox contains two heme groups (heme a and a3) and two Cu2+ centres (Cu2+ A and Cu2+ B) in the catalytic centre and uses more than 90% of the oxygen consumed by cells and tissues [44].
Cox transforms O2 into water in the reduction site that contains Fea3 and CuB, each of which accepts one electron. O2 bounds to the reduction site when both metals are in the reduced state (Fea32+ and CuB1+). The fully reduced state is named R-intermediate.
The bond of O2 to the R intermediate forms Fea33+–O2− (called intermediate A). This is converted into intermediate P without electron transfer from low-potential metal sites (Fea and CuA). When the P intermediate forms, the bound O2 accepts four electrons (two from Fea3, one from CuB, and the fourth from a neighbouring OH tyrosine). In the intermediate P, an oxide (O2−) and a hydroxide (OH−) bond to Fea3 and CuB, respectively. Subsequently, intermediate P receives four electrons from cytochrome c through the low potential metal sites, CuA and Fea (heme a). These equivalents are added one at a time, forming the intermediates F−, O−, and E, and finally reproducing the intermediate R. Each electron transfer from heme a couples with the pumping of one proton in the intermembrane space of the mitochondria, creating the driving force for ATP synthesis [43].
According to this view, the first three phases of the reaction generate, in sequence, the superoxide ion (O2•−), peroxide ion (O22−), which immediately protonates to hydrogen peroxide (H2O2), and the hydroxyl radical (•OH). The addition of the fourth electron leads to the hydroxyl ion, which is transformed into the water by the addition of an H+.
Cox, firmly bound in its active site, holds all partially reduced intermediates of O2 until complete reduction.
These intermediates of oxygen reduction do not have the kinetic impediments of O2 in the ground state and are much more reactive. For this reason, they are referred to as reactive oxygen species (ROS).
However, Cox is not a site of ROS release in the cells. Several experimental pieces of evidence suggest that other cellular sites are responsible for the formation of the ROS.
In the ROP, the ROS generated during the hyperoxia determine the destruction arrest of the development of the neurovascular retina as well as abnormal neovascularisation during a vulnerable developmental retinal stage [24,25].
5. Reactive Oxygen Species and Their Sources
ROS encompass a group of molecules derived from molecular oxygen, which are formed by reduction–oxidation (redox) reactions or by electronic excitation [45]. They include, other than superoxide, hydrogen peroxide, hydroxyl radical, as well as atoms such as alkoxy radicals (RO•) and peroxyl radicals (ROO•), and radical species such as singlet oxygen and ozone (O3) (Table 1).

Table 1.
Main reactive oxygen species, their reactivity and half-life [46].
The reactivity of the different ROS is very variable. Superoxide is generally the first ROS generated. It is moderately reactive and exhibits a short half-life of 2–4 μs [46]. Therefore, it is not damaging by itself, and its harmful effects depend on the products formed after dismutation. O2•− dismutates to H2O2 and O2 spontaneously or owing to the intervention of superoxide dismutases. Therefore, it is the main source of H2O2. Due to high electrostatic attraction, O2•− oxidises Fe–S clusters at a high rate. This oxidation leads to iron release. While O2•− diffuses slowly in the membrane due to its negative charge, its protonated form perhydroxyl radical (HO2) is uncharged and may diffuse in lipids and produce a carbon-centred radical of polyunsaturated lipids. O2•− reacts efficiently with nitric oxide (NO•), which leads to the formation of peroxynitrite (ONOO−), a tyrosine-nitrating agent.
Notwithstanding, H2O2 is a strong two-electron oxidant, and the high activation energy restricts its reactivity to a few biological targets [45]. Hydrogen peroxide is relatively stable and shows a half-life of 1 ms. With glutathione, cysteine, and methionine, H2O2 shows a low reactivity while the reaction with cysteine in specific proteins can be very high. Reactivity with cysteine depends on the structure and environment of the protein, which could explain the selectivity and specificity of H2O2 in redox signalling [45].
The most reactive ROS is hydroxyl radical, which oxidises biological molecules at a diffusion-controlled rate which renders it a non-specific oxidant. Its formation is due to the reduction reaction of hydrogen peroxide in the Fenton reaction and requires the presence of a Fe or Cu metal in the reduced state [47,48]. •OH reacts as soon as produced with sites adjacent to the production site. For this reason, the localisation of Fe2+ is a determining factor for the site of toxicity. •OH is an initiator of lipid peroxidation.
Peroxyl radical (ROO·) forms in the initial phases of lipid peroxidation and propagate free radical chain reactions by abstracting a proton from another polyunsaturated fatty acid, thereby creating a lipid hydroperoxide and another carbon radical. Alkoxyl radical (RO•) forms as an intermediate in lipid peroxidation during metal-catalysed decomposition of lipid hydroperoxides and can amplify lipid peroxidation chain reactions.
ROS are generated by various sources which can either be endogenous or exogenous.
6. Exogenous Sources
Exogenous or environmental sources of ROS include electromagnetic and corpuscular radiations and various types of xenobiotics.
6.1. Radiation
Various types of radiation can exert effects on living organisms, resulting in a series of biological effects that affect life processes to varying degrees. Radiations regulate several physiological processes such as photosynthesis, photoperiodism, circadian rhythms, vision, and vitamin D formation [49]. On the other hand, radiations can also be responsible for a series of pathological events in which free radicals are sometimes involved.
Ultraviolet radiation can induce biological damage involving radical intermediates. This effect is due to the capacity of some molecules to absorb quanta of energy of the ultraviolet radiation passing to an excited state, from which they can decay, dissociate, and form free radicals.
Ionising radiation can be divided into corpuscular radiation (electrons, protons, neutrons, and particles α and β) and electromagnetic radiation (X-rays and γ rays). They possess sufficiently high energy to move an electron away from a molecule, thus forming an ion that can dissociate and generate a free radical or recapture the electron passing to an excited state and then undergoing homolytic cleavage [50].
Exposure of living organisms to ionising radiation can give rise to primary radicals by transferring their energy to cellular substances, especially to the main component of cells, namely water. Following the absorption of energy, water undergoes ionisation and excitation. The electron hydrates, the radical ion decomposes, and the excited molecule splits homolytically and generates the hydrogen atom and the hydroxyl radical.
The primary radicals, which include •OH, H•, and e−aq, can diffuse and undergo secondary reactions with various cellular solutes, including organic macromolecules, generating new chemical species [51].
Ionising radiation, unlike ultraviolet radiation, penetrates deeply into the irradiated tissues so that damage can also occur in deeply located cells [49].
Biological systems are much more sensitive to radiation when irradiated in the presence of O2 (oxygen effect) [52]. Radiosensitivity decreases at low levels (hypoxia) or in the absence (anoxia) of O2. The study of the dependence of radiosensitivity on O2 concentration has shown a sharp decrease in radiosensitivity when the pO2 voltage drops to values below 15 mm Hg, while for higher values, the radiosensitivity is almost constant [53]. Since in mammals the pO2 of arterial blood is about 70 mm Hg, and that of venous blood is about 40 mm Hg, it is evident that most normal tissues show the highest degree of sensitivity towards the oxygen effect.
The oxygen effect found an initial explanation in the possibility that the primary radicals H• and e−aq react with O2, forming the hydroperoxyl radical and superoxide, respectively.
In this way, new free radicals are produced, amplifying the effect of the radiation. On the other hand, a free radical generated by a biological molecule, either due to the direct result of radiation or by extraction of hydrogen by the primary radical •OH, can bind to O2, forming a peroxyl radical which can, in turn, extract hydrogen from a biological molecule, causing a chain reaction.
6.2. Environmental Pollutants
Numerous environmental agents, such as photochemical pollutants, pesticides, solvents, anaesthetics, components of cigarette smoke and automobile exhaust gases, and aromatic hydrocarbons, can cause damage to cells.
Studies aimed at determining the mechanism underlying the deleterious effects of air pollution have shown that, although various substances are harmful to the body after their absorption, they are converted into radical species by intracellular detoxification processes, and many other environmental pollutants already exist as free radicals [54].
6.3. Nitrogen Dioxide
One of the most widespread radicals in the environment is nitrogen dioxide, a red-brown gas that irritates the mucous membranes and is responsible for specific pathologies affecting the respiratory system (irritation, bronchitis, allergies). It arises from the reaction of N2 with O2 during the combustion of organic material [55]. Unlike nitric oxide, dioxide is very reactive, and is emitted into the air by car and factory exhausts.
Healthy men can tolerate brief exposures at levels as low as 4 ppm without obvious lung damage. However, in asthmatics, even levels below 0.3 ppm can cause an increase in bronchial reactivity.
The dioxide is also found inside homes, where it can be produced by gas cookers, wood stoves, coal, kerosene, and by smokers, reaching levels above 1 ppm.
Nitrogen dioxide reacts with water to form nitric oxide and nitric acid. The dangerous effects of nitrogen dioxide are also related to its ability to act as a precursor to other harmful gases, such as ozone [56,57].
6.4. Ozone
Ozone forms from a photochemical reaction between O2 and NO2•, other than for O2 photodissociation. Solar radiation and high temperatures activate the reaction [58]. In a simplified way, it is composed of two phases. In the first, which requires sunlight, the photochemical splitting of the dioxide into nitric oxide and atomic oxygen takes place. In the second phase, atomic oxygen reacts with molecular oxygen forming ozone.
In highly polluted urban areas during the day, there is a significant formation of ozone which is the leading cause of the so-called photochemical smog [59]. During winter, because the low temperatures inhibit the reaction, the formation of ozone is a summer-linked problem.
Parallel to the ozone synthesis reaction, there is an opposite ozone elimination reaction in which the ozone reacts with nitric oxide, reforming the starting products. This reaction is independent of light. Therefore, an almost total elimination of ozone occurs during the night. In rural areas, where the air is cleaner, nocturnal ozone depletion is much lower [59].
The gas, which in the upper layers of the atmosphere (stratosphere) is useful for protecting the Earth’s surface from excessive irradiation of ultraviolet rays, in the lower layers of the atmosphere (troposphere) is instead a direct cause of damage to vegetation, buildings, and health. Ozone, being strongly oxidising, may: (i) cause the oxidation of metals and the depolymerisation and degradation of plastics and rubbers; (ii) interfere with the function of chlorophyll and with plant growth; (iii) cause irritation and inflammation of the eyes and the upper respiratory tract.
These phenomena, dependent on short-term exposures, rapidly cease with the cessation of exposure. Conversely, prolonged exposure to high levels of ozone can cause irreversible damage to the structure of the respiratory and cardiovascular systems.
6.5. Cigarette Smoke Radicals
Tobacco smoke is a mixture of around 3000 compounds, many of which are toxic. These include nicotine, carbon monoxide, benzene, hydrogen cyanide, free radicals, cytotoxic aldehydes, and carcinogens such as nitrosamines and benzopyrene [60].
The smoke can be separated by using special filters in a gas phase and in a particulate phase; the tar residue. A smoker inhales both the gaseous phase and a large part of the particulate fraction that penetrates the body whatever filter is used. Both the gas phase and the tar are highly oxidising and are sources of free radicals. The tar, in addition to many aromatic compounds, contains about 1017 radicals per gram, most of which are very stable and persist for hours [61]. There are at least four distinct species of radicals, including the semiquinone, that undergo redox interconversions with quinones and hydroquinones. Aqueous tar extracts generate the superoxide radical, probably by the reaction of the semiquinones with O2. On the other hand, diphenol/quinone systems can function as scavengers of superoxide in dependence of the equilibrium position and pH [60].
The gaseous phase contains radicals that are both less stable and less concentrated. These include alkoxy radicals, peroxyl radicals, and, to a lesser extent, carbon-centred radicals. It also contains aerosols, carbon particles containing adsorbed carcinogenic hydrocarbons, and other toxins, including polyphenols and quinones, which can generate ROS [61]. These particles are part of the so-called fine dust, also referred to as a particulate matter of the environment (PM), in which particulate matter is the set of all solid or liquid particles suspended in the air, excluding pure water, with microscopic dimensions [61].
PM10 is the atmospheric particulate with a diameter equal to or less than 10 µm, while PM2.5 is the finest fraction of PM10, made up of particles with a diameter equal to or less than 2.5 µm. The particle diameter is the most important parameter for characterising the physical behaviour of atmospheric particulate matter. Thus, PM2.5 is the most dangerous particulate for health and the environment as it can remain suspended in the atmosphere for days or weeks. The larger particles (from 2.5 to 10 µm) remain in the atmosphere for a few hours to a few days and are significantly less harmful to health and the environment. Moreover, the measurement of PM10 (expressed in µg/m3) as a method of assessing particulate pollution provides incomplete information since it does not distinguish coarse particles from dangerous PM2.5. Paradoxically, a high value of PM10 can correspond to the presence of a few PM 2.5-type particles and many of a larger size, a more acceptable situation than a lower-value PM10 with few coarse particles and many smaller than 2.5 µm. The observations made available by the scanning electron microscope are therefore important, for they allow us to see the particles, count them, distinguish the various families, and study their chemical composition. Cigarette smoke as well as respirable fibres and dust act synergistically in increasing the production of damaging hydroxyl radicals [61].
7. Endogenous ROS Generation Sites
There is no doubt that, although the body’s exposure to ROS resulting from exogenous sources can be remarkably high, exposure to endogenous sources is much more important and continuous. Free radicals are only occasionally formed by environmental factors, while they are continuously generated throughout the life span of every cell in the body, as by-products of normal cellular metabolism.
Several cellular sites of ROS generation have been identified, located on the plasma membrane, in the cytoplasm, in the peroxisomes, and on the membranes of the mitochondria and endoplasmic reticulum. However, under non-pathological conditions, most of these species appear to arise from reactions involving components from the mitochondrial and microsomal electron transfer chains.
7.1. Mitochondria
Reactive oxygen species can be produced in the mitochondria by components of the respiratory chain located on the inner membrane, as well as by enzymes, located both on the inner membrane and on the outer membrane.
7.2. Mitochondrial Respiratory Chain
About 95% of the oxygen that animals breathe is used by the mitochondria, where the synthesis of ATP occurs. The electrons, deriving from the electron carriers, reach Cox and are used to operate a tetravalent reduction in O2. Cox has a very high affinity for O2 which allows it to work even at oxygen concentrations lower than one millimetre of mercury (1 mm Hg) and when the mitochondrial energy production is at low oxygen concentrations [62]. This aspect is important because the oxygen concentration at the mitochondria level can be very low. Indeed, while in the fully oxygenated blood that leaves the lungs, the partial pressure of O2 (pO2) is about 100 mm Hg, pO2 rapidly falls into the tissues when oxyhaemoglobin releases O2. Cells in a tissue may be in the presence of extracellular O2 concentrations that provide a pO2 as low as 5–15 mm Hg and a further drop in the local pO2 around individual oxygen-consuming mitochondria verifies [62].
Cox’s ability to work at very low O2 concentrations and the fact that its activity is not associated with a ROS release may represent a means of minimising cellular O2 damage [63].
Although Cox does not release oxygen radicals in the solution, evidence of H2O2 production by submitochondrial particles has been available since 1966 [64]. The observation that mitochondria contain Mn-dependent superoxide dismutase (SOD) [65] and produce O2•− [66] suggested that mitochondria production of H2O2 is due to superoxide dismutation.
Some components of the respiratory chain, although they transfer a large mass of electrons to the next component in the chain, they also transfer a small number of electrons directly to O2 molecules, causing its reduction into superoxide (univalent reduction). Since these components are unable to retain partially reduced forms of oxygen in their sites, the superoxide radical is released and partially transformed into H2O2 by spontaneous or catalysed dismutation by mitochondrial SOD [67].
Theoretical and experimental reasons suggest that the production of H2O2 by the mitochondria is the result of the dismutation of superoxide rather than the direct transfer of two electrons to O2. The bivalent reduction in O2, although being thermodynamically favoured compared to the univalent one, is kinetically disadvantaged since it requires the inversion of the spin. O2•− production is detectable only using submitochondrial particles which do not contain SOD [66]. Part of the O2•− produced by intact mitochondria appears on the cytosolic side of the inner membrane and can be measured by following the oxidation of adrenaline or by stimulating the formation of H2O2 with the addition of SOD [68].
Moreover, O2•− can also undergo other types of reactions besides dismutation. Indeed, the reaction of O2•− with nitric oxide (NO∙), which leads to the formation of the highly reactive peroxynitrite, can effectively compete with SOD-induced dismutation [69], having a rate constant of one order of magnitude higher than that of the SOD. Normally, due to the low concentration of NO∙, the likelihood of this reaction occurring is low. A higher concentration of NO∙ may produce mitochondrial nitrosative damage which may be exacerbated by the MnSOD damage due to peroxynitrite [69].
The product of the superoxide dismutation, the H2O2, can also be detected in intact mitochondria, and in this case the measured H2O2 is only a fraction of that produced and metabolised by enzymes (catalase, thioredoxin system, glutathione peroxidase) or removed by the reaction with mitochondrial components containing iron complexes which transforms it into an •OH radical via the Fenton reaction [67].
Thus, the measurement of H2O2 generation with intact mitochondria reflects the net result of the O2•− production, its dismutation, its reaction with NO•, and the competition between H2O2 destruction and diffusion. Despite these limitations, the measurements of the quantity of peroxide, which can diffuse through the mitochondrial membrane, can provide indications of the relative levels of ROS production in different metabolic conditions [70]. While Cox activity saturates at low O2 concentration [62], the rate of electronic loss (and therefore the O2•− production) increases at high concentrations of O2 [71]. In slices of rat lungs exposed to pure oxygen, about 90% of the total O2 consumed leads to ROS formation, but when the concentration is lowered even up to 85%, ROS formation lowers to 18% of the total O2 consumed [71].
Although the numbers of radicals produced by mitochondria in vivo and in vitro is not easily determined, it is nevertheless clear that mitochondria produce ROS at rates that depend on their metabolic state [72]. The production rate is higher during basal respiration, i.e., in the presence of respiratory substrates but in the absence of adenosine diphosphate (state 2), characterised by a low oxygen consumption, than during ADP stimulated respiration (state 3), characterised by a higher rate of oxygen consumption [72].
The rate of formation of ROS in the respiratory chain is controlled by the law of mass action and, with the same O2 concentration, increases when the flow of electrons slows down, which increases the concentration of electron carriers in the reduced form capable of donating an electron in oxygen [72].
As electrons flow through the respiratory chain, the released energy converts into a proton gradient across the mitochondrial membrane. This gradient, in turn, is dissipated through the ATP synthetase complex that leads to the synthesis of ATP [72]. In the absence of ADP, the movement of H+ through the ATP synthetase ceases, and the gradient of H+ increases, causing a slowing of electron flow, an increase in the degree of reduction in the respiratory chain, and the generation of superoxide [72].
Physiological uncoupling occurs in brown adipose tissue (BAT) mitochondria, in which a mitochondrial membrane protein, named uncoupling protein 1 (UCP1), participates in the dissipation of the energy associated with the proton gradient as heat, uncoupling electron flow from ATP synthesis. This protein decreases both mitochondrial membrane potential and H2O2 generation [73].
At a constant pressure of O2, the production rate of ROS increases as a function of the degree of reduction in the auto oxidable electronic carriers.
Univalent oxygen reduction sites were initially identified in Complex I [74,75] and Complex III [76]. Subsequently, ROS production sites were identified in mitochondrial complex II [77,78] and in various mitochondrial enzymes [79,80]. Nowadays, the exact sites of mitochondrial ROS generation and their contribution in vivo are not a settled question.
7.3. Mitochondrial Oxidoreductases
Given the moderate redox potential of the O2•−/O2 pair (E° = −0.33 V), the univalent reduction reaction of O2 is thermodynamically favourable for numerous mitochondrial oxidoreductases. A measurable production of ROS by at least nine enzymes has been reported, the best known of which are monoamine oxidase [81,82], α-glycerophosphate dehydrogenase [83], and aconitase [84,85]. The enzymes are ubiquitously present in mammalian mitochondria, but their expression and their ability to produce ROS vary widely between tissues and animal species. Although they are sources of ROS in experiments with isolated enzymes or with mitochondria, their contribution to mitochondrial ROS production under physiological conditions is not known.
7.4. Endoplasmic Reticulum
In the endoplasmic reticulum (ER), the production of ROS is linked to the activity of two electronic transport systems with distinct functions and the folding process of proteins.
7.5. Metabolism of Xenobiotics
The first electronic transport system participates in the metabolism of xenobiotics which can be verified in two phases. The first phase (Phase I) involves a flavoprotein (NADPH-cytochrome P450 reductase), each molecule of which contains a flavin adenine mononucleotide and a flavin adenine dinucleotide, as well as cytochromes collectively known as cytochrome P450 [86]. It has been shown that subcellular fractions isolated from various tissues containing endoplasmic reticulum (microsomal fractions) rapidly produce H2O2 when incubated with NADPH [87,88].
Cytochrome P450 enzymes (CYPs) are a heterogeneous group of monooxygenase that play a crucial in the oxidative metabolism of many lipophilic compounds, xenobiotics, biosynthesis of sterols, fatty acids eicosanoids vitamins, etc. [89,90], and their high concentrations are found in the endoplasmic reticulum of the liver where many chemicals are metabolised [91,92]. Significant concentrations are also found in the lungs [93], adrenal medulla [94], kidneys [95], and small intestine [96]. In any case, one oxygen atom binds to the substrate, and the other forms water, so this reaction is known as a monooxygenase reaction or mixed-function oxygenase reaction. Phase I reactions introduce a polar group into a lipophilic substrate using an electron transport system, increasing the substrates’ polarity, and aiding in excretion. The process is necessary so that insecticides, hydrocarbons, and drugs are solubilised in water and then excreted in the urine or so they undergo conjugation and are excreted in the bile. During the CYP catalytic cycle, two “shunts”, which can generate ROS and interrupt substrate oxidation, exist [97]. The first ROS released is O2− and the second is H2O2 [98]. Many factors can affect the coupling efficiency of a given CYP, among which the reaction with the substrate and the pH or O2 concentration play an important role [98]. Moreover, the different CYP isoforms have different rates of reaction uncoupling, and they can also be dependent on the substrate [90].
The reactions of the second phase (Phase II) are conjugation reactions, in which an endogenous molecule is added to the product of the Phase I reaction, sometimes directly to the xenobiotic [86]. Phase II reactions, for example, are conjugations with glutathione (GSH) catalysed by glutathione-S-transferase (GST), and conjugations with glucuronic acid, catalysed by glucuronyl transferase. Other examples are sulfation (addition of a sulphate group catalysed by sulfotransferase), methylation (addition of a methyl group, usually provided by S-adenosyl-methionine), and acetylation (addition of an acetyl group catalysed by N-acetyltransferase).
Generally, the products of the reaction of the CYPs are less toxic than the substrates. However, the reaction often determines the transient formation of oxidative intermediates, such as epoxides, or the generation of highly reactive products. For example, the drug acetaminophen via CYP oxidation converts into a reactive intermediate, N-acetyl-p-benzo-quinone-imine (NAPQI), that is conjugated with glutathione, by glutathione S-transferase (GST), that inactivates NAPQI [99]. Acetaminophen overdose and toxicity are caused by the generation of excessive amounts of NAPQI that are not detoxified by GST, due to glutathione exhaustion, and can be treated with the administration of N-acetylcysteine [98].
7.6. Unsaturation of Fatty Acids
The second electronic transport system, in which the flow of electrons proceeds from NADH to cytochrome b5, operates the desaturation of fatty acids, i.e., the introduction of C=C double bonds into these substances.
The system consists of three proteins: a flavoprotein (NADH-cytochrome b5 reductase), a cytochrome b5, and the desaturase enzyme [100]. The first protein, which contains FAD as a prosthetic group, catalyses the transfer of electrons from NADH to cytochrome b5 through the flavoprotein, and the reduced cytochrome b5 donates electrons to the desaturase enzyme. This enzyme, also named cyanide sensitive factor (CSF) because it is inhibited by cyanide, contains only one non-heme iron per polypeptide chain and can desaturate acyl-CoA to enoyl-CoA using one molecule of O2 and produce two molecules of H2O.
Both cytochrome b5 and flavoprotein can transfer electrons to O2 to form O2−, and this may be an additional source of in vivo ROS [101].
7.7. Protein Folding
The conformation of most of the proteins synthesised in the endoplasmic reticulum (ER) is stabilised by the formation of intramolecular disulphide bonds, a process that requires the oxidation of free sulfhydryl groups. The intraluminal medium of the ER seems to ensure the realisation of the oxidative folding of proteins maintaining a GSH/GSSG ratio lower than that of the cytosol [102,103]. The stability and functionality of the newly synthesised proteins are guaranteed by the formation, reduction, and isomerisation (i.e., the exchange of -SH groups with -S-S- groups) of disulphide bonds between pairs of cysteines. The enzyme that catalyses these reactions is the protein disulphide isomerase (PDI), a family of ER proteins characterised by the presence of thioredoxin-like domains [104].
How the ER disposes of the electrons deriving from the oxidative formation of disulphides remained unknown for a long time, until the discovery, in S. cerevisiae, and subsequently in mammals, of a protein that transfers disulphide bonds to PDI (Ero1p in eukaryotes and DsbB in bacteria) [105].
Ero1p functions as a PDI oxidase: oxidising equivalents flow from Ero1p to the substrate protein via PDI, through the direct dithiol–disulphide exchange between PDI and Ero1p [105]. Furthermore, Ero1p has an FAD group and is very sensitive to small changes in the physiological levels of free FAD [105]. The oxidative folding of proteins does not use oxidised glutathione (GSSG) as a source of oxidant equivalents. This role is played by molecular oxygen, which is used by Ero1p as its preferred electron terminal acceptor. Consequently, Ero1p directly couples the disulphide formation to the consumption of O2, while its activity is modulated by the luminal levels of free FAD [105].
However, in bacteria, the oxidative folding is conveniently coupled to O2 via the respiratory chain, while in eukaryotes, where the oxidative winding and respiration are confined to separate organelles, Ero1p uses an FAD-dependent reaction to pass electrons directly to O2 [106]. As a result, the activity of Ero1p can generate reactive oxygen species and, thus, provide an additional source of oxidative stress, suggesting that its activity must be regulated according to the wind load [107]. Using theoretical calculations, it has been estimated that the oxidation of protein thiols via PDI and Ero1p in the endoplasmic reticulum could account for up to 25% of cellular ROS produced during protein synthesis [108].
7.8. Peroxisomes
Peroxisomes, organelles present in almost all eukaryotic cells, are other sources of ROS [109]. They can reduce oxygen to water through a two-phase mechanism that involves H2O2 as an intermediate [110]. In the first phase, the substrates (electron donors) include uric acid, amino acids, glycolic acid, and α-hydroxy acids according to the cell type. The electron donors in the second phase are either an H2O2 molecule, which is oxidised to oxygen, or a substrate such as methanol, ethanol, formaldehyde, phenol, or nitrite ions that can be oxidised by catalase [110].
Over time, more than 50 enzymes have been discovered in peroxisomes. These enzymes are involved in various metabolic pathways, including cholesterol biosynthesis, β-oxidation of fatty acids, detoxification of xenobiotics, and ROS [111]. Peroxisomes contain superoxide dismutase, which metabolises superoxide, glutathione peroxidase, which metabolises both H2O2 and membrane hydroperoxides.
Peroxisomes contain high concentrations of catalase, so whether a loss of H2O2 from these organelles contributes significantly to the intracellular concentration of the peroxide under normal conditions is not known. One of the characteristics of peroxisomes is their inducibility by xenobiotics which leads to their remarkable proliferation with a concomitant synthesis of some of their enzymes, particularly those of β-oxidation. Peroxisome proliferators include structurally diverse compounds, including hypolipidemic drugs, such as fibrates, free fatty acids, herbicides, and many environmental pollutants. The selective transcription of peroxisomal genes by these substances is mediated by the peroxisomal proliferator-activated receptor α (PPAR-α), which belongs to the family of nuclear transcription factors [112]. While the expression of β-oxidation genes is significantly induced (10–30 times), the maximum induction of catalase does not exceed 1–2 times. Therefore, it has been suggested that the increase in H2O2 release linked to these disproportionate inductions is one of the causes of the increased onset of liver tumours recorded following prolonged exposure of rodents to peroxisome proliferators [113].
7.9. Cytoplasm
There is a small number of cytoplasmic oxidases capable of producing the superoxide radical. These include xanthine oxidase [114], tryptophan dioxygenase, and indolamine dioxygenase [115].
One of the most studied is xanthine oxidase, an enzyme involved in the breakdown of purines. In mammals, this enzyme is ubiquitously expressed and shows the highest specific activity in the liver and mucosa of the small intestine. The enzyme converts hypoxanthine into xanthine and xanthine into uric acid, and electrons are transferred from the substrates to the O2, forming O2•− as well as, probably, singlet oxygen [114]. In vivo, the enzyme is present as xanthine dehydrogenase, which transfers electrons from substrates to NAD+. Thus, the metabolism of xanthine or hypoxanthine does not produce the superoxide ion normally. However, xanthine dehydrogenase is converted to xanthine oxidase by oxidation of cysteine residues or irreversibly by limited proteolysis involving Ca2+-stimulated proteases in oxidatively damaged tissues [116].
7.10. Cell Membranes
The plasma membrane is a critical site of radical reactions for several reasons. Free radicals generated outside the cells must cross the plasma membrane before reacting with other cellular components, and some of them can initiate toxic reactions at the membrane level. Hydrogen peroxide can cross membranes as easily as water. The charged superoxide molecule can cross membranes and enter cells through transmembrane anion channels. Furthermore, the superoxide can react with the H+ ion forming the hydroxy peroxyl radical HO2•, which is a stronger oxidant than O2−• and is soluble in lipids. Thus, it can be expected to exert toxic effects on lipids and on the hydrophobic core of membrane proteins.
7.11. Lipoxygenase and Cyclooxygenase
Enzymes associated with membranes, such as lipoxygenases and cyclooxygenases, involved in the metabolism of arachidonic acid (C20:4, ω-6, AA) and of other polyunsaturated acids, can produce ROS.
The metabolism of AA leads to the formation of a large and complex family of biologically active lipids produced by almost all mammalian cells, with the exception of red blood cells. The metabolites include prostaglandins, prostacyclins, thromboxanes, and leukotrienes. These compounds are collectively known as eicosanoids, as they are all 20-carbon compounds [117]. The cytosolic phospholipase A2 mediates the release of AA from glycerolphospholipids of the nuclear envelope and plasma membrane. AA undergoes insertion of O2 through a radical mediated mechanism by lipoxygenase (LOX, linear pathway) and cyclooxygenase (Cox, cyclic pathway) [118].
AA is metabolised by LOXs, which are non-heme iron-containing enzymes, into hydroperoxyeicosatetraenoic acids (HpETEs). HpETEs are reduced into hydroxyeicosatetraenoic acids (HETEs) [119].
The major cyclooxygenase-derived eicosanoids are prostaglandins (PGs) and thromboxane (TX). Cox enzymes catalyse the first two steps of the enzymatic reaction, including cyclooxygenase (dioxygenase) and peroxidase activity, to convert AA into PGH2 [120]. PGH2 is the precursor of PGs and TX, whose production depends on the differential expression of isomerases and PG synthases in different tissues [121]. Cox includes two isozymes: COX-1 and COX-2. The first is the constitutive isoform, responsible for the low PGs synthesis required for cell homeostasis, and the latter is inducible by many extracellular stimuli, including cytokines and growth factors, during chronic inflammation [122,123].
The oxidation of arachidonic acid by Cox produces an oxygen (probably a haemoprotein) radical.
The metabolism of arachidonic acid by LOX also produces carbon-centred radical intermediates, which are due to the extraction of methylene hydrogen, and involved in the degradation processes of membrane lipids.
7.12. NADPH Oxidase
Currently, there is a growing body of evidence that a family of membrane-bound cellular enzymes called NADPH oxidase (NOX) is a significant source of ROS [124,125].
The primary function of NOX is to catalyse the transfer of electrons across the membrane from NADPH to molecular oxygen by generating superoxide or hydrogen peroxide.
NOX2, also known as phagocytic NADPH, is the prototype of NOX and has the role of defence against bacteria that are killed by superoxide. In addition to its presence in phagocytes, NOX2 is expressed at lower levels in many cell types, including cardiomyocytes, endothelial cells [126], neurons [127], and pancreatic β cells.
During activation, which occurs following the phosphorylation of the p47phox regulatory subunit [128,129], the G protein binds GTP, migrating to the plasma membrane with the cytosolic components forming the active complex. It removes electrons from the NADPH, oxidising it to NADP+ inside the cell and transferring them to the FAD. Subsequently, it releases the electrons of the two heme groups, which eventually transfers them to the oxygen on the opposite side of the membrane, forming the superoxide radical.
Some isoforms of NOX, such as NOX4 which is strongly expressed in the kidney but is also present in vascular cells and neurons [130], and DUOX1 and DUOX2 which are particularly important in the thyroid [131] although they are also found in the epithelia of the digestive and respiratory tracts, mainly produce H2O2, while the primary product may be superoxide, which undergoes rapid dismutation to hydrogen peroxide. Furthermore, NOX4 is distinguished from the rest of the family since it appears to be constitutively active, and its activity is therefore mainly regulated by the level of expression.
8. ROS-Induced Oxidative Damage
Biological systems have an integrated antioxidant system to counteract the damaging effects of ROS (Figure 2). This system includes low molecular weight enzymes and antioxidants. The discovery of the superoxide dismutase (SOD) enzyme by McCord and Fridovich [132] allowed us to define the mechanisms underlying oxygen toxicity. SOD transforms O2−• into H2O2, the latter being metabolised by various enzymes such as catalase, glutathione peroxidase (GPX), and peroxiredoxin (Prx) [72].
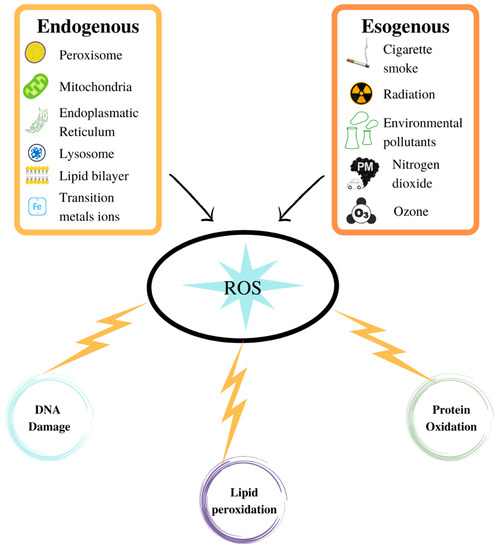
Figure 2.
ROS production in aerobic organisms can be better verified via an exogenous source than through normal physiological processes. ROS can damage the biological macromolecules.
GPX and Prx metabolise most of the H2O2 and their activities depend on the thiol groups of the cysteine residues of reduced glutathione (GSH) and thioredoxin (Trx), respectively, to reduce H2O2 [72]. The enzymes glutathione reductase (GR) and thioredoxin reductase (TrxR) serve to maintain the sulfhydryl groups of GSH and Trx in the reduced form and require NADPH as a source of electrons for their activity [72]. Low molecular weight antioxidants include endogenous molecules and antioxidants that must be introduced with food. These molecules can be lipo- or hydro-soluble and comprise endogenously produced molecules such as GSH, or molecules introduced with food such as vitamin E and C polyphenols [72]. Vitamin E is the main liposoluble antioxidant that is localised in all cell membranes, including the mitochondrial one, protecting it from oxidative damage in several physio pathological conditions [133].
A low ROS production is important because ROS are also signalling molecules, but their high production can determine a reduction in redox signalling and/or damage to biomolecules [134]. Oxidative stress originates from the imbalance between ROS production and the ability of the antioxidant system to counteract their oxidative action. In this case, the redox signalling is altered and oxidative damage to macromolecules increases [134]. ROS can damage lipid proteins and DNA [135].
Mono- and poly-unsaturated lipids are the molecules more prone to oxidation, which results in the formation of lipid hydroperoxides (LOOHs) as primary nonradical-reaction products. LOOHs may undergo degradation into various products that have been implicated in vital biological reactions, and thus in the pathogenesis of various diseases [136]. The molecular changes caused by the reactions of products of lipid peroxidation alter the structure and function of proteins and DNA and are responsible for the cytotoxicity induced by these molecules [136].
ROS can oxidise DNA, modifying the structure of nucleotides and inducing double-stranded break formation which can favour genomic instability [137]. Moreover, ROS can directly oxidise nucleoside bases (e.g., formation of 8-oxo guanine) [138], which can determine G-T or G-A transversions if unrepaired. The base excision repair pathway recognises and repairs oxidised bases, but when oxidation occurs simultaneously on opposing strands, the base excision repair system can determine the generation of a DNA double-strand break which may be mutagenic due to chromosomal rearrangements or loss of genetic information [139]. ROS accumulation also induces mitochondrial DNA lesions, strand breaks, and degradation of mitochondrial DNA [140].
ROS can directly or indirectly affect protein functionality. Direct action of ROS determines the nitrosylation, carbonylation, disulphide bond formation, and glutathionylation of proteins [141]. Indirectly oxidative damage to proteins consists of their conjugation with products of the breakdown of oxidised fatty acids [133]. Oxidatively damaged proteins undergo site-specific modifications of amino acids, fragmentation of the peptide chain, aggregation of cross-linked reaction products, altered electric charge, and increased susceptibility to proteolysis [141].
9. ROS as Regulators of Important Physiological Processes
The discovery that a hydroxyl radical induces the activation of guanylate cyclase and increases the levels of cyclic guanosine mono phosphate [142], as well as the fact that hydrogen peroxide mimics the signalling activity of insulin [143], suggested that ROS can not only be dangerous but also involved in the regulation of important physiological processes.
More insights into this idea have been obtained thanks to the discovery in the adipose cell membrane of the enzyme NADPH oxidase, whose activity is stimulated by insulin [144] which produces the superoxide from which H2O2 is generated. The discovery suggested that insulin, by stimulating H2O2 production, induces the oxidation of membrane sulfhydryl groups that play a role in activating the insulin signalling pathway [145].
Subsequently, low levels of O2−• and H2O2 proved to activate the proliferation of both hamster [146] and human [147] fibroblasts in culture. From these initial data, several experimental pieces of evidence have suggested that ROS, other than acting as insulin-activated second messengers and stimulating cell proliferation in different cell types [148], also regulate a wide range of physiological functions. They control the activation of genes acting through the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [149], the biosynthesis of prostaglandins [150], and embryonic development [151]. Moreover, they also function as signalling molecules inside the cell and between cells throughout life [152].
ROS are also involved in muscle contraction, as suggested by the observation that they are present at low levels in skeletal muscle under basal conditions [153] and that the antioxidant supplementation to diaphragmatic fibre bundles reducing their content determines the inhibition of muscle contraction [154].
ROS also contribute to the regulation of blood pressure [155] and are involved in the maintenance of cognitive function [156]. It has been also evidenced that, even if the increase in the rate of formation of ROS is considered an important factor involved in the determination of life span [157], some interventions able to increase longevity are linked to enhancement in the ROS production that can induce cellular pathways that, in turn, reduce cellular degeneration and promote healthy aging [158]. All these observations showed that the living being has not only adapted to the presence of free radicals but has also developed useful mechanisms to advantageously use these species in a myriad of physiological functions.
In all organisms ranging from bacteria to mammals, ROS can regulate signalling pathways initiated by factors sensitive to the cellular redox state that can induce the increased expression of antioxidant enzymes. This increase in antioxidant defences allows cells to survive to exposure to oxidants that would be lethal under normal conditions. For example, it has been found that the irradiation of cells and tissues of mice [159] and rats [160] induces the expression of antioxidant enzymes, thus activating an adaptive response that increases their resistance to radiation by increasing the antioxidant capacity.
Bacteria present a sophisticated molecular mechanism that allows them to check the cellular level of oxidant and to induce the expression of the genes coding for the antioxidant enzymes, if necessary. The regulation of genes in response to oxidative stress has been extensively studied using the bacterium Escherichia coli as a model. These studies have shown that the fundamental principles of the response to oxidants are conserved from bacteria to eukaryotes, even if the molecules and mechanisms involved in ROS sensing are variable according to the organism as well as the severity and conditions of stress.
In E. coli, some systems stimulate an increase in the cellular content of enzymes that metabolise superoxide or peroxides, repair DNA and proteins, and reduce the deleterious effects of oxidative mutagens [161,162,163]. The induction of these systems is an adaptive response to oxidative stress due to exposure to oxidising agents at a non-toxic level or to ionising radiation, which makes the organism more resistant to further stress even with levels of oxidants that would be toxic under basal conditions.
In Salmonella typhimurium, the oxyR gene has been identified as a regulator of acquired resistance to oxidants. oxyR is involved in the expression of nine proteins, including catalase, alkyl hydroperoxide reductase, glutathione reductase, and MnSOD, in which levels increase upon exposure to hydrogen peroxide to sublethal levels [164].
In mammals, ROS can activate the transcription of genes that determine cell survival in two ways: employing transcription factors, which interact with specific DNA motifs on the promoters of target genes, or by activating cascades of mitogen-activated protein kinases, which activate transcription factors which ultimately activate the transcription of the target gene [165]. Clear evidence shows that the cellular level of ROS is correlated to the cellular antioxidant levels [166]. One of the main factors involved in this relationship is the factor 2-related erythroid nuclear factor 2 (Nrf2), which regulates the expression of genes encoding antioxidant and detoxifying enzymes [167]. Nrf2 links to a protein named small musculoaponeurotic fibrosarcoma (Maf) and acts through the binding to the antioxidant response element (ARE) of the promoter region of the target genes [168]. Its activity is linked to its subcellular localisation that, in turn, depends, at least in part, on the binding with specific residues of the cysteine of the protein Kelch ECH associating protein 1 (Keap1) [169].
Keap1 is the major negative regulator of Nrf2, regulating its ubiquitination and degradation [170,171]. The increase in ROS levels or the presence of Nrf2 activating compounds induce the oxidation of cysteine residues in Keap1, thus inducing the loss of its ubiquitin ligase activity and the release of Nrf2. Nrf2 is then phosphorylated and reaches the nucleus where it binds to the small protein of muscle aponeurotic fibrosarcoma (Maf) forming a heterodimer capable of binding to the ARE located in the promoter regions of genes regulated by Nrf2 [168,172] Interestingly, the glycogen synthase kinase-3 (GSK-3β), a serine/threonine protein kinase that participates in glycogen metabolism, cell proliferation, stem cell renewal, apoptosis, and development, regulates the relocation of Nrf2 to the cytosol and its degradation [173]. The activation of GSK-3β is induced by hydrogen peroxide [174]. Thus, GSK-3β regulation of Nrf2 activity is oxidant-sensitive as well.
ROS can activate many cellular processes, among which the proliferation and survival, interacting with signalling molecules among which mitogen-activated protein kinases (MAPKs), phosphoinositide 3-kinases (PI3K), phosphatase and tensing homolog (PTEN), and protein tyrosine phosphatases [175].
9.1. Redox Signalling
Although the role of ROS as cell-damaging and signal-transduction agents is defined, some aspects of these roles are still under study.
Variations in the concentration of ROS seem to be at the basis of their role as molecules capable of regulating cellular functions or as harmful substances. ROS act as regulators of signalling processes at low concentrations, while they act as damaging agents as they can damage and, therefore, inactivate cellular molecules at moderate or high concentrations. Shifting concentrations from positive to damaging effects is an unsolved question.
Moreover, how the specificity of ROS signalling is obtained remains unknown. In fact, ROS show indiscriminate reactivity while cell signalling verifies through the non-covalent bond between the ligand and its receptor due to the complementarity of the molecule shapes. ROS act in signalling by reactions with specific atoms of target proteins that result in the covalent modifications of the proteins. Thus, ROS recognise their targets at the atomic and non-macromolecular level, and this extends the number of potential ROS receptors. Therefore, it is not known how the specificity of ROS signalling is achieved.
9.2. Source of ROS Acting as Signalling Agents
It is not easy to identify a single source of ROS which act as signalling agents. On the other hand, it is also not easy to identify the main cell source of ROS. Mitochondria are considered to be the most important cellular source of ROS in both physiological and pathological states [176]. They seem to play a role in various diseases and in the ageing process [177], even if the demonstration that they really are the principal source of ROS is absent [178]. On the other hand, the available data show a strong interaction between the cellular ROS sources [179,180,181], which makes it difficult to understand which is the main one. The control of signalling by ROS requires the colocalization of their sources and targets. This colocalization allows for the fine control of the signalling reaction kinetic by controlling the local concentrations of the reagents. For example, NOX that affects the tyrosine kinase receptor-linked signalling colocalized in the cell membrane together with its targets, phosphatases, kinases, and oxidation of other molecules, avoids, i.e., the nuclear or mitochondrial nucleotides [182,183]. The compartmentalization of ROS producers that play a role in signalling has been identified in various cell types [184]. Localization in the endoplasmic reticulum of NOX4 and of its target, the protein tyrosine phosphatase 1B (PTP1B) [185], and the generation in the phagosomes of hypochlorous acid from myeloperoxidase for defence against pathogens [186], are other examples of colocalization of ROS sources and targets.
9.3. ROS Species and Signalling
Due to their high instability and the action of the cellular antioxidant defence system, most ROS do not spread far from the production site. The hydroxyl radical, whose half-life is about 10−9 s for its high reactivity, has a small range of action (about 30 A). Hydrogen peroxide has a half-life of about 1 ms and can diffuse over longer distances [187]. Thus, a hydroxyl radical generated within the mitochondrion is unlikely to exert effects on other cell sites.
Another important aspect is that while the hydroxyl radical exhibits a broad reactivity towards all biological molecules, O2−• and H2O2 appear reactive towards specific preferential biological targets. Despite having a generalized reactivity, the hydroxyl radical has been involved in the SOD-induced activation of the enzyme guanylate cyclase [188]. It has been suggested that the formation of H2O2 produces •OH due to the reaction with superoxide and that •OH can activate guanylate cyclase [188]. The involvement of the hydroxyl radical in the activation of guanylate cyclase by SOD has been later questioned, and it has been shown that this activation [188] depends on the elimination of superoxide. Excluding this example of •OH as a signalling molecule, the current idea is that this radical does not play a specific role in signalling.
9.4. H2O2
The major signalling molecules are O2−• or H2O2.
Superoxide can oxidize thiols into thiyl radicals, and this can initiate a chain reaction showing a low-rate constant (approximately 103 M−1 s−1 at pH 7.4) [189], which is negligible compared to the reaction rate constant catalysed by both cytosolic and mitochondrial SOD (about 109 M−1 s−1) [190]. O2−• generated outside the cell from membrane NOX2, or endothelial NOX2, passes into the cell and acts as a signalling molecule [191]. Once inside the cell, superoxide is rapidly metabolized by SOD to form H2O2 and O2. No superoxide target has been identified so far and it appears to act only as a precursor of H2O2. The latter can act as a second messenger due to its production and degradation, which provides specificity for the time and place of production, as well as its chemistry which provides specificity for thiol oxidation, which is finely controlled.
H2O2 can initiate cell signalling by modifying either molecule in target proteins or the intracellular redox state [192], although the distinction between these mechanisms is not clear.
The thiol groups of cysteine residues are the main targets of H2O2, and their oxidation is well described. The oxidation of sulfhydryl groups determines the genesis of some different products, including sulfenic acid derivatives (-SOH) [193], which can transform by subsequent oxidation into sulfinic (-SO2H) and possibly sulfonic (-SO3H) acid. Furthermore, they can determine the formation of disulphide bonds with adjacent cysteines (-S-S-) [193,194] and generate some adducts that react with NO• (S-nitrosylation) or GSH (S-glutathionylation) [195]. The modifications can be cancelled by reducing systems such as thioredoxin and peroxiredoxins, apart from sulfonic and, to a lesser extent, sulfinic acid [196].
The formation of sulfinic acid requires a further reaction with H2O2 that possibly occurs in oxidative stress conditions and not during physiological signalling. This reaction is also verified for sulfonic acid which does not exist under biological conditions. It can be argued that only the sulfenic acid derivative and the disulphide are relevant to signalling the oxidation state of cysteine.
The changes induced by the oxidation of the thiol groups cause structural and functional changes in the proteins and may modify the activity of an enzyme whether the critical cysteine is in the catalytic domain [197] or the affinity of a transcription factor for DNA if the critical cysteine is in the DNA binding domain [198]. Redox processes regulate many proteins, including transcription factors, molecular chaperones, and protein tyrosine phosphatases. Therefore, the redox state of the thiol groups constitutes a molecular switch that can reversibly activate or deactivate the function of a protein. This action is like the phosphorylative regulation of proteins due to the binding of a phosphate group by a protein kinase and its cleavage by a protein phosphatase [199].
Moreover, the two signalling mechanisms crosstalk each other and show a sequential partnership in some circumstances of cell regulation. The study of the system Trx/apoptosis signal-regulating kinase-1 (ASK1) provided proof of the link between the cellular redox state and phosphorylative process [200]. ASK1, a member of the family of mitogen-activated protein kinase kinase kinases (MAPKKK), is implicated in the activation of stress-activated protein kinases, among which p38 and JNK [201]. The antioxidant protein Trx forms a complex with ASK1 and inhibits its activity [200]. The increase in the levels of ROS due to tumour necrosis factor stimulation induces the dissociation of Trx from ASK1 and the increase in ASK1 activity [201].
Regarding the way changes in the cellular redox state initiate cell signalling, it should be underlined that, with respect to the extracellular environment, the cytosol is usually maintained under “reduction” conditions. Under normal conditions, the maintenance of the redox state of the cytosol is due to the ability of redox buffer systems of intracellular thiols, such as GSH and Trx, which counteract cellular oxidative stress by reducing H2O2.
The activities of the enzymes GSH reductase and Trx reductase serve to keep the high content of the reduced forms of GSH and Trx, respectively. GSH and Trx participate in cell signalling processes. Indeed, GSH regulates redox signalling through the modification in both total GSH levels and the ratio between its reduced and oxidised forms, while Trx regulates the activity of specific proteins by directly binding them [192].
10. Conclusions
The evolution of aerobic organisms has required the concomitant evolution of an antioxidant defence system to counteract the toxic effects of oxygen and the metabolites produced during its metabolism.
Within the organism, there are multiple sources of reactive oxygen species. Some of these are constitutively expressed, such as mitochondria and NADPH oxidase, while others are inducible. Additionally, some sources are physiological, while others, such as xanthine oxidase, are generated by pathological conditions, such as hypoxia–reperfusion.
However, it is now known that reactive oxygen species are not only harmful molecules for the organism but also necessary for the proper functionality of the biochemical machinery and communication inside and among the cells. Generally, the physiological effects of ROS depend on their low content.
There are important implications for ROS as physiological signalling molecules that impact therapeutic strategies. Antioxidants have long been considered potential therapeutics for several conditions involving oxidative stress. In general, trials using antioxidants likely failed, in part, due to the unintentional inhibition of important basal and adaptive ROS signalling pathways. It has even been argued that taking antioxidants as daily dietary supplements might also disturb these ROS pathways in ways that are not beneficial nor harmful [202].
The idea that is currently emerging is that while the administration of antioxidants in basal conditions can prevent the induction of beneficial adaptations for the organism, the administration of antioxidants under conditions of oxidative stress can prevent or mitigate the deleterious effects due to oxidative stress. For example, antioxidant supplementation to rats during a program of physical training can interfere with the training-induced metabolic adaptations in several tissues including the liver [203], muscle [204], and heart [205]. Conversely, antioxidant supplementation in a condition of oxidative stress such as those induced by thyroid hormone administration reduces the thyroid hormone-induced alteration of heart rate [206] and prevents the alteration in insulin sensitivity due to the hormones in the liver [207].
Cancer cells show an elevated level of oxidative stress, and this has been proposed to orchestrate tumorigenesis and tumour progression through direct or indirect mechanisms [208]. However, high doses of antioxidants have shown to contribute to tumour progression [209]. Cancer cells may be more sensitive to ROS than normal cells and it has been suggested that increasing oxidative stress by exogenous ROS generation therapy that selectively kills cancer cells without affecting normal cells could be a strategy against cancer progression [210]. However, the role of ROS in the cancer biology is very complex as they play both pro-tumorigenic and anti-tumorigenic roles [211].
Significant steps in the research field of antioxidant and ROS actions are being undertaken, as a lot remains to be understood. Certainly, antioxidants are not the panacea for all ills and their careful use is necessary to avoid incurring cell, organ, and whole organism dysfunctions.
Author Contributions
Conceptualisation, writing—original draft preparation, P.V.; writing—review and editing, P.V., G.N. and G.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: New York, NY, USA, 2015. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Khademian, M.; Imlay, J.A. How Microbes Evolved to Tolerate Oxygen. Trends Microbiol. 2021, 29, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Blankenship, R.E. Molecular Mechanisms of Photosynthesis, 2nd ed.; Wiley-Blackwell: Chichester, UK, 2014. [Google Scholar]
- Overmann, J.; Garcia-Pichel, F. The phototrophic way of life. In The Prokaryotes: Prokaryotic Communities and Ecophysiology; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 203–257. [Google Scholar]
- Bolton, J.R.; Hall, D.O. Photochemical conversion and storage of solar energy. Annu. Rev. Energy 1979, 4, 353–401. [Google Scholar] [CrossRef]
- Blankenship, R.E. Early evolution of photosynthesis. Plant Physiol. 2010, 154, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.W.; Hemp, J.; Johnson, J.E. Evolution of oxygenic photosynthesis. Annu. Rev. Earth Planet. Sci. 2016, 44, 647–683. [Google Scholar] [CrossRef]
- Berkner, L.V.; Marshall, L.C. On the Origin and Rise of Oxygen Concentration in the Earth’s Atmosphere. J. Atmos. Sci. 1965, 22, 225–261. [Google Scholar] [CrossRef]
- Knoll, A.H.; Nowak, M.A. The timetable of evolution. Sci. Adv. 2017, 3, 1–14. [Google Scholar] [CrossRef]
- Raymond, J.; Segre, D. The effect of oxygen on biochemical networks and the evolution of complex life. Science 2006, 311, 1764–1767. [Google Scholar] [CrossRef]
- Davies, K.J. Oxidative stress: The paradox of aerobic life. Biochem. Soc. Symp. 1995, 61, 1–31. [Google Scholar] [CrossRef]
- Moan, J.; Juzenas, P. Singlet oxygen in photosensitization. J. Environ. Pathol. Toxicol. Oncol. 2006, 25, 29–50. [Google Scholar] [CrossRef]
- Bert, P. La Pression Barométrique. Recherches de Physiologie Expérimentelle (Barometric Pressure: Researches in Experimental Physiology); Masson, G., Ed.; Hitchcock, M.A.; Hitchcock, F.A., Translators; Librairie de L’académie de Médecine: Paris, France, 1878; College Book Company: Columbus, OH, USA, 1943. [Google Scholar]
- Smith, J.L. The pathological effects due to increase of oxygen tension in the air breathed. J. Physiol. 1899, 24, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Chang, M. Optimal oxygen saturation in premature infants Korean. J. Pediatr. 2011, 54, 359–362. [Google Scholar]
- Hartnett, M.E.; Penn, J.S. Mechanisms and management of retinopathy of prematurity. N. Engl. J. Med. 2012, 367, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- Askie, L.M.; Darlow, B.A.; Davis, P.G.; Finer, N.; Stenson, B.; Vento, M.; Whyte, R. Effects of targeting lower versus higher arterial oxygen saturations on death or disability in preterm infants. Cochrane Database Syst. Rev. 2017, 4, CD011190. [Google Scholar] [CrossRef] [PubMed]
- Terry, T.L. Fibroblastic overgrowth of persistent tunica vasculosa lentis in infants born prematurely, II. Report of cases-clinical aspects. Trans. Am. Ophthalmol. Soc. 1942, 40, 262–284. [Google Scholar]
- Campbell, K. Intensive oxygen therapy as a possible cause of retrolental fibroplasia; a clinical approach. Med. J. Aust. 1951, 2, 48–50. [Google Scholar] [CrossRef]
- Patz, A.; Hoeck, L.E.; De La Cruz, E. Studies on the effect of high oxygen administration in retrolental fibroplasia I. Nursery observations. Am. J. Ophthalmol. 1952, 35, 1248–1253. [Google Scholar] [CrossRef]
- Ashton, N.; Ward, B.; Serpell, G. Effect of oxygen on developing retinal vessels with particular reference to the problem of retrolental fibroplasia. Br. J. Ophthalmol. 1954, 38, 397–432. [Google Scholar] [CrossRef]
- Smith, L.E.; Hard, A.L.; Hellstrom, A. The biology of retinopathy of prematurity: How knowledge of pathogenesis guides treatment. Clin. Perinatol. 2013, 40, 201–214. [Google Scholar] [CrossRef]
- Moskowitz, A.; Hansen, R.M.; Fulton, A.B. Retinal, visual, and refractive development in retinopathy of prematurity. Eye Brain 2016, 8, 103–111. [Google Scholar] [CrossRef][Green Version]
- Rivera, J.C.; Holm, M.; Austeng, D.; Morken, T.S.; Zhou, T.E.; Beaudry-Richard, A.; Sierra, E.M.; Dammann, O.; Chemtob, S. Retinopathy of prematurity: Inflammation, choroidal degeneration, and novel promising therapeutic strategies. J. Neuroinflamm. 2017, 14, 1–14. [Google Scholar] [CrossRef]
- Whyte, R.K.; Nelson, H.; Roberts, R.S.; Schmidt, B. Benefits of oxygen saturation targeting trials: Oximeter calibration software revision and infant saturations. J. Pediatr. 2017, 182, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Bowen, F.W., Jr.; Abbasi, S.; Herrmann, N.; Weston, M.; Sacks, L.; Porat, R.; Stahl, G.; Peckham, G.; Delivoria-Papadopoulos, M.; et al. Relationship of prolonged pharmacologic serum levels of vitamin E to incidence of sepsis and necrotizing enterocolitis in infants with birth weight 1500 grams or less. Pediatrics 1985, 75, 619–638. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, T.; Mino, M. Vitamin E and preterm infants. Free Radic. Biol. Med. 2022, 180, 13–32. [Google Scholar] [CrossRef]
- Thomson, L.; Paton, J. Oxygen toxicity. Paediatr. Respir. Rev. 2014, 15, 120–123. [Google Scholar] [CrossRef]
- Chawla, A.; Lavania, A.K. OXYGEN TOXICITY. Med. J. Armed Forces India 2001, 57, 131–133. [Google Scholar] [CrossRef]
- Clark, J.M.; Lambertsen, C.J. Pulmonary oxygen toxicity: A review. Pharmacol. Rev. 1971, 23, 37–133. [Google Scholar]
- Bitterman, H. Bench-to-bedside review: Oxygen as a drug. Crit. Care 2009, 13, 205. [Google Scholar] [CrossRef]
- Caldwell, P.R.; Lee, W.L., Jr.; Schildkraut, H.S.; Archibald, E.R. Changes in lung volume, diffusing capacity, and blood gases in men breathing oxygen. J. Appl. Physiol. 1966, 21, 1477–1483. [Google Scholar] [CrossRef]
- Huber, G.L.; Drath, D.B. Pulmonary oxygen toxicity. In Oxygen and Living Processes; Gilbert, D.L., Ed.; Springer: New York, NY, USA, 1981; pp. 273–342. [Google Scholar]
- Pratt, P.C. The relation of the human lung to enriched oxygen atmosphere. Ann. N. Y. Acad. Sci. 1965, 121, 809–822. [Google Scholar] [CrossRef]
- Balentine, J.D. Pathology of Oxygen Toxicity; Academic Press: New York, NY, USA, 1982. [Google Scholar]
- Haugaard, N. Cellular mechanisms of oxygen toxicity. Physiol. Rev. 1968, 48, 311–373. [Google Scholar] [CrossRef] [PubMed]
- Frank, L.; Massaro, D. Oxygen toxicity. Am. J. Prev. Med. 1980, 69, 117–126. [Google Scholar] [CrossRef]
- Gerschman, R. Historical Introduction to the “Free Radical Theory” of Oxygen Toxicity. In Oxygen and Living Processes: An Inter-disciplinary Approach; Gilbert, D.L., Ed.; Springer: Berlin/Heidelberg, Germany, 1981. [Google Scholar]
- Taube, H. Mechanisms of oxidation with oxygen. J. Gen. Physiol. 1965, 49, 29–50. [Google Scholar] [CrossRef]
- Agnez-Lima, L.F.; Melo, J.T.; Silva, A.E.; Oliveira, A.H.; Timoteo, A.R.; Lima-Bessa, K.M.; Martinez, G.R.; Medeiros, M.H.; Di Mascio, P.; Galhardo, R.S.; et al. DNA damage by singlet oxygen and cellular protective mechanisms. Mutat. Res. Rev. Mutat. Res. 2012, 751, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Oxygen: How do we stand it? Med. Princ. Pract. 2013, 22, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, S.; Shimada, A. Reaction mechanism of cytochrome c oxidase. Chem. Rev. 2015, 115, 1936–1989. [Google Scholar] [CrossRef]
- Srinivasan, S.; Avadhani, N.G. Cytochrome c oxidase dysfunction in oxidative stress. Free Radic. Biol. Med. 2012, 53, 1252–1263. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Hider, R.H. Iron and redox cycling. Do’s and don’ts. Free Radic. Biol. Med. 2019, 133, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, M.A.; Nihal, M.; Wood, G.S.; Ahmad, N. Skin, reactive oxygen species, and circadian clocks. Antioxid. Redox Signal. 2014, 20, 2982–2996. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Ionizing Radiation, Health Effects and Protective Measures; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Le Caër, S. Water Radiolysis: Influence of Oxide Surfaces on H2 Production under Ionizing Radiation. Water 2011, 3, 235–253. [Google Scholar] [CrossRef]
- Hutchinson, F. Sulfhydryl Groups and the Oxygen Effect on Irradiated Dilute Solutions of Enzymes and Nucleic Acids. Radiat. Res. 1961, 14, 721–731. [Google Scholar] [CrossRef]
- Liu, C.; Lin, Q.; Yun, Z. Cellular and molecular mechanisms underlying oxygen-dependent radiosensitivity. Radiat. Res. 2015, 183, 487–496. [Google Scholar] [CrossRef]
- Suzuki, T.; Hidaka, T.; Kumagai, Y.; Yamamoto, M. Environmental pollutants and the immune response. Nat. Immunol. 2020, 21, 1486–1495. [Google Scholar] [CrossRef]
- Chauhan, A.J.; Krishna, M.T.; Frew, A.J.; Holgate, S.T. Exposure to nitrogen dioxide (NO2) and respiratory disease risk. Rev. Environ. Health 1998, 13, 73–90. [Google Scholar]
- Mollenhauer, K.; Tschöke, H. Handbook of Diesel Engines; Springer: Berlin/Heidelberg, Germany, 2010; pp. 445–446. ISBN 978-3540890829. [Google Scholar]
- Omidvarborna, H.; Kumar, A.; Kim, D.S. NOx emissions from low-temperature combustion of biodiesel made of various feedstocks and blends. Fuel Process. Technol. 2015, 140, 113–118. [Google Scholar] [CrossRef]
- Sukhodolov, T.; Rozanov, E.; Ball, W.T.; Bais, A.; Tourpali, K.; Shapiro, A.I.; Telford, P.; Smyshlyaev, S.; Fomin, B.; Sander, R.; et al. Evaluation of simulated photolysis rates and their response to solar irradiance variability. J. Geophys. Res. Atmos. 2016, 121, 6066–6084. [Google Scholar] [CrossRef]
- D’Amato, G. Effects of climatic changes and urban air pollution on the rising trends of respiratory allergy and asthma. Multidiscip. Respir. Med. 2011, 6, 28–37. [Google Scholar] [CrossRef]
- Church, D.F.; Pryor, W.A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 1985, 64, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Budinger, G.R.S.; Choe, S.H.; Schumacker, P.T. Cellular respiration during hypoxia: Role of cytochrome oxidase as the oxygen sensor in hepatocytes. J. Biol. Chem. 1997, 272, 111–112. [Google Scholar] [CrossRef] [PubMed]
- Mik, E.G.; Stap, J.; Sinaasappel, M.; Beek, J.F.; Aten, J.A.; van Leeuwen, T.G.; Ince, C. Mitochondrial PO2 measured by delayed fluorescence of endogenous protoporphyrin IX. Nat. Methods 2006, 3, 939–945. [Google Scholar] [CrossRef]
- Jensen, P.K. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles. I. pH dependency and hydrogen peroxide formation. Biochim. Biophys. Acta Enzymol. Biol. Oxid. 1966, 122, 157–166. [Google Scholar] [CrossRef]
- Weisiger, R.A.; Fridovich, I. Superoxide dismutase: Organelle specificity. J. Biol. Chem. 1973, 248, 3582–3592. [Google Scholar] [CrossRef]
- Loschen, G.; Azzi, A.; Richter, C.; Flohé, L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974, 42, 68–72. [Google Scholar] [CrossRef]
- Napolitano, G.; Fasciolo, G.; Di Meo, S.; Venditti, P. Mitochondrial redox biology: Reactive species production and antioxidant defences. In Mitochondrial Physiology and Vegetal Molecules; de Oliveira, M.R., Ed.; Academic Press: Cambridge, MA, USA, 2021; Chapter 4; pp. 335–344. ISBN 9780128215623. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Venditti, P.; De Rosa, R.; Di Meo, S. Effect of thyroid state on H2O2 production by rat liver mitochondria. Mol. Cell. Endocrinol. 2003, 205, 185–192. [Google Scholar] [CrossRef]
- Freeman, B.A.; Crapo, J.D. Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria. J. Biol. Chem. 1981, 256, 10986–10992. [Google Scholar] [CrossRef]
- Napolitano, G.; Fasciolo, G.; Venditti, P. Mitochondrial Management of Reactive Oxygen Species. Antioxidants 2021, 10, 1824. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- Takeshige, K.; Minakami, S. NADH- and NADPH-dependent formation of superoxide anion by bovine heart submitochon-drial particle and NADH-ubiquinone reductase preparation. Biochem. J. 1979, 180, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F.; Boveris, A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 1980, 191, 421–427. [Google Scholar] [CrossRef]
- Loschen, G.; Flohé, L.; Chance, B. Respiratory chain linked H2O2 production in pigeon heart mitochondria. FEBS Lett. 1971, 18, 261–264. [Google Scholar] [CrossRef]
- Paranagame, M.P.; Sakamotop, K.; Amino, H.; Awano, M.; Myoshi, J.H.; Kita, K. Contribution of the FAD and quinine binding sites to the production of reative oxygen species from Ascaris suum mitochondrial complex II. Mitochondrion 2010, 10, 158–165. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can gen-erate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Virzi, V. Generation of reactive oxygen species in the reaction catalyzed by α-ketoglutarate dehydrogenase. J. Neurosci. 2004, 24, 7771–7778. [Google Scholar] [CrossRef]
- Starkov, A.A.; Fiskum, G.; Chinipoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 2004, 24, 7779–7788. [Google Scholar] [CrossRef]
- Edmondson, D.E. Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: Biological implications. Curr. Pharm. Des. 2014, 20, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Santin, Y.; Resta, J.; Parini, A.; Mialet-Perez, J. Monoamine oxidases in age-associated diseases: New perspectives for old enzymes. Ageing Res. Rev. 2021, 66, 101256. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Adam-Vizi, V. High Ca2+ load promotes hydrogen peroxide generation via activation of α-glycerophosphate dehydrogenase in brain mitochondria. Free Radic. Biol. Med. 2012, 53, 2119–2130. [Google Scholar] [CrossRef]
- Cantu, D.; Schaack, J.; Patel, M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS ONE 2009, 4, e7095. [Google Scholar] [CrossRef]
- Bota, D.A.; Davies, K.J. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 2002, 4, 674–680. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Goasduff, T.; Cederbaum, A.I. NADPH-dependent microsomal electron transfer increases degradation of CYP2E1 by the proteasome complex: Role of reactive oxygen species. Arch. Biochem. Biophys. 1999, 370, 258–270. [Google Scholar] [CrossRef]
- Bondy, S.C.; Naderi, S. Contribution of hepatic cytochrome P450 systems to the generation of reactive oxygen species. Biochem. Pharmacol. 1994, 48, 155–159. [Google Scholar] [CrossRef]
- Isin, E.M.; Guengerich, F.P. Complex reactions catalyzed by cytochrome p450 enzymes. Biochim. Biophys. Acta 2007, 1770, 314–329. [Google Scholar] [CrossRef]
- Guengerich, F.P. Intersection of the roles of cytochrome p450 enzymes with xenobiotic and endogenous substrates: Relevance to toxicity and drug interactions. Chem. Res. Toxicol. 2017, 30, 2–12. [Google Scholar] [CrossRef]
- Bromek, E.; Daniel, W.A. The regulation of liver cytochrome P450 expression and activity by the brain serotonergic system in different experimental models. Expert Opin. Drug Metab. Toxicol. 2021, 17, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Sanders, J.L.; Porubsky, P.R.; Lushington, G.H.; Stout, C.D.; Scott, E.E. Structure of the human lung cytochrome P450 2A13. J. Biol. Chem. 2007, 282, 17306–17313. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, M.C.E.; Melvin, W.T.; Murray, G.I. Regional distribution of individual forms of cytochrome P450 mRNA in normal adult human brain. Biochem. Pharmacol. 1998, 55, 825–830. [Google Scholar] [CrossRef]
- Simpson, A.E. The cytochrome P450 4 (CYP4) family. Gen. Pharmacol. 1997, 28, 351–359. [Google Scholar] [CrossRef]
- Thelen, K.; Dressman, J.B. Cytochrome P450-mediated metabolism in the human gut wall. J. Pharm. Pharmacol. 2009, 61, 541–558. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome p450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of cytochrome p450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef]
- Gonzalez, F.J. The 2006 bernard b. Brodie award lecture Cyp2e1. Drug Metab. Dispos. 2007, 35, 1–8. [Google Scholar] [CrossRef]
- Elahian, F.; Sepehrizadeh, Z.; Moghimi, B.; Mirzaei, S.A. Human cytochrome b5 reductase: Structure, function, and potential applications. Crit. Rev. Biotechnol. 2014, 34, 134–143. [Google Scholar] [CrossRef]
- Samhan-Arias, A.K.; Gutierrez-Merino, C. Cytochrome B5 as a pleitropic metabolic modulator in mammalian cells. In Cytochromes B and C: Biochemical Properties, Biological Functions and Electrochemical Analysis; Rurik, T., Ed.; Nova Publishers: New York, NY, USA, 2014; ISBN 978-1-63117-467-4. [Google Scholar]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthi, S.; Jessop, C.E.; Bulleid, N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006, 7, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Ellgaard, L. The human PDI family: Versatility packed into a single fold. Biochim. Biophys. Acta 2008, 1783, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.P.; Weissman, J.S. The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell 2002, 10, 983–994. [Google Scholar] [CrossRef]
- Shergalis, A.G.; Hu, S.; Bankhead, A., III; Neamati, N. Role of the ERO1-PDI interaction in oxidative protein folding and disease. Pharmacol. Ther. 2020, 210, 107525. [Google Scholar] [CrossRef]
- Sevier, C.S.; Kaiser, C.A. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim. Biophys. Acta 2008, 1783, 549–556. [Google Scholar] [CrossRef]
- Del Río, L.A.; López-Huertas, E. ROS Generation in Peroxisomes and its Role in Cell Signaling. Plant Cell Physiol. 2016, 57, 1364–1376. [Google Scholar] [CrossRef]
- De Duve, C.; Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R. Peroxisomal disorders: The single peroxisomal enzyme deficiencies. Biochim. Biophys. Acta 2006, 1763, 1707–1720. [Google Scholar] [CrossRef]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.S.; Reddy, J.K. An overview of peroxisome proliferator-induced hepatocarcinogenesis. Environ. Health Perspect. 1991, 93, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase-Derived Reactive Species: Physiological and Pathological Effects. Oxid. Med. Cell. Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Golfinopoulos, S.; Liakopoulos, V.; Stefanidis, I. Role of indoleamine 2,3-dioxygenase in ischemia-reperfusion injury of renal tubular epithelial cells. Mol. Med. Rep. 2021, 23, 472. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Okamoto, K.; Kawaguchi, Y.; Hori, H.; Matsumura, T.; Eger, B.T.; Pai, E.F.; Nishino, T. Mechanism of the conversion of xanthine dehydrogenase to xanthine oxidase: Identification of the two cysteine disulfide bonds and crystal structure of a non-convertible rat liver xanthine dehydrogenase mutant. J. Biol. Chem. 2005, 280, 24888–24894. [Google Scholar] [CrossRef]
- Cho, K.J.; Seo, J.M.; Kim, J.H. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol. Cells 2011, 32, 1–5. [Google Scholar] [CrossRef]
- Kim, C.; Kim, J.Y.; Kim, J.H. Cytosolic phospholipase A(2), lipoxygenase metabolites, and reactive oxygen species. BMB Rep. 2008, 41, 555–559. [Google Scholar] [CrossRef]
- Wang, M.H.; Hsiao, G.; Al-Shabrawey, M. Eicosanoids and Oxidative Stress in Diabetic Retinopathy. Antioxidants 2020, 9, 520. [Google Scholar] [CrossRef]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. [Google Scholar] [CrossRef]
- Harris, R.C. COX-2 and the kidney. J. Cardiovasc. Pharmacol. 2006, 47, 37–42. [Google Scholar] [CrossRef]
- Luo, P.; Wang, M.H. Eicosanoids, beta-cell function, and diabetes. Prostaglandins Other Lipid Mediat. 2011, 95, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.F.; Harris, R.C. Cyclooxygenases, the kidney, and hypertension. Hypertension 2004, 43, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Liu, R.; Lin, N.; Luo, H.; Tang, J.; Huang, Q.; Sun, H.; Tang, L. NADPH Oxidase Hyperactivity Contributes to Cardiac Dysfunction and Apoptosis in Rats with Severe Experimental Pancreatitis through ROS-Mediated MAPK Signaling Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 4578175. [Google Scholar] [CrossRef]
- Meza, C.A.; La Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Cachia, O.; Benna, J.E.; Pedruzzi, E.; Descomps, B.; Gougerot-Pocidalo, M.A.; Leger, C.L. alpha-tocopherol inhibits the respiratory burst in human monocytes. Attenuation of p47(phox) membrane translocation and phosphorylation. J. Biol. Chem. 1998, 273, 32801–32805. [Google Scholar]
- Calvisi, D.F.; Ladu, S.; Hironaka, K.; Factor, V.M.; Thorgeirsson, S.S. Vitamin E down-modulates iNOS and NADPH oxidase in c-Myc/TGF-alpha transgenic mouse model of liver cancer. J. Hepatol. 2004, 41, 815–822. [Google Scholar] [CrossRef]
- Jiang, J.; Huang, K.; Xu, S.; Garcia, J.G.N.; Wang, C.; Cai, H. Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol. 2020, 36, 101638. [Google Scholar] [CrossRef]
- Szanto, I.; Pusztaszeri, M.; Mavromati, M. H2O2 Metabolism in Normal Thyroid Cells and in Thyroid Tumorigenesis: Focus on NADPH Oxidases. Antioxidants 2019, 8, 126. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for eithrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Napolitano, G.; Fasciolo, G.; Di Meo, S.; Venditti, P. Vitamin E Supplementation and Mitochondria in Experimental and Functional Hyperthyroidism: A Mini-Review. Nutrients 2019, 11, 2900. [Google Scholar] [CrossRef]
- Sies, H. On the history of oxidative stress: Concept and some aspects of current development. Curr. Opin. Toxicol. 2017, 7, 122–126. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar] [PubMed]
- Kodali, S.T.; Kauffman, P.; Kotha, S.R.; Yenigalla, A.; Veeraraghavan, R.; Pannu, S.R.; Hund, T.J.; Satoskar, A.R.; McDaniel, J.C.; Maddipati, R.K.; et al. Oxidative Lipidomics: Analysis of Oxidized Lipids and Lipid Peroxidation in Biological Systems with Relevance to Health and Disease. In Measuring Oxidants and Oxidative Stress in Biological Systems; Berliner, L.J., Parinandi, N.L., Eds.; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Kregel, K.C.; Zhang, H.J. An integrated view of oxidative stress in aging: Basic mechanisms, functional effects, and pathological considerations. Am. Physiol. Soc. J. 2007, 292, 18–36. [Google Scholar] [CrossRef] [PubMed]
- Salehi, F.; Behboudi, H.; Kavoosi, G.; Ardestani, S.K. Oxidative DNA damage induced by ROS-modulating agents with the ability to target DNA: A comparison of the biological characteristics of citrus pectin and apple pectin. Sci. Rep. 2018, 8, 13902. [Google Scholar] [CrossRef]
- Cannan, W.J.; Tsang, B.P.; Wallace, S.S.; Pederson, D.S. Nucleosomes suppress the formation of double-strand DNA breaks during attempted base excise, on repair of clustered oxidative damages. J. Biol. Chem. 2014, 289, 19881–19893. [Google Scholar] [CrossRef]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef]
- Sharma, P.; Jha, A.B.; Dubey, R.M.; Pessarakli, M. Reactive Oxygen Species, Oxidative Damage, and Antioxidative Defense Mechanism in Plants under Stressful Conditions. J. Bot. 2012, 2012, 217037. [Google Scholar] [CrossRef]
- Mittal, C.K.; Murad, F. Activation of guanylate cyclase by superoxide-dismutase and hydroxyl radical—Physiological regulator of guanosine 3’,5’-monophosphate formation. Proc. Natl. Acad. Sci. USA 1977, 74, 4360–4364. [Google Scholar] [CrossRef]
- Czech, M.P.; Lawrence, J.C.; Lynn, W.S. Evidence for electron transfer reactions involved in Cu2+-dependent thiol activation of fat cell glucose utilization. J. Biol. Chem. 1974, 249, 1001–1006. [Google Scholar] [CrossRef]
- Mukherjee, S.P.; Lynn, W.S. Reduced nicotinamide adenine dinucleotide phosphate oxidase in adipocyte plasma membrane and its activation by insulin. Possible role in the hormone’s effects on adenylate cyclase and the hexose monophosphate shunt. Arch. Biochem. Biophys. 1977, 184, 69–76. [Google Scholar] [CrossRef]
- May, J.M.; de Haen, C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J. Biol. Chem. 1979, 254, 2214–2220. [Google Scholar] [CrossRef]
- Burdon, R.H.; Rice-Evans, C. Free radicals and the regulation of mammalian cell proliferation. Free Radic. Res. Commun. 1989, 6, 345–358. [Google Scholar] [CrossRef]
- Murrell, G.A.C.; Francis, M.J.O.; Bromley, L. Oxygen free radicals stimulate fibroblast proliferation. Biochem. Soc. Trans. 1989, 17, 484. [Google Scholar] [CrossRef]
- Burdon, R.H. Free radicals and Cell proliferation. In Free Radical Damage and Its Control; Burdon, R.H., Rice-Evans, C., Eds.; Elsevier: Amsterdam, The Netherlands, 1994; pp. 155–188. [Google Scholar]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, F.A., Jr.; Egan, R.W. Prostaglandins, arachidonic acid, and inflammation. Science 1980, 210, 978–984. [Google Scholar] [CrossRef]
- Saugstad, O.D. Update on oxygen radical disease in neonatology. Curr. Opin. Obstet. Gynecol. 2001, 13, 147–153. [Google Scholar] [CrossRef]
- Benhar, M.; Engelberg, D.; Levitzki, A. ROS, stress activated kinases and stress signaling in cancer. EMBO Rep. 2002, 34, 420–425. [Google Scholar] [CrossRef]
- Reid, M.B.; Khawli, F.A.; Moody, M.R. Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J. Appl. Physiol. 1993, 75, 1081–1087. [Google Scholar] [CrossRef]
- Reid, M.B.; Moody, M.R. Dimethyl sulfoxide depresses skeletal muscle contractility. J. Appl. Physiol. 1994, 76, 2186–2190. [Google Scholar] [CrossRef]
- Wolin, M.S. Interactions of oxidants with vascular signaling systems. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Pao, M.; Wiggs, E.A.; Anastacio, M.M.; Hyun, J.; DeCarlo, E.S.; Miller, J.T.; Anderson, V.L.; Malech, H.L.; Gallin, J.I.; Holland, S.M. Cognitive function in patients with chronic granulomatous disease: A preliminary report. Psychosomatics 2004, 45, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Origin and evolution of the free radical theory of aging: A brief personal history, 1954–2009. Biogerontology 2009, 10, 773–781. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef]
- Oberley, L.W.; Clair, D.K.S.; Autor, A.P.; Oberley, T.D. Increase in manganese superoxide dismutase activity in the mouse heart after X-irradiation. Arch. Biochem. Biophys. 1987, 254, 69–80. [Google Scholar] [CrossRef]
- Summers, R.W.; Maves, B.V.; Reeves, R.D.; Arjes, L.J.; Oberley, L.W. Irradiation increases superoxide dismutase in rat intestinal smooth muscle. Free Radic. Biol. Med. 1989, 6, 261–270. [Google Scholar] [CrossRef]
- Hassan, H.M.; Fridovich, I. Regulation of the synthesis of superoxide dismutase in Escherichia coli. Induction by methyl viologen. J. Biol. Chem. 1977, 252, 7667–7672. [Google Scholar] [CrossRef]
- Demple, B.; Halbrook, J. Inducible repair of oxidative DNA damage to Escherichia coli. Nature 1983, 304, 466–468. [Google Scholar] [CrossRef]
- Chan, E.; Weiss, B. Endonuclease IV of Escherichia coli is induced by paraquat. Proc. Natl. Acad. Sci. USA 1987, 84, 3189–3193. [Google Scholar] [CrossRef]
- Christman, M.F.; Morgan, R.W.; Jacobson, F.S.; Ames, B.N. Positive control of a regulon for defenses against oxidative stress and some heat-shock proteins in Salmonella typhimurium. Cell 1985, 41, 753–762. [Google Scholar] [CrossRef]
- Scandalios, J.G. Oxidative stress: Molecular perception and transduction of signals triggering antioxidant gene defenses. Braz. J. Med. Biol. Res. 2005, 38, 995–1014. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- O’Connell, M.A.; Hayes, J.D. The Keap1/Nrf2 pathway in health and disease: From the bench to the clinic. Biochem. Soc. Trans. 2015, 43, 687–689. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 3, 76–86. [Google Scholar] [CrossRef]
- Kansanen, E.; Kivelä, A.M.; Levonen, A.L. Regulation of Nrf2-dependent gene expression by 15-deoxy-Delta12,14-prostaglandin J2. Free Radic. Biol. Med. 2009, 47, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and-independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef]
- Silva-Palacios, A.; Ostolga-Chavarría, M.; Zazueta, C.; Königsberg, M. Nrf2: Molecular and epigenetic regulation during aging. Ageing Res. Rev. 2018, 47, 31–40. [Google Scholar] [CrossRef]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar] [PubMed]
- Dai, D.-F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 1–22. [Google Scholar] [CrossRef]
- Bonekamp, N.A.; Delille, H.K.; Schrader, M. Organelle dynamics and dysfunction: A closer link between peroxisomes and mitochondria. J. Inherit. Metab. Dis. 2009, 32, 163–180. [Google Scholar]
- Vannuvel, K.; Renard, P.; Raes, M.; Arnould, T. Functional and morphological impact of ER stress on mitochondria. J. Cell Physiol. 2013, 228, 1802–1818. [Google Scholar] [CrossRef]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of Nox1. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef]
- Brown, G.C.; Borutaite, V. There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 2012, 12, 1–4. [Google Scholar] [CrossRef]
- Camões, F. Nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar]
- Ushio-Fukai, M. Localizing NADPH oxidase–derived ROS. Sci. STKE 2006, 2006, re8. [Google Scholar] [CrossRef]
- Mishina, N.M.; Tyurin-Kuzmin, P.A.; Markvicheva, K.N.; Vorotnikov, A.V.; Tkachuk, V.A.; Laketa, V.; Schultz, C.; Lukyanov, S.; Belousov, V.V. Does cellular hydrogen peroxide diffuse or act locally? Antioxid. Redox Signal. 2011, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.F.; Ma, Z.; Liu, Z.; Terada, L.S. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol. Cell Biol. 2010, 30, 3553–3568. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, F. The distance that a radical formed by ionizing radiation can diffuse in a yeast cell. Radiat. Res. 1957, 7, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Schultz, G.; Koesling, D. Stimulation of soluble guanylate cyclase by superoxide dismutase is mediated by NO. Biochem. J. 1998, 335, 527–531. [Google Scholar] [CrossRef]
- Winterbourn, C. Reaction of superoxide with glutathione and other thiols. Methods Enzymol. 1995, 251, 81–86. [Google Scholar] [PubMed]
- Forman, H.J.; Fridovich, I. Superoxide dismutase: A comparison of rate constants. Arch. Biochem. Biophys. 1973, 158, 396–400. [Google Scholar] [CrossRef]
- Hawkins, B.J.; Madesh, M.; Kirkpatrick, C.J.; Fisher, A.B. Superoxide flux in endothelial cells via the chloride channel-3 mediates intracellular signaling. Mol. Biol. Cell 2007, 18, 2002–2012. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, 1005–1028. [Google Scholar] [CrossRef]
- Denu, J.M.; Tanner, K.G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulphenic acid intermediate and implications for redox regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef]
- Zheng, M.; Åslund, F.; Storz, G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 1998, 279, 1718–1721. [Google Scholar] [CrossRef]
- Ji, Y.; Akerboom, T.P.; Sies, H.; Thomas, J.A. S-nitrosylation and S-glutathiolation of protein sulfhydryls by S-nitroso glutathione. Arch. Biochem. Biophys. 1999, 362, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Roos, G.; Messens, J. Protein sulfenic acid formation: From cellular damage to redox regulation. Free Radic. Biol. Med. 2011, 51, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Barrett, W.C.; DeGnore, J.P.; Keng, Y.F.; Zhang, Z.Y.; Yim, M.B.; Chock, P.B. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J. Biol. Chem. 1999, 274, 34543–34546. [Google Scholar] [CrossRef] [PubMed]
- Abate, C.; Patel, L.; Rauscher, F.J.; Curran, T. Redox regulation of fos and jun DNA-binding activity in vitro. Science 1990, 249, 1157–1161. [Google Scholar] [CrossRef]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Disulfide relays and phosphorylative cascades: Partners in redox- mediated signaling pathways. Cell Death Differ. 2005, 12, 1555–1563. [Google Scholar] [CrossRef]
- Gotoh, Y.; Cooper, J.A. Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-alpha signal transduction. J. Biol. Chem. 1998, 273, 17477–17482. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E.; Irie, K.; Dijke, P.T.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Venditti, P.; Napolitano, G.; Barone, D.; Di Meo, S. Effect of training and vitamin E administration on rat liver oxidative metabolism. Free Radic. Res. 2014, 48, 322–332. [Google Scholar] [CrossRef]
- Venditti, P.; Napolitano, G.; Barone, D.; Di Meo, S. Vitamin E supplementation modifies adaptive responses to training in rat skeletal muscle. Free Radic. Res. 2014, 48, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Napolitano, G.; Barone, D.; Pervito, E.; Di Meo, S. Vitamin E-enriched diet reduces adaptive responses to training determining respiratory capacity and redox homeostasis in rat heart. Free Radic. Res. 2016, 50, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; De Leo, T.; Di Meo, S. Vitamin E administration attenuates the tri-iodothyronine-induced modification of heart electrical activity in the rat. J. Exp. Biol. 1997, 200, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Fasciolo, G.; Napolitano, G.; Aprile, M.; Cataldi, S.; Costa, V.; Ciccodicola, A.; Di Meo, S.; Venditti, P. Hepatic Insulin Resistance in Hyperthyroid Rat Liver: Vitamin E Supplementation Highlights a Possible Role of ROS. Antioxidants 2022, 11, 1295. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, Z.; Huang, Z.; Nice, E.; Zou, B.; Huang, C. Revisiting cancer hallmarks: Insights from the interplay between oxidative stress and non-coding RNAs. Mol. Biomed. 2020, 1, 1–24. [Google Scholar] [CrossRef]
- Bekhet, O.H.; Eid, M.E. The interplay between reactive oxygen species and antioxidants in cancer progression and therapy: A narrative review. Transl. Cancer Res. 2021, 10, 4196–4206. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Z. Increased Oxidative Stress as a Selective Anticancer Therapy. Oxid. Med. Cell. Longev. 2015, 2015, 294303. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).