Abstract
The evolution of the MADS-box gene family is essential for the rapid differentiation of floral organs and fruit types in angiosperms. Two key processes drive the evolution of gene families: gene duplication and functional differentiation. Duplicated copies provide the material for variation, while advantageous mutations can confer new functions on gene copies. In this study, we selected the Rosaceae family, which includes a variety of fruit types and flower organs, as well as species that existed before and after whole-genome duplication (WGD). The results indicate that different fruit types are associated with different copies of MADS-box gene family duplications and WGD events. While most gene copies derived from WGD have been lost, MADS-box genes not only retain copies derived from WGD but also undergo further gene duplication. The sequences, protein structures, and expression patterns of these gene copies have undergone significant differentiation. This work provides a clear example of MADS-box genes in the context of gene duplication and functional differentiation, offering new insights into the evolution of fruit types and floral organs.
1. Introduction
Whole-genome duplication (WGD) events have been crucial in the adaptive evolution and diversification of angiosperms [1,2,3]. Approximately 100 to 120 million years ago (Ma), several ancient polyploidy events occurred in angiosperms [4], with four significant events detected in major clades: a whole-genome triplication in the common ancestor of core eudicots (γ) [5] and WGDs in monocots (τ) [6], magnoliids (λ) [7], and the Nymphaeales lineage (π) [8]. Numerous species-rich families provide strong evidence of WGD, with recent polyploidizations identified in Arabidopsis thaliana [9], Populus trichocarpa [10], and Glycine max [11]. The Maleae tribe within the Rosaceae family has undergone a recent WGD event [12], leading to the development of pome fruit. Studying WGD’s role in the evolution of diverse fruit types is highly valuable.
The Rosaceae family comprises over 3000 species [13]. Recent molecular phylogenetic analyses have classified Rosaceae species into three subfamilies, Dryadoideae, Rosoideae, and Amygdaloideae, with Maleae belonging to Amygdaloideae [14]. Both Amygdaloideae and Rosoideae include numerous species that are vital for human nutrition and ornamental purposes. These subfamilies exhibit wide phenotypic diversity in fruit types, such as the fleshy fruit known as a pome (found in apples and pears), which is formed from the fusion of the hypanthium and ovary; the drupe (seen in peaches and cherries), a fleshy fruit with a hard endocarp enclosing a seed; and the achenetum and drupetum (typical of strawberries and raspberries), which are fleshy fruits derived from the receptacle and/or multiple achenes [15,16].
Generally, fruits develop from floral tissue following pollination; thus, the diversity of fruits is closely linked to the structural variations in floral organs [17]. Both apple and peach flowers possess a hypanthium, a cup-like structure formed by the fusion of the lower portions of the sepals, petals, and stamens [18,19]. However, in peaches, the fruit is formed solely from the ovary. Strawberry and raspberry flowers feature raised stem tips known as receptacles, which connect to the bases of numerous carpels. Each carpel has the potential to develop into a single small, one-seeded fruit after fertilization [15,20,21]. These structural differences in floral organs directly influence the physiological processes of fruit development.
The MADS-box family genes are well known for their role in regulating the morphogenesis of floral organs [22]. According to the latest model of flower development, the identity of floral organs is determined by the combinatorial actions of five classes of MADS-box genes, named A, B, C, D, and E, most of which belong to the type II group (also known as the MIKC type) [23,24]. Specifically, class A genes include APETALA1 (AP1) and AP2; class B genes include AP3 and PISTILLATA (PI); class C genes include AGAMOUS (AG); class D genes include SEEDSTICK (STK), SHATTERPROOF1 (SHP1), and SHP2; and class E genes include SEPALLATA1 (SEP1), SEP2, and SEP3 [25,26,27,28,29]. Class A and E genes determine sepal formation; class A, B, and E genes determine petal formation; class B, C, and E genes determine stamen formation; class C and E genes determine carpel formation; and class C, D, and E genes control ovule formation and development [30,31].
The MADS-box family genes are widely present in angiosperms and have a large number of members, many of which originated from duplications of a common ancestor gene [22,32,33]. For instance, following the γ event, multiple copies of the AP1, AP3, AG, SEP1, AGL6, and SOC1 genes remain [34]. After genome polyploidy, the retention of priority genes and subsequent divergence have contributed to the development of new traits. There has been a significant expansion in the number of MADS-box genes in the Malus and Pyrus genera, comprising 132 and 74 genes, respectively [35,36]. In contrast, the Prunus genus, which did not experience the recent WGD, has an average of 37 MADS-box genes [37,38,39]. Although there is evidence indicating the expression of duplicated MADS-box genes in plant tissue [35,36], it is still unclear how this leads to the evolution of the pome. Further analysis is needed to elucidate the functional differentiation after WGD in Maleae.
In this study, we reconstructed the phylogenetic tree and the ancestral Rosaceae karyotype (ARK) with nine ancient chromosomes, encompassing the Rosoideae subfamily, the Prunus genus, and the Maleae tribe, both before and after the WGD. Karyotype evolution analysis revealed that the Maleae tribe has continuously experienced chromosome breakage and rearrangement following the WGD, and it was inferred that this event occurred at approximately 14.69 Ma. We found that approximately 30.71% of the gene copies derived from the WGD are still expressed in plant tissue. Among these, the orthologous groups of MADS18 and MADS6 contain both WGD and single gene duplication copies. These copies undergo sequence differential evolution, affecting their physical and chemical properties and protein structure, and exhibit significant differential expression in apple fruits at different developmental stages and in different floral organs, providing new insights into the evolution of fruit types and floral organs.
2. Results
2.1. The Expansion in the Number of MADS-Box Genes across Different Fruit Types Is Correlated with WGD in Rosaceae
To investigate the role of the recent whole-genome duplication (WGD) in the evolution of pome fruit, we selected four species from the Maleae tribe, which experienced the recent WGD event, and eight species from the Prunus genus and the Rosoideae subfamily, which did not undergo the recent WGD event, to perform a comparative genomic analysis (Table S1).
Crataegus pinnatifida, Eriobotrya japonica, Pyrus pyrifolia, and Malus × domestica of the Maleae tribe are all pome fruits with a chromosome number of x = 17. Prunus avium, Prunus persica, Prunus armeniaca, and Prunus salicina in the Prunus genus are all drupe fruits with a chromosome number of x = 8. Fragaria × ananassa, Rubus argutus, Rubus occidentalis, and Rubus idaeus in the Rosoideae are all drupetum/achenetum fruits with a chromosome number of x = 7 (Figure 1A). The chromosome number in the Maleae tribe is about twice that of the Prunus genus and the Rosoideae.
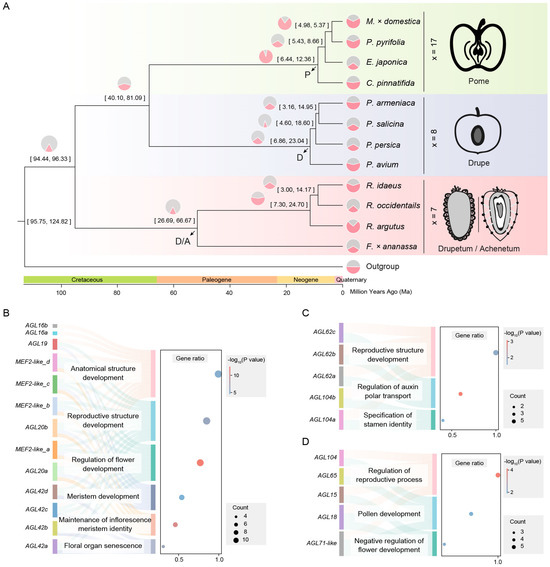
Figure 1.
Expansion analysis of MADS-box gene family in different fruit types of Rosaceae. (A) The phylogenetic analysis of 12 Rosaceae species uses V. vinifera as the outgroup. The Rosaceae family is divided into drupetum/achenetum (pink), drupe (light blue), and pome (light green). The numbers at the branch node indicate the divergence time with a 95% confidence interval. The pie charts show the relative sizes of expansion (light pink) and contraction (light gray) of the gene family. We define the MRCA of pome fruits as the P node, the MRCA of drupe fruits as the D node, and the MRCA of drupeum/achenetum fruits as the D/A node. (B) GO enrichment results of expanded MADS-box genes at the P node. The lowercase suffix letters represent gene copies within the same orthologous group (the same below). (C) GO enrichment results of expanded MADS-box genes at the D node. (D) GO enrichment results of expanded MADS-box genes at the D/A node.
The crown age of the Rosaceae family is estimated to be 94.46–96.36 million years (Ma) [14]. Combined with phylogenetic analysis, we estimate that Maleae and Prunus diverged between 40.10 and 81.09 Ma, and the most recent common ancestor (MRCA) of Maleae emerged between 6.44 and 12.36 Ma. The MRCA of Prunus emerged between 6.86 and 23.04 Ma, and the MRCA of Rosoideae emerged between 26.69 and 66.67 Ma (Figure 1A). We define the MRCA of Maleae as the P (pome) node based on the type of fruit, the MRCA of Prunus as the D (drupe) node, and the MRCA of Rosoideae as the D/A (drupeum/achenetum) node (Figure 1A).
The results showed that the gene families underwent significant expansion and contraction in the Rosaceae family. In the Maleae tribe, the number of gene family expansions was significantly greater than the number of contractions, with the average number of expansions being 2355, which was 1.53 times the number of contractions, except for E. japonica. In contrast, the average number of gene family expansions in Prunus and Rosoideae was 1175.5, which was 0.89 times the number of contractions (Figure 1A).
Specifically, the expansion of the MADS-box gene family was detected at the P, D, and D/A nodes, with gene numbers of 13, 5, and 5, respectively. These genes include multiple AGAMOS-like (AGL) genes and their homologs, as well as myocyte enhancer factor 2 (MEF2)-like genes and their homologs. The gene ontology (GO) enrichment results showed that these genes are mainly involved in the regulation of reproductive processes and the development of reproductive structures. In the Maleae tribe, they are also involved in the regulation of flower development, meristem development, and floral organ senescence (Figure 1B–D). The correlation between the number of MADS-box genes at the P, D, and D/A nodes and their expanded functions may be related to the recent WGD event.
2.2. The Retention of Duplicated Genes in Maleae following WGD and Subsequent Chromosome Rearrangement
The results showed that the species in the Prunus genus and Rosoideae subfamily had strong collinearity and few structural variations (SV) (Figures S1 and S2), while those in the Maleae tribe had many SVs and inferior collinearity (Figure S3). To investigate the evolutionary process of chromosomes in Maleae, we reconstructed the ARK with nine ancient chromosomes and subsequently analyzed the karyotype evolution. The karyotypes of F. × ananassa and P. avium can be directly formed by seven fissions/nine fusions and seven fissions/eight fusions of the ARK, respectively (Figure 2A). Chromosomes 4 and 7 of F. × ananassa and chromosomes 8, 3, 5, and 2 of P. avium are identical to the ARK, similar to previous findings [12,40].
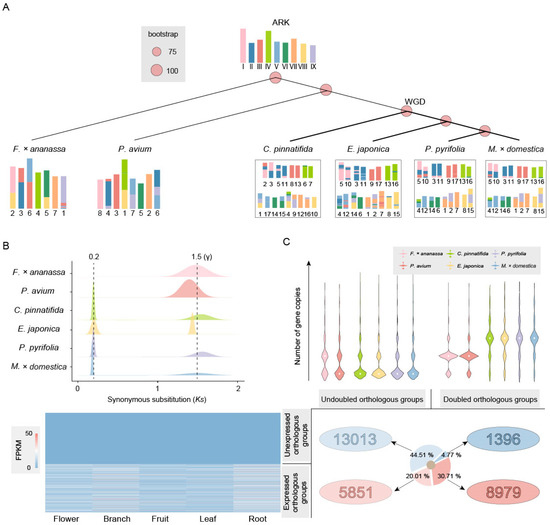
Figure 2.
Chromosome karyotype and gene evolution after WGD in Rosaceae. (A) Evolutionary scenarios of chromosome karyotype changes between the ARK and F. × ananassa, P. avium, C. pinnatifida, E. japonica, P. pyrifolia, and M. × domestica. (B) Ks peaks of homologous genes in F. × ananassa, P. avium, C. pinnatifida, E. japonica, P. pyrifolia, and M. × domestica. A Ks value of 1.5 corresponds to the γ event that occurred in the common ancestor of core eudicots, about 117 ± 1 Ma. The peak at Ks = 0.2 corresponds to the recent WGD event, which occurred at approximately 14.69 Ma. (C) Preservation and expression of gene copies after WGD. All genes are divided into four orthologous groups: those with an undoubled number of genes not expressed in tissue, those with an undoubled number of genes expressed in tissue, those with a doubled number of genes not expressed in tissue, and those with a doubled number of genes expressed in tissue.
The Maleae tribe has undergone duplication and complex chromosome evolution. After duplication, the karyotypes of C. pinnatifida, E. japonica, P. pyrifolia, and M. × domestica experienced 28 fissions/20 fusions, 59 fissions/51 fusions, 33 fissions/25 fusions, and 29 fissions/21 fusions, respectively (Figure 2A).
The distribution of synonymous substitutions (Ks) of homologous genes showed significant peaks in all Rosaceae species at around Ks = 1.5, corresponding to the γ event that occurred in the common ancestor of core eudicots. Additionally, another peak was detected in Maleae near Ks = 0.2, corresponding to the recent WGD event, which occurred at approximately 14.69 Ma (Figure 2B), consistent with the phylogenetic inference results.
We filtered out data with gene copies present only in one species or only in the Maleae tribe, leaving 29,239 orthologous groups present in both Maleae and Prunus and Rosoideae. Undoubled orthologous groups indicate that the number of genes in Maleae is less than twice that of other species, accounting for 64.52% of the total. Additionally, 44.51% of these orthologous groups show no expression in the flowers, branches, fruits, leaves, and roots. Doubled orthologous groups indicate that the number of genes in Maleae is more than twice that of other species, with 8979 orthologous groups expressed in plant tissue, which is more than six times that of the unexpressed orthologous groups (1396) (Figure 2C).
2.3. Four MADS-Gene Orthologous Groups Are Expressed in Plant Tissues
The genes in the orthologous groups that have doubled in quantity and are expressed are involved in basic biological processes, cellular components, and molecular functions in plants, including metabolic processes, organelle parts, membrane and cytoplasmic parts, nucleic acid binding, and catalytic activity (Figure S4). To explore the functional differentiation of genes after duplication, we filtered out 2420 tissue-specific expressed genes (Figure 3A), among which four orthologous groups were related to MADS-box genes, with the numbers of these genes in Maleae being about twice those in Prunus and Rosoideae.
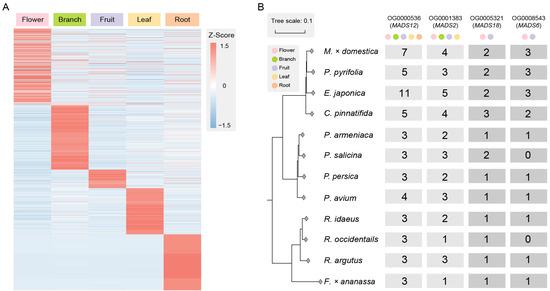
Figure 3.
Exploring tissue-specific expressed genes and MADS-gene orthologous groups. (A) Tissue-specific expressed genes in doubled orthologous groups include those expressed in tissue types such as flowers, branches, fruits, leaves, and roots. (B) There are four orthologous groups of MADS-box genes. The number of gene copies shows a fold relationship between Maleae and other species in Rosaceae. Some gene copies from MADS12 and MADS2 are expressed in multiple tissue types, while the gene copies from MADS18 and MADS6 are only expressed in the flowers and fruits.
Among them, OG0000536 is an orthologous group of MADS12, and MADS12 is an AP1-like gene involved in specifying the identity of perianth organs [41]. In this study, gene copies in OG0000536 were expressed in the flowers, branches, fruits, leaves, and roots. OG0001383 is an orthologous group of MADS2, with some gene copies expressed in the flowers, branches, fruits, and leaves. OG0005321 is an orthologous group of MADS18, which is a homologous gene of AGL9 [42]. OG0008543 is an orthologous group of MADS6, which is a homologous gene of CMB1-like (Figure 3B). They both have two and three gene copies, respectively, in Maleae and are only expressed in the flowers and fruits.
2.4. The MADS18 Genes Derived from WGD in M. × domestica Have Undergone Differential Evolution
The phylogenetic analysis results of genes in OG0005321 are consistent with the whole-genome analysis results. CpMADS18b, CpMADS18c, EjMADS18a, EjMADS18b, PpMADS18a, PpMADS18b, MdMADS18a, and MdMADS18b are all derived from the WGD event. CpMADS18a and CpMADS18b are the result of gene duplication (Figure 4A).
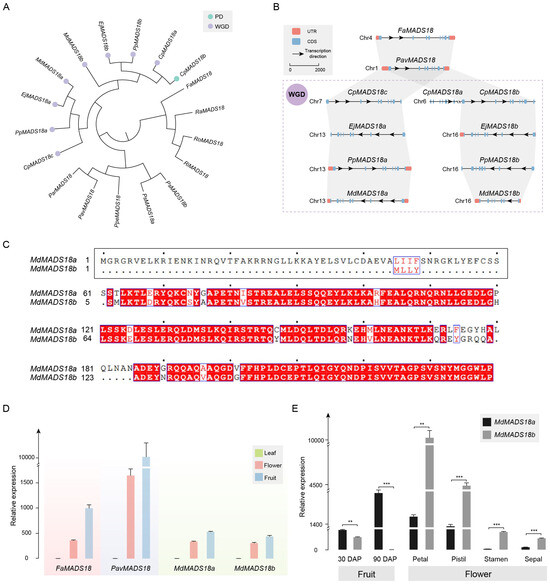
Figure 4.
Analysis of MADS18 orthologous group. (A) Phylogenetic analysis of MADS18 gene copies in Rosaceae. The duplication events are classified into WGD or segmental (WGD) and proximal duplications (PD). (B) In the collinearity analysis of MADS18 gene copies, rectangles represent gene structures, arrows indicate transcription directions, the reference line represents a length of 2000 bp, and the purple dashed box represents species that have undergone WGD. (C) Analysis of protein sequence similarity between MdMADS18a and MdMADS18b; the N-terminus of MdMADS18b specifically lacks 60 amino acids. The black border represents the sequence with a specific deletion at the beginning of MdMADS18a in this article. The blue border and red font indicate that more than 70% of the amino acids at this position have similar physico-chemical properties. The red background with white font indicates that the amino acids at this position are highly conserved. The black font represents standard amino acids, and the center point represents the sequence position, with each point indicating the next 10 amino acids. (D) Relative expression of the MADS18 gene copies in the leaf, flower, and fruit by qRT-PCR. (E) Relative expression of MdMADS18a and MdMADS18b genes in fruits at different developmental stages and in different flower organs. Statistical significance was determined using Student’s t-test, where ** indicates p < 0.01, and *** indicates p < 0.001.
The results of the collinearity and physical and chemical properties support that CpMADS18c, EjMADS18a, PpMADS18a, and MdMADS18a are orthologous genes that emerged after the species differentiated (Figure 4B). These genes have an almost identical size, molecular weight (Mw), isoelectric point (pI), instability index (II), and grand average of hydropathicity (GRAVY). The MADS18b genes from Maleae are also orthologous genes that emerged after the species differentiated, sharing a similar gene structure. However, MdMADS18b has undergone significant evolution, containing only seven coding sequence (CDS) regions. The encoded protein sequence has become shorter and more unstable, with a reduced Mw and pI (Table S2).
The identity of the protein sequences between MdMADS18a and MdMADS18b is only 85.23% (Figure 4C). The N-terminus of MdMADS18b specifically lacks 60 amino acids, and this polymorphic segment encodes a MEF2-like domain (Figure S5A), ultimately resulting in the absence of at least one α-helix and two β-sheets in its protein structure (Figure S5B).
To investigate the impact of sequence differences on their expression levels, we tested the expression of MdMADS18a and MdMADS18b in different tissue types, using the expression of FaMADS18 and PavMADS18 as controls for the ancestral status. The results showed that all four genes were expressed in flower and fruit tissue, but not in leaf tissue (Figure 4D).
Specifically, in the flesh tissue 30 days after pollination (DAP), the expression level of MdMADS18b was significantly lower than that of MdMADS18a. In the flesh tissue at 90 DAP, the expression level of MdMADS18b had decreased to zero, while the expression level of MdMADS18a continued to increase (Figure S6A). The expression patterns of the two genes in flower organs are also different: MdMADS18a is almost absent in the stamen and sepal, whereas MdMADS18b is expressed in almost all flower organs, and the expression level of MdMADS18b is significantly higher than that of MdMADS18a (Figure S6A and Figure 4E).
2.5. The MADS6 Genes Derived from Both WGD and Tandem Duplication in M. × domestica Exhibit Differential Evolution
The phylogenetic analysis results of genes in OG0008543 are consistent with the whole-genome analysis results (Figure 5A). Among them, the two gene copies in C. pinnatifida, E. japonica, P. pyrifolia, and M. × domestica were all derived from the WGD event. Additionally, MADS6b originated from the gene duplication of MADS6a in Maleae (Figure 5B).
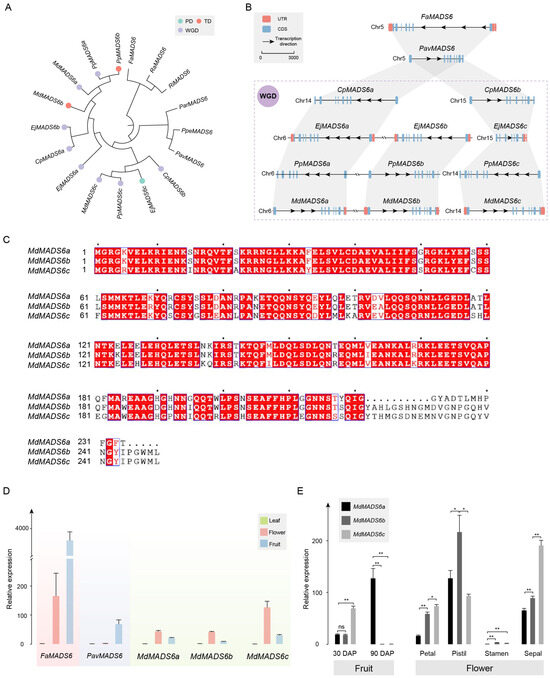
Figure 5.
Analysis of MADS6 orthologous group. (A) Phylogenetic analysis of MADS6 gene copies in Rosaceae. The duplication events are classified into WGD or segmental (WGD), tandem (TD), and proximal duplications (PD). (B) In the collinearity analysis of MADS6 gene copies, rectangles represent gene structures, arrows indicate transcription directions, the reference line represents a length of 2000 bp, and the purple dashed box represents species that have undergone WGD. (C) Analysis of protein sequence similarity among MdMADS6a, MdMADS6b, and MdMADS6c. Compared to MdMADS6b, MdMADS6a has specifically deleted or mutated 27 amino acids at the C-terminus, while MdMADS6c has a specific mutation of 41 amino acids. The blue border and red font indicate that more than 70% of the amino acids at this position have similar physico-chemical properties. The red background with white font indicates that the amino acids at this position are highly conserved. The black font represents standard amino acids, and the center point represents the sequence position, with each point indicating the next 10 amino acids. (D) Relative expression of the MdMADS6 gene copies in the leaf, flower, and fruit by qRT-PCR. (E) Relative expression of MdMADS6a and MdMADS6b genes in fruits at different developmental stages and in different flower organs. Statistical significance was determined using Student’s t-test, where * indicates p < 0.05 and ** indicates p < 0.01.
The physical and chemical properties indicate that the MADS6 genes in Maleae have undergone different evolutionary changes (Table S3). Taking M. × domestica as an example, MdMADS6a and MdMADS6b are derived from gene duplication, but MdMADS6a has specifically deleted or mutated 27 amino acids at the C-terminus, resulting in a shorter protein sequence with a smaller Mw and pI. Compared with MdMADS6b, MdMADS6c has a specific mutation of 41 amino acids, and its encoded protein sequence length remains unchanged, but both its pI and II are reduced. The identity of the protein sequences of MdMADS6a and MdMADS6b is 85.14%, while the identity of the protein sequences of MdMADS6c and MdMADS6b is 83.53% (Figure 5C). The protein structures of the three are not significantly different in composition (Figure S7).
We investigated the expression of MdMADS6a, MdMADS6b, and MdMADS6c in different tissue types, using the expression of FaMADS6 and PavMADS6 as controls for the ancestral status. The results showed that four genes were expressed in flower and fruit tissue, but not in leaf tissue, with the exception of PavMADS6, which was only expressed in fruits (Figure 5D). The expression levels of FaMADS6 and PavMADS6 in fruits were significantly higher than those in flowers. In contrast, the expression levels of MdMADS6a, MdMADS6b, and MdMADS6c in fruits were lower than those in flowers.
Specifically, in the flesh tissue at 30 DAP, the expression levels of MdMADS6a and MdMADS6b were similar but lower than that of MdMADS6c. In the flesh tissue at 90 DAP, the expression level of MdMADS6a had significantly increased, while MdMADS6b and MdMADS6c were almost absent. In both petal and sepal tissue, the expression patterns of the three gene copies were similar: the expression level of MdMADS6c was the highest, followed by MdMADS6b, and the expression level of MdMADS6a was the lowest, with significant differences among the three genes. The expression level of MdMADS6b in the pistil was significantly higher than that of MdMADS6a and MdMADS6c. The expression levels of the three genes in the stamen were very low (Figure 5E and Figure S6).
3. Discussion
3.1. Changes in Chromosomes and Genes Following the WGD Event in Rosaceae
The connection between whole-genome polyploidization and angiosperm evolution has been confusing among researchers [1,4]. Many studies have confirmed that the Maleae tribe experienced a recent WGD event, while other species in Rosaceae did not [12,43]. In addition, many species in Rosaceae are closely related to humans, such as apples, peaches, and strawberries, which are important economic crops and essential sources of various nutrients, such as vitamins and dietary fiber [44,45,46]. The Rosaceae family contains abundant and easily accessible genomic resources [47]. To explore the relationship between WGD and the differentiation of the Rosaceae family, we chose the genomes of the Maleae tribe, which experienced a recent WGD event, and Prunus and Rosoideae, which did not.
We inferred that the recent WGD event experienced by Maleae likely occurred between 6.44–12.36 Ma and 40.10–81.09 Ma, with the specific time possibly around 14.69 Ma, through the reconstruction of the phylogenetic trees and the analysis of the species divergence times (Figure 1A and Figure 2B).
Compared with other species, the Maleae tribe not only doubled its number of chromosomes but also experienced complex chromosome breakage and rearrangement events (Figure 1A and Figure 2A). Moreover, many structural variations occurred after the differentiation of C. pinnatifida, E. japonica, P. pyrifolia, and M. × domestica, resulting in weaker collinearity compared to Prunus, suggesting that the Maleae are still undergoing rapid evolution (Figures S1–S3).
Many studies have shown that multiple ancient polyploidization events and gene duplications in angiosperms have made significant contributions to their adaptation to different environments [5,6]. For example, HSF transcription factors, which play an important role in the plant heat stress response, rapidly expanded during ancient polyploidization, with the expansion closely related to the warm and arid climate [48,49]. The C-repeat binding factors (CBFs) are the main regulatory factors for cold acclimation in many angiosperm plants. All CBF transcription factors are derived from the duplication and functional differentiation of the same DREB III gene, and these gene duplication events were concentrated during the ice ages, when the global average temperature significantly decreased, helping plants to adapt to the global cooling [50,51,52].
3.2. The Expansion of the MADS-Box Gene Family Is Associated with Different Fruit Types in Rosaceae
The rapid diversification of angiosperms relies on the evolution of diverse floral organs and fruit types [17]. Coincidentally, the Rosaceae family includes various fruit types: pome, drupe, and drupetum/achenetum fruits. Correspondingly, Rosaceae also exhibit flower organs with significant morphological differences [15,16].
The MADS-box gene family is deeply involved in various stages of flower organ morphogenesis and fruit development. Some studies have shown that the differentiation of Arabidopsis flowers and fruits is controlled by two duplication genes, SHATTERPROOF1 (SHP1) and SHP2. The expression of these two genes is also necessary in promoting ovule identity [28,29].
In addition, there is a reproductive strategy where seeds develop without fertilization, known as apomixis [53], which is also influenced by MADS-box genes. The overexpression of ZaMADS80 in Zanthoxylum armatum triggered precocity and parthenocarpy, and the interaction between ZaMADS80 and ZaMADS67 (AGL32-like) may contribute to apomixis [54]. In the ovule of Brachiaria brizantha, the differential expression of BbrizAGL6 is the main difference between sexual and apomictic reproduction. Transcripts of BbrizAGL6 are located in the megaspore mother cells of the ovaries from both apomictic and sexually reproducing plants, as well as in the nucelli of apomictic plants, in the region where aposporic initial cells differentiate [55]. The MADS-box genes can facilitate asexual reproduction by bypassing the need for fertilization; however, there is still a lack of relevant research in Rosaceae.
We found that the MADS-box gene family members underwent expansion at the MRCA nodes of different fruit types, and the difference in the number of expansions was associated with WGD (Figure 1B–D). Subsequently, we specifically filtered the MADS-box genes that were duplicated and preserved during the WGD event and found two typical examples: the orthologous groups of MADS18 and MADS6 (Figure 3B).
3.3. MADS18 and MADS6 Are Expanded through WGD and Gene Duplication
In these two examples, the Maleae tribe contains two gene copies derived from WGD, while Prunus and Rosoideae mostly contain one gene copy. In addition, C. pinnatifida has three copies of MADS18, while other species in Maleae only have two copies (Figure 4A,B). Based on the phylogenetic and collinearity analysis, it is speculated that this gene duplication event only exists in C. pinnatifida. C. pinnatifida has two copies of MADS6, while other species in Maleae have three copies. These gene copies have undergone various sequence differentiations in different species, so it is speculated that this gene duplication event occurred in the ancestors of Maleae, underwent differentiation among species, and lost one copy in C. pinnatifida (Figure 5A,B).
3.4. MADS18 and MADS6 Have Different Expression Levels in Apple Fruits at Different Stages and Different Flower Organs
F. × ananassa and P. avium both have one copy of MADS18 and MADS6 and have not experienced WGD, so their expression is considered to represent the Maleae ancestors before the WGD. All genes were expressed in both mixed fruit and mixed flower tissue. These genes were not expressed in leaf tissue, which served as a negative control, and transcriptome data showed that they were not expressed in the roots and branches but were specifically expressed in the flowers and fruits (Figure 4D and Figure 5D).
We found that sequence differentiation occurs between MdMADS18a and MdMADS18b, as well as among MdMADS6a, MdMADS6b, and MdMADS6c, which affects their physical and chemical properties and protein structures (Figure 4C and Figure 5C, Tables S2 and S3). We divided the fruit samples into two stages, 30 DAP and 90 DAP, and divided the flower organs into the petal, pistil, stamen, and sepal. Surprisingly, the expression patterns of these genes have undergone significant changes in a more detailed structure, with some even no longer expressed (Figure 4E and Figure 5E). These genes are likely to play a crucial role in the evolution of the fruit types and floral organs of pomes.
4. Materials and Methods
4.1. Collection of Publicly Available Genomic and Transcriptome Data
We collected genomic and gene annotation information that is public, including that on Crataegus pinnatifida var. major N.E. Br., Eriobotrya japonica (Thunb.) Lindl., Pyrus pyrifolia Nakai, Malus × domestica Borkh., Prunus avium L., Prunus persica Batsch, Prunus armeniaca L., Prunus salicina Lindl., Fragaria × ananassa Duchesne ex Rozier, Rubus argutus Link, Rubus occidentalis L., Rubus idaeus L., and Vitis vinifera L. (Table S1). In addition, we downloaded the raw transcriptome sequence data for different plant tissue types, including the flower, leaf, root, fruit, and branch from M. × domestica (Table S1).
4.2. Phylogenetic Analysis and Time Inference
A total of 351 single-copy orthologous genes were selected from 13 species using OrthoFinder2 (https://github.com/davidemms/OrthoFinder, accessed on 29 October 2021) [56], with V. vinifera as the outgroup. All protein sequences were aligned with MUSCLE (https://www.drive5.com/muscle, accessed on 15 August 2021) [57] and Gblocks (https://www.biologiaevolutiva.org/jcastresana/Gblocks.html, accessed on 15 August 2021) [58]. All sites including missing and gap data were used to construct the phylogenetic tree with the maximum likelihood method using RAxML (https://github.com/stamatak/standard-RAxML/archive/master.zip, accessed on 16 August 2021) [59], applying the PROTGAMMAJTTF model and 1000 bootstraps.
The divergence times were estimated based on the phylogenetic tree using an approximate likelihood method implemented in MCMCtree of the PAML 4.9 package (http://abacus.gene.ucl.ac.uk/software/paml4.9j.tgz, accessed on 10 August 2022) [60] with the options “normal approximation” and the “JC69” model, using the parameters “burnin = 400,000; sampfreq = 10; nsample = 100,000”.
4.3. GO Term Enrichment
We extracted sequence IDs using in-house Python (https://www.python.org/, accessed on 16 March 2020) scripts and sorted and deduplicated them. Then, we submitted them to the HortGenome Search Engine (http://hort.moilab.net/#/, accessed on 15 July 2024) [61]. The result was visualized using R scripts (https://www.r-project.org/, accessed on 16 March 2020).
4.4. Gene Family Expansion and Contraction
The orthologous group analysis was performed using OrthoFinder2 and the phylogenetic tree as mentioned above. Expansions and contractions of all 13 species were detected using CAFÉ (https://github.com/hahnlab/CAFE5.git, accessed on 10 July 2024) [62] with the default parameters. The expanded and contracted gene families were subjected to statistical analysis and visualization using Excel (https://www.microsoftstore.com.cn/software/office, accessed on 25 April 2019).
4.5. Collinearity and Sequence Similarity Analysis
Whole-genome collinearity analysis was performed with JCVI (https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version), accessed on 15 October 2020). Credible collinear blocks were obtained using GFF files and protein sequence files, filtered with the parameter “--cscore=.99”, and visualized with the parameter “-m jcvi.graphics.karyotype”, and segments of chromosomes were visualized with the parameter “-m jcvi.graphics.synteny”.
Protein sequence alignment and similarity analysis were performed with MUSCLE as mentioned above. The visualization of gene structures was performed using TBtools (https://github.com/CJ-Chen/TBtools-II/releases, accessed on 15 March 2024) [63].
4.6. Ancestral Chromosome Karyotype Reconstruction and Evolution
We reconstructed the ancestral Rosaceae karyotype (ARK), and the standard process of WGDI (https://github.com/SunPengChuan/wgdi, accessed on 2 March 2023) [64] was used to infer karyotypes. First, BLASTP (https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/, accessed on 28 August 2020) was used to search for homologs, with the parameter “-evalue 1e-5 -max_target_seqs 5 -outfmt 6”. Second, the shared “synteny blocks” with telomeres (chromosome-like) and intact chromosomes across extant genomes were explored. Third, shared blocks, including small syntenic blocks, were deleted and the remaining parts were merged into an ancestor length file, using the parameter “-ak” to produce an ancestor annotation file. Fourth, an additional round of “synteny exploration” was performed for each extant genome to extract all protochromosomes until there were no genomic blocks left, following the parameter “-icl”, “-bi” and “-d”.
4.7. Analysis of WGD and Gene Duplication
WGDI was used to infer duplication events, the parameter “-icl” was used to classify homologs, the parameter “-ks” was used to calculate the Ks between homologs, the parameter “-bi” was used to merge the collinearity message and Ks values, the parameter “-bk” was used to visualize the results, and the parameters “-kp” and “-pf” were used to fit the homologs generated by the same duplication event into a peak. The time at which the WGD events occurred was calculated using “T = Ks/2r” (where “T” represents the time in millions of years and “r” represents the nucleotide replacement rate).
We used MCScanX (https://github.com/wyp1125/MCScanX, accessed on 15 August 2022) [65] to classify the duplication events into WGD or segmental (WGD), tandem (TD), and proximal duplications (PD).
4.8. Transcriptome Analysis
We verified the completeness and correctness of the downloaded raw transcriptome data following the parameter “vdb-validate”. We aligned the RNA-seq data to the reference genomes using HISAT2 (https://daehwankimlab.github.io/hisat2/, 20 August 2022) [66] and calculated the expression level for each gene using StringTie (https://github.com/gpertea/stringtie, 20 August 2022) [66].
4.9. Conserved Domain Search and Protein Structure Prediction
All protein sequences of MADS18 and MADS6 were submitted to the conserved domain search service of NCBI to identify the conserved domains (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi, accessed on 19 July 2024).
The protein sequences of MdMADS18a, MdMADS18b, MdMADS6a, MdMADS6b, and MdMADS6c were submitted to the AlphaFold Server (https://golgi.sandbox.google.com/, accessed on 19 July 2024) [67] generate highly accurate protein structures.
4.10. Plant Material and qRT-PCR
The tested M. × domestica plants were grown at the Shangzhuang experimental station of China Agricultural University in Beijing. Fresh leaves, fresh fruits at 30 DAP and 90 DAP, and fresh flowers at the late pink bud stage were collected. The tested P. avium plants were grown at the Beijing Academy of Agriculture and Forestry Sciences in Beijing. Fresh leaves, fresh fruits at 30 DAP, and fresh flowers were collected. The tested F. × ananassa plants were grown in the greenhouse in China Agricultural University in Beijing. Fresh leaves, fresh fruits at 30 DAP, and fresh flowers were collected. The RNA of tissue was extracted using the Promega RNG extraction kit (Promega Biotech, Beijing, China). cDNA was synthesized from 0.5 μg of total RNA using the ReverTra Ace™ qPCR RT Master Mix with gDNA Remover (Toyobo, Japan).
qRT-PCR was performed in a 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using TB Green Premix Ex Taq™ (TaKaRa, Dalian, China), according to the manufacturer’s instructions. The primers were designed using NCBI Primer BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/, 19 July 2024) under the following principles: specific primers were designed in the non-conserved regions of each sequence, with a length of 18–25 bp and a Tm value of around 60 °C, avoiding secondary structures and repetitive sequences. The product length was approximately 80–200 bp. For each sequence, 3–5 pairs of primers were designed, and their effectiveness was assessed by agarose gel electrophoresis. Finally, an optimal pair of primers was selected. The experiments were carried out in a total volume of 20 μL, containing 10 μL of 2× SYBR Green qPCR Mix (Beijing, China), 0.1 μM of each specific primer, and 100 ng of template cDNA. The reaction mixtures were heated to 95 °C for 10 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 32 s, 95 °C for 15 s, 60 °C for 60 s, increased by 0.3 °C each time. We measured the melt curve until the end. Three biological replicates were performed for each sample and normalized using the actin gene as an internal control. The 2−ΔΔCt method [68] was used to calculate the relative expression levels. Detailed information about the actin gene and primers is shown in (Table S4).
5. Conclusions
In this study, we selected the genomes of Rosaceae species from before and after WGD and focused on the differences in the fruit types and flower organs. We reconstructed the phylogenetic tree and the ancestral Rosaceae karyotype (ARK) with nine ancient chromosomes. The results revealed that the Maleae tribe has continuously experienced chromosome breakage and rearrangement following the WGD, and it was inferred that this event occurred at approximately 14.69 Ma. We identified approximately 30.71% of genes that underwent duplication and functional differentiation following the WGD. Among these, the orthologous groups of MADS18 and MADS6 contained both WGD and single gene duplication copies. These copies underwent sequence differential evolution and exhibited significant differential expression in apple fruits at different developmental stages and in different floral organs, providing new insights into and solid evidence for the impact of WGD on species evolution.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25168962/s1. References [12,43,69,70,71,72,73,74,75,76,77,78,79,80,81] are cited in the Supplementary Materials.
Author Contributions
B.W., W.L. and T.L. designed the experiments. B.W. performed the analyses. Y.X. performed the qRT-PCR experiment. B.W., Y.Z., W.L. and T.L. wrote the manuscript together. M.Y. and W.F. performed material collection. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (32102323) and the Project of the Joint Research Institute of China Agricultural University in Aksu.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in FigShare at 10.6084/m9.figshare.26425204, reference number https://doi.org/10.6084/m9.figshare.6025748, accessed on 1 August 2024.
Acknowledgments
We would like to thank Jinyao Ouyang (China Agricultural University) and ChuanBao Wu (Beijing Academy of Agriculture and Forestry Sciences) for their plant materials. We would also like to express our appreciation to the providers of the public data used in this article. Their generous sharing of the data facilitated our research.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ren, R.; Wang, H.; Guo, C.; Zhang, N.; Zeng, L.; Chen, Y.; Ma, H.; Qi, J. Widespread Whole Genome Duplications Contribute to Genome Complexity and Species Diversity in Angiosperms. Mol. Plant 2018, 11, 414–428. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 2016, 30, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 2005, 8, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, S.; Chang, X.; Wang, X.; Zhao, Y.; Xia, Y.; Trigiano, R.N.; Jiao, Y.; Chen, F. The ancient wave of polyploidization events in flowering plants and their facilitated adaptation to environmental stress. Plant Cell Environ. 2020, 43, 2847–2856. [Google Scholar] [CrossRef]
- Jiao, Y.N.; Leebens-Mack, J.; Ayyampalayam, S.; Bowers, J.E.; McKain, M.R.; McNeal, J.; Rolf, M.; Ruzicka, D.R.; Wafula, E.; Wickett, N.J.; et al. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 2012, 13, R3. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.N.; Li, J.P.; Tang, H.B.; Paterson, A.H. Integrated Syntenic and Phylogenomic Analyses Reveal an Ancient Genome Duplication in Monocots. Plant. Cell 2014, 26, 2792–2802. [Google Scholar] [CrossRef]
- Chen, Y.C.; Li, Z.; Zhao, Y.X.; Gao, M.; Wang, J.Y.; Liu, K.W.; Wang, X.; Wu, L.W.; Jiao, Y.L.; Xu, Z.L.; et al. The Litsea genome and the evolution of the laurel family. Nat. Commun. 2020, 11, 1675. [Google Scholar] [CrossRef]
- Zhang, L.S.; Chen, F.; Zhang, X.T.; Li, Z.; Zhao, Y.Y.; Lohaus, R.; Chang, X.J.; Dong, W.; Ho, S.; Liu, X.; et al. The water lily genome and the early evolution of flowering plants. Nature 2020, 577, 79. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.E.; Chapman, B.A.; Rong, J.; Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 2003, 422, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Qiao, Q.; Du, X.; Zhang, X.; Hou, Y.; Wei, X.; Sun, C.; Zhang, R.; Yun, Q.; Crabbe, M.J.C.; et al. Cultivated hawthorn (Crataegus pinnatifida var. major) genome sheds light on the evolution of Maleae (apple tribe). J. Integr. Plant Biol. 2022, 64, 1487–1501. [Google Scholar] [CrossRef] [PubMed]
- Potter, D.; Eriksson, T.; Evans, R.C.; Oh, S.; Smedmark, J.E.E.; Morgan, D.R.; Kerr, M.; Robertson, K.R.; Arsenault, M.; Dickinson, T.A.; et al. Phylogeny and classification of Rosaceae. Plant Syst. Evol. 2007, 266, 5–43. [Google Scholar] [CrossRef]
- Zhang, S.D.; Jin, J.J.; Chen, S.Y.; Chase, M.W.; Soltis, D.E.; Li, H.T.; Yang, J.B.; Li, D.Z.; Yi, T.S. Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics. New Phytol. 2017, 214, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, H.; Jung, S.; Main, D.; Guo, L. Developmental Mechanisms of Fleshy Fruit Diversity in Rosaceae. Annu. Rev. Plant Biol. 2020, 71, 547–573. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Huang, C.; Hu, Y.; Wen, J.; Li, S.; Yi, T.; Chen, H.; Xiang, J.; Ma, H. Evolution of Rosaceae Fruit Types Based on Nuclear Phylogeny in the Context of Geological Times and Genome Duplication. Mol. Biol. Evol. 2017, 34, 262–281. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhang, T.; Zhao, Y.; Dong, H.; Chen, H.; Hu, Y.; Huang, C.H.; Xiang, J.; Ma, H. Angiosperm-wide analysis of fruit and ovary evolution aided by a new nuclear phylogeny supports association of the same ovary type with both dry and fleshy fruits. J. Integr. Plant Biol. 2024, 66, 228–251. [Google Scholar] [CrossRef]
- Su, W.B.; Zhu, Y.M.; Zhang, L.; Yang, X.H.; Gao, Y.S.; Lin, S.Q. The cellular physiology of loquat (Eriobotrya japonica Lindl.) fruit with a focus on how cell division and cell expansion processes contribute to pome morphogenesis. Sci. Hortic. 2017, 224, 142–149. [Google Scholar] [CrossRef]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.Y.; Darwish, O.; Geretz, A.; Shahan, R.; Alkharouf, N.; Liu, Z.C. Genome-Scale Transcriptomic Insights into Early-Stage Fruit Development in Woodland Strawberry Fragaria vesca. Plant. Cell 2013, 25, 1960–1978. [Google Scholar] [CrossRef]
- Mezzetti, B.; Landi, L.; Pandolfini, T.; Spena, A. The defH9-iaaM auxin-synthesizing gene increases plant fecundity and fruit production in strawberry and raspberry. BMC Biotechnol. 2004, 4, 4. [Google Scholar] [CrossRef]
- Becker, A.; Theissen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Theißen, G.; Melzer, R.; Rümpler, F. MADS-domain transcription factors and the floral quartet model of flower development: Linking plant development and evolution. Development 2016, 143, 3259–3271. [Google Scholar] [CrossRef]
- Becker, A.; Kaufmann, K.; Freialdenhoven, A.; Vincent, C.; Li, M.A.; Saedler, H.; Theissen, G. A novel MADS-box gene subfamily with a sister-group relationship to class B floral homeotic genes. Mol. Genet. Genom. 2002, 266, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.L.; Smyth, D.R.; Meyerowitz, E.M. The ABC model of flower development: Then and now. Development 2012, 139, 4095–4098. [Google Scholar] [CrossRef] [PubMed]
- Causier, B.; Schwarz-Sommer, Z.; Davies, B. Floral organ identity: 20 years of ABCs. Semin. Cell Dev. Biol. 2010, 21, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Theissen, G.; Melzer, R. Combinatorial Control of Floral Organ Identity by MADS-Domain Transcription Factors; Wiley: Hoboken, NJ, USA, 2006; pp. 253–265. [Google Scholar]
- Favaro, R.; Pinyopich, A.; Battaglia, R.; Kooiker, M.; Borghi, L.; Ditta, G.; Yanofsky, M.F.; Kater, M.M.; Colombo, L. MADS-box protein complexes control carpel and ovule development in Arabidopsis. Plant. Cell 2003, 15, 2603–2611. [Google Scholar] [CrossRef]
- Pinyopich, A.; Ditta, G.S.; Savidge, B.; Liljegren, S.J.; Baumann, E.; Wisman, E.; Yanofsky, M.F. Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature 2003, 424, 85–88. [Google Scholar] [CrossRef]
- Battaglia, R.; Brambilla, V.; Colombo, L.; Stuitje, A.R.; Kater, M.M. Functional analysis of MADS-box genes controlling ovule development in Arabidopsis using the ethanol-inducible alc gene-expression system. Mech. Dev. 2006, 123, 267–276. [Google Scholar] [CrossRef]
- Laitinen, R.; Broholm, S.; Albert, V.A.; Teeri, T.H.; Elomaa, P. Patterns of MADS-box gene expression mark flower-type development in Gerbera hybrida (Asteraceae). BMC Plant Biol. 2006, 6, 11. [Google Scholar] [CrossRef]
- Eckardt, N.A. MADS monsters: Controlling floral organ identity. Plant. Cell 2003, 15, 803–805. [Google Scholar] [CrossRef]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef]
- Vekemans, D.; Proost, S.; Vanneste, K.; Coenen, H.; Viaene, T.; Ruelens, P.; Maere, S.; Van de Peer, Y.; Geuten, K. Gamma paleohexaploidy in the stem lineage of core eudicots: Significance for MADS-box gene and species diversification. Mol. Biol. Evol. 2012, 29, 3793–3806. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Dong, Q.; Ji, Z.; Chi, F.; Cong, P.; Zhou, Z. Genome-wide identification and analysis of the MADS-box gene family in apple. Gene 2015, 555, 277–290. [Google Scholar] [CrossRef]
- Meng, D.; Cao, Y.; Chen, T.; Abdullah, M.; Jin, Q.; Fan, H.; Lin, Y.; Cai, Y. Evolution and functional divergence of MADS-box genes in Pyrus. Sci. Rep. 2019, 9, 1266. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhu, G.; Zhao, H.; Liu, H.; Luo, Y.; Xu, W.; Huang, M.; Wu, Y.; Wang, L. Genome-wide Identification of MADS-box Gene Family and Expression Analysis in Prunus sibirica. Mol. Plant Breed. 2020, 18, 6575–6585. [Google Scholar]
- Wells, C.E.; Vendramin, E.; Tarodo, S.J.; Verde, I.; Bielenberg, D.G. A genome-wide analysis of MADS-box genes in peach [Prunus persica (L.) Batsch]. BMC Plant Biol. 2015, 15, 41. [Google Scholar] [CrossRef]
- Nie, C.R.; Xu, X.G.; Zhang, X.Q.; Xia, W.S.; Sun, H.B.; Li, N.; Ding, Z.Q.; Lv, Y.M. Genome-Wide Identified MADS-Box Genes in Prunus campanulata ‘Plena’ and Theirs Roles in Double-Flower Development. Plants 2023, 12, 3171. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Cestaro, A.; Troggio, M.; Main, D.; Zheng, P.; Cho, I.; Folta, K.M.; Sosinski, B.; Abbott, A.; Celton, J.M.; et al. Whole genome comparisons of Fragaria, Prunus and Malus reveal different modes of evolution between Rosaceous subfamilies. BMC Genom. 2012, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Shi, X.; Lin, X.; Liu, Y.; Chong, K.; Theißen, G.; Meng, Z. The ABCs of flower development: Mutational analysis of AP1/FUL-like genes in rice provides evidence for a homeotic (A)-function in grasses. Plant J. 2017, 89, 310–324. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Chiu, Y.F.; Wu, J.W.; Yang, C.H. Four orchid (Oncidium Gower Ramsey) AP1/AGL9-like MADS box genes show novel expression patterns and cause different effects on floral transition and formation in Arabidopsis thaliana. Plant Cell Physiol. 2009, 50, 1425–1438. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, A.M.; Dandekar, A.M. Apple. In Transgenic Crops V. Biotechnology in Agriculture and Forestry; Springer: Berlin/Heidelberg, Germany, 2007; pp. 241–282. [Google Scholar] [CrossRef]
- Shulaev, V.; Korban, S.S.; Sosinski, B.; Abbott, A.G.; Aldwinckle, H.S.; Folta, K.M.; Iezzoni, A.; Main, D.; Arus, P.; Dandekar, A.M.; et al. Multiple models for Rosaceae genomics. Plant Physiol. 2008, 147, 985–1003. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Miao, D.; Miao, R.; Fan, D.; Meng, J.; Liu, X.; Cheng, T.; Zhang, Q.; Sun, L. Advances in the omics research of Rosaceae. Ornam. Plant Res. 2024, 4, e013. [Google Scholar] [CrossRef]
- Jung, S.; Lee, T.; Cheng, C.H.; Buble, K.; Zheng, P.; Yu, J.; Humann, J.; Ficklin, S.P.; Gasic, K.; Scott, K.; et al. 15 years of GDR: New data and functionality in the Genome Database for Rosaceae. Nucleic Acids. Res. 2019, 47, D1137–D1145. [Google Scholar] [CrossRef]
- Jacob, P.; Hirt, H.; Bendahmane, A. The heat-shock protein/chaperone network and multiple stress resistance. Plant Biotechnol. J. 2017, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Heimhofer, U.; Hochuli, P.A.; Burla, S.; Dinis, J.; Weissert, H. Timing of Early Cretaceous angiosperm diversification and possible links to major paleoenvironmental change. Geology 2005, 33, 141–144. [Google Scholar] [CrossRef]
- Nie, Y.; Guo, L.; Cui, F.; Shen, Y.; Ye, X.; Deng, D.; Wang, S.; Zhu, J.; Wu, W. Innovations and stepwise evolution of CBFs/DREB1s and their regulatory networks in angiosperms. J. Integr. Plant Biol. 2022, 64, 2111–2125. [Google Scholar] [CrossRef]
- Thomashow, M.F. Plant cold acclimation: Freezing tolerance genes and regulatory mechanisms. Annu. Rev. Plant Biol. 1999, 50, 571–599. [Google Scholar] [CrossRef]
- Park, S.; Lee, C.M.; Doherty, C.J.; Gilmour, S.J.; Kim, Y.; Thomashow, M.F. Regulation of the Arabidopsis CBF regulon by a complex low-temperature regulatory network. Plant J. 2015, 82, 193–207. [Google Scholar] [CrossRef]
- Chaudhury, A.M.; Craig, S.; Dennis, E.S.; Peacock, W.J. Ovule and embryo development, apomixis and fertilization. Curr. Opin. Plant Biol. 1998, 1, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Cao, Z.Y.; Wu, P.Y.; Zhang, X.; Lou, J.; Liu, Y.N.; Wang, Q.Y.; Hu, Y.; Si, S.; Sun, X.F.; et al. Genome-wide identification, interaction of the MADS-box proteins in Zanthoxylum armatum and functional characterization of ZaMADS80 in floral development. Front. Plant Sci. 2022, 13, 1038828. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, L.A.; Dusi, D.; Masiero, S.; Resentini, F.; Gomes, A.; Silveira, E.D.; Florentino, L.H.; Rodrigues, J.; Colombo, L.; Carneiro, V. BbrizAGL6 Is Differentially Expressed During Embryo Sac Formation of Apomictic and Sexual Brachiaria brizantha Plants. Plant Mol. Biol. Rep. 2013, 31, 1397–1406. [Google Scholar] [CrossRef][Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Wang, S.; Wei, S.; Deng, Y.; Wu, S.; Peng, H.; Qing, Y.; Zhai, X.; Zhou, S.; Li, J.; Li, H.; et al. HortGenome Search Engine, a universal genomic search engine for horticultural crops. Hortic. Res. 2024, 11, uhae100. [Google Scholar] [CrossRef]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2020, 36, 5516–5518. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Sun, P.C.; Jiao, B.B.; Yang, Y.Z.; Shan, L.X.; Li, T.; Li, X.N.; Xi, Z.X.; Wang, X.Y.; Liu, J.Q. WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant 2022, 15, 1841–1851. [Google Scholar] [CrossRef]
- Wang, Y.P.; Tang, H.B.; DeBarry, J.D.; Tan, X.; Li, J.P.; Wang, X.Y.; Lee, T.H.; Jin, H.Z.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, Q.; Yan, X.; Wu, X.; Yang, F.; Li, J.; Wei, J.; Ni, J.; Ahmad, M.; Bai, S.; et al. High-quality genome assembly of ‘Cuiguan’ pear (Pyrus pyrifolia) as a reference genome for identifying regulatory genes and epigenetic modifications responsible for bud dormancy. Hortic. Res. 2021, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Jing, Y.; Lin, S.; Yue, Z.; Yang, X.; Xu, J.; Wu, J.; Zhang, Z.; Xia, R.; Zhu, J.; et al. Polyploidy underlies co-option and diversification of biosynthetic triterpene pathways in the apple tribe. Proc. Natl. Acad. Sci. USA 2021, 118, e2101767118. [Google Scholar] [CrossRef]
- The International Peach Genome Initiative; Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Le Dantec, L.; Girollet, N.; Gouzy, J.; Sallet, E.; Fouche, M.; Quero-Garcia, J.; Dirlewanger, E. An improved assembly of the diploid ‘Regina’ Sweet cherry genome. In Proceedings of the International Plant & Animal Genome XXVII, San Diego, CA, USA, 12–16 January 2019. [Google Scholar]
- Groppi, A.; Liu, S.; Cornille, A.; Decroocq, S.; Bui, Q.T.; Tricon, D.; Cruaud, C.; Arribat, S.; Belser, C.; Marande, W.; et al. Population genomics of apricots unravels domestication history and adaptive events. Nat. Commun. 2021, 12, 3956. [Google Scholar] [CrossRef]
- Huang, Z.; Shen, F.; Chen, Y.; Cao, K.; Wang, L. Chromosome-scale genome assembly and population genomics provide insights into the adaptation, domestication, and flavonoid metabolism of Chinese plum. Plant J. 2021, 108, 1174–1192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, M.; Li, Y.; Xiao, Y.; Luo, X.; Gao, S.; Ma, Z.; Sadowski, N.; Timp, W.; Dardick, C.; et al. Comparison of red raspberry and wild strawberry fruits reveals mechanisms of fruit type specification. Plant Physiol. 2023, 193, 1016–1035. [Google Scholar] [CrossRef] [PubMed]
- VanBuren, R.; Wai, C.M.; Colle, M.; Wang, J.; Sullivan, S.; Bushakra, J.M.; Liachko, I.; Vining, K.J.; Dossett, M.; Finn, C.E.; et al. A near complete, chromosome-scale assembly of the black raspberry (Rubus occidentalis) genome. Gigascience 2018, 7, giy094. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Aryal, R.; Dudchenko, O.; Sargent, D.J.; Mead, D.; Buti, M.; Cavallini, A.; Hytönen, T.; Andres, J.; Pham, M.; et al. A chromosome-length genome assembly and annotation of blackberry (Rubus argutus, cv.“Hillquist”). G3 Genes|Genomes|Genetics 2023, 13, jkac289. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Peng, Y.; Liu, L.; Li, G.; Zhao, X.; Wang, X.; Cao, S.; Muyle, A.; Zhou, Y.; Zhou, H. Phased gap-free genome assembly of octoploid cultivated strawberry illustrates the genetic and epigenetic divergence among subgenomes. Hortic. Res. 2023, 11, uhad252. [Google Scholar] [CrossRef] [PubMed]
- Massonnet, M.; Cochetel, N.; Minio, A.; Vondras, A.M.; Lin, J.; Muyle, A.; Garcia, J.F.; Zhou, Y.; Delledonne, M.; Riaz, S.; et al. The genetic basis of sex determination in grapes. Nat. Commun. 2020, 11, 2902. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, J.; Han, X.; Li, J.; Gao, Y.; Richards, C.M.; Zhang, C.; Tian, Y.; Liu, G.; Gul, H.; et al. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 2019, 10, 1494. [Google Scholar] [CrossRef]
- Duan, Y.; Ma, S.; Chen, X.; Shen, X.; Yin, C.; Mao, Z. Transcriptome changes associated with apple (Malus domestica) root defense response after Fusarium proliferatum f. sp. malus domestica infection. BMC Genom. 2022, 23, 484. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).