Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance

 , ,
, , 
Abstract
:1. Introduction
2. Epidemiology of HCC
3. Molecular Pathogenesis of HCC
4. Oxidative Stress and Inflammation in HCC
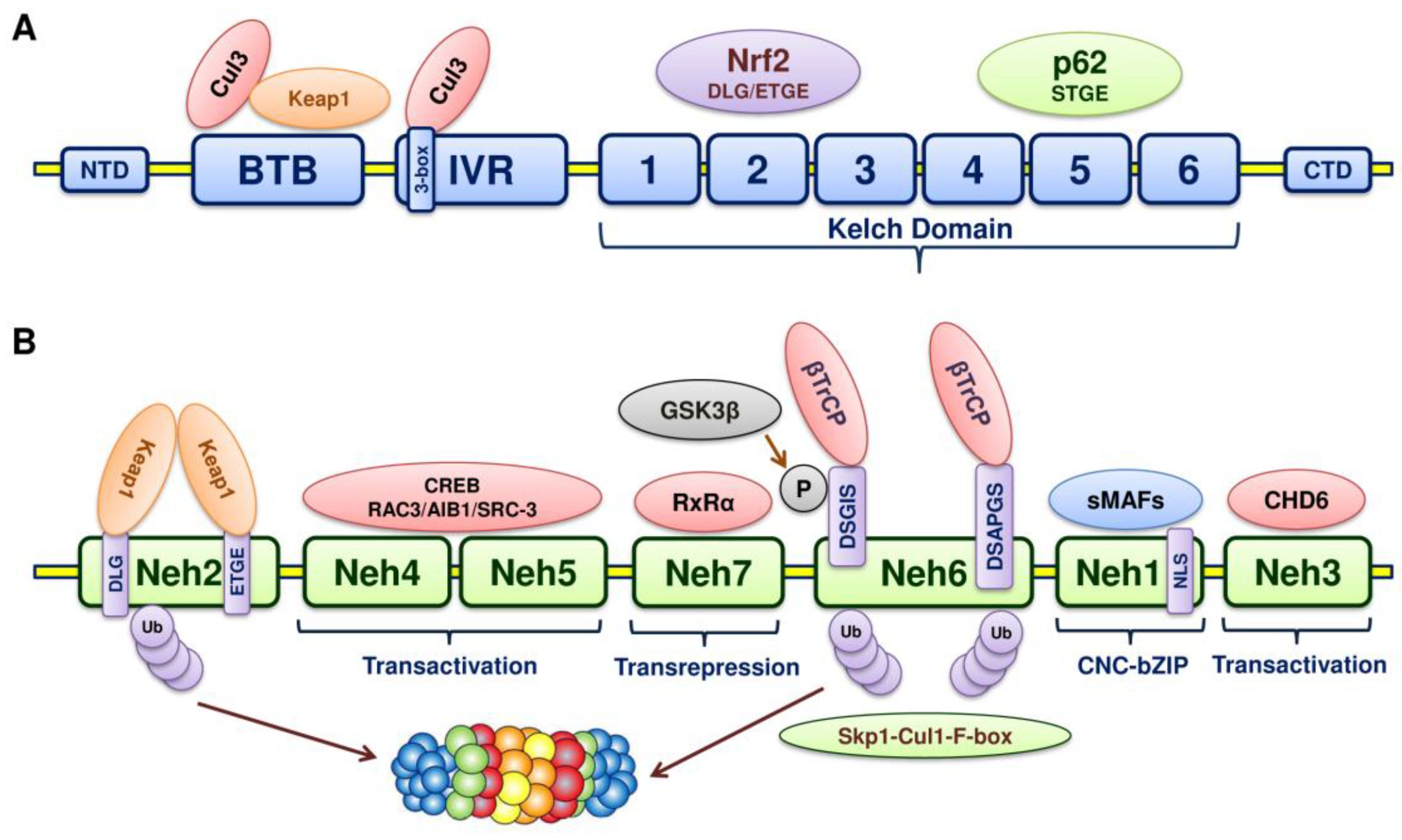
5. Structure of Nrf2
6. Structure of Keap1
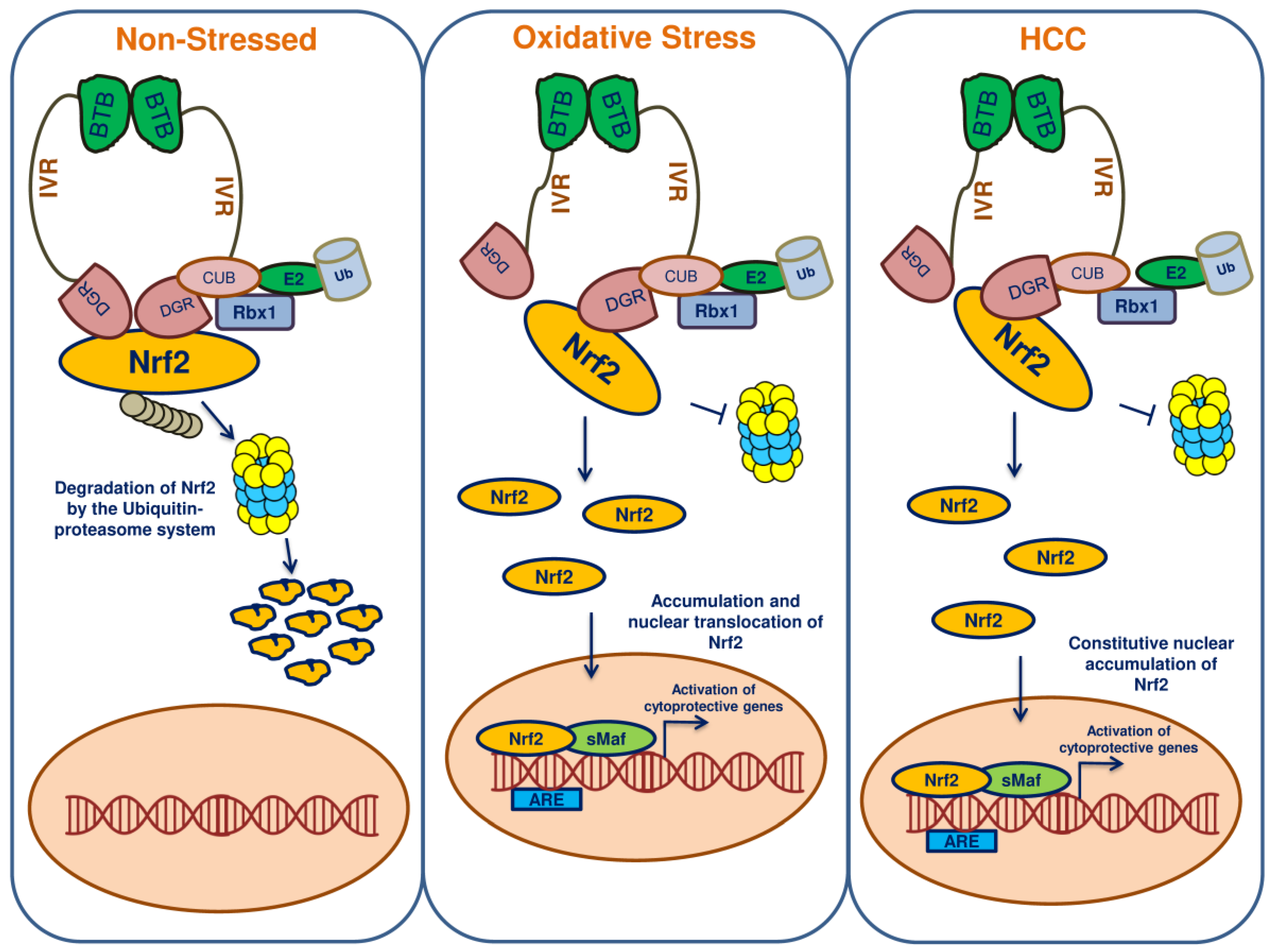
7. Keap1-Nrf2-ARE Pathway
8. Nrf2 and Keap1 Mutations Lead to HCC
9. Nrf2 in HCC
10. Autophagy, p62 and Nrf2 in HCC
11. Phytochemicals/Molecules Can Elicit Activation of Nrf2 in HCC
12. Phytochemicals/Molecules Sensitizing Resistant HCC through Nrf2 Signalling
13. Endoplasmic Reticulum Stress and Nrf2 Signalling in HCC
14. Metal Complexes and Nrf2 in HCC
15. miRs Regulation/Dysregulation of Nrf2 in HCC
16. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Heimbach, J.K.; Kulik, L.M.; Finn, R.S.; Sirlin, C.B.; Abebcassis, M.M.; Roberts, L.R.; Zhu, A.X.; Murad, M.H.; Marrero, J.A. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018, 67, 358–380. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Takaki, A.; Yamamoto, K. Control of oxidative stress in hepatocellular carcinoma: Helpful or harmful? World J. Hepatol. 2015, 7, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Diaber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Turati, F.; La Vecchia, C. Hepatocellular carcinoma epidemiology. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 753–770. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts and Figures; American Cancer Society Inc.: Atlanta, GA, USA, 2018. [Google Scholar]
- Ozakyol, A. Global epidemiology of hepatocellular carcinoma (HCC epidemiology). J. Gastrointest. Cancer 2017, 48, 238–240. [Google Scholar] [CrossRef]
- Tang, A.; Hallouch, O.; Chernyak, V.; Kamaya, A.; Sirlin, C.B. Epidemiology of hepatocellular carcinoma: Target population for surveillance and diagnosis. Abdom. Radiol. 2018, 43, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Nault, J.C.; Llovet, J.M. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 2015, 149, 1226–1239. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Petrick, J.L.; London, W.T. Global epidemiology of hepatocellular carcinoma: An emphasis on demographic and regional variability. Clin. Liver Dis. 2015, 19, 223–238. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma: Consider the population. J. Clin. Gastroenterol. 2013, 47, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular carcinoma in the absence of cirrhosis in united states veterans is associated with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Han, Y.; Xu, C.; Xiao, T.; Wang, B. Effect of type 2 diabetes mellitus on the risk for hepatocellular carcinoma in chronic liver diseases: A meta-analysis of cohort studies. Eur. J. Cancer Prev. 2015, 24, 89–99. [Google Scholar] [CrossRef]
- Lee, Y.C.; Cohet, C.; Yang, Y.C.; Stayner, L.; Hashibe, M.; Straif, K. Meta-analysis of epidemiologic studies on cigarette smoking and liver cancer. Int. J. Epidemiol. 2009, 38, 1497–1511. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Nishimura, T.; Nagasaka, T.; Ikai, I.; Goael, A.; Boland, C.R. Extensive methylation is asscociated with beta-catenin mutations in hepatocellular carcinoma: Evidence for two distinct pathways of human hepatocarcinogenesis. Cancer Res. 2007, 67, 4586–4594. [Google Scholar] [CrossRef]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef] [Green Version]
- Calado, R.T.; Brudno, J.; Mehta, P.; Kovacs, J.J.; Zago, M.A.; Chanock, S.J.; Boyer, T.D.; Young, N.S. Constitutional telomerase mutations are genetis risk factors for cirrhosis. Hepatology 2011, 53, 1600–1607. [Google Scholar] [CrossRef]
- Cleary, S.P.; Jeck, W.R.; Zhao, X.; Chen, K.; Selitsky, S.R.; Savich, G.L.; Tan, T.X.; Wu, M.C.; Getz, G.; Lawrence, M.S.; et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology 2013, 58, 1693–1702. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.W.; Klimstra, D.S.; Mongeau, M.E.; Tatem, J.L.; Boyartchuk, V.; Lewis, B.C. Loss of p53 and Ink4a/Arf cooperate in a cell autonomous fashion to induce metastasis of hepatocellular carcinoma cells. Cancer Res. 2007, 67, 7589–7596. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.B.; Nagae, G.; Midorikawa, Y.; Yagi, K.; Tsutsumi, S.; Yamamoto, S.; Hasegawa, K.; Kokudo, N.; Aburatani, H.; Kaneda, A. Identification of genes preferentially methylated in hepatitis C virus-related hepatocellular carcinoma. Cancer Sci. 2010, 101, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Tischoff, I.; Tannapfe, A. DNA methylation in hepatocellular carcinoma. World J. Gastroenterol. 2008, 14, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- EI-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Moon, R.T.; Bowerman, B.; Boutros, M.; Perrimon, N. The promise and perils of Wnt signaling through beta-catenin. Science 2002, 296, 1644–1646. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Newell, P.; Chiang, D.Y.; Friedman, S.L.; Llovet, J.M. Genomics and signaling pathways in hepatocellular carcinoma. Semin. Liver Dis. 2007, 27, 55–76. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, EA.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 117–1128. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.; Olivarez, M.; Waris, G. Activation of transcription factor Nrf2 by hepatitis C virus induces the cell survivial pathway. J. Gen. Virol. 2010, 91, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Schaedler, S.; Krause, J.; Himmelsbach, K.; Carvajal-Yepes, M.; Lieder, F.; Klingel, K.; Nassal, M.; Weiss, T.S.; Werner, S.; Hildt, E. Hepatitis B virus induces expression of antioxidant response element regulated genes by activation of Nrf2. J. Biol. Chem. 2010, 285, 41074–41086. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Cederbaum, A.I. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology 2006, 43, 144–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.T.; Arjumand, W.; Nafees, S.; Seth, A.; Ali, N.; Rashid, S.; Sultana, S. Hesperidin alleviates acetaminophen induced toxicity in wistar rats by abrogation of oxidative stress, apoptosis and inflammation. Toxicol. Lett. 2012, 208, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role of STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Sutter, T.R.; Kensler, T.W. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol. Med. 2001, 7, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, C.; Zhang, L.; Yang, Q.; Zhou, S.; Wen, Q.; Wang, J. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer 2015, 15, 531. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Yepes, M.; Himmelsbach, K.; Schaedler, S.; Ploen, D.; Krause, J.; Ludwig, L.; Weiss, T.; Klingel, K.; Hildt, E. Hepatitis C virus impairs the induction of cytoprotective Nrf2 target genes by delocalization of small Maf proteins. J. Biol. Chem. 2011, 286, 8941–8951. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Chouteau, P.; Lerat, H. Liver let die: Oxidative DNA damage and hepatotropic viruses. J. Gen. Virol. 2014, 95, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Tsai, W.L.; Shao, R.X.; Wu, G.; Peng, L.F.; Barlow, L.L.; Chung, W.J.; Zhang, L.; Zhao, H.; Jang, J.Y.; et al. Hepatitis C virus regulates transforming growth factor beta 1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gasteroenterology 2010, 138, 2509–2518. [Google Scholar] [CrossRef]
- Malik, A.N.; Czajka, A. Is mitochondria DNA content a potential biomarker of mitochondrial dysfunction. Mitochondrion 2013, 13, 481–492. [Google Scholar] [CrossRef]
- Lambert, M.P.; Paliwal, A.; Vaissiere, T.; Chemin, I.; Zoulum, F.; Tommasino, M.; Hainaut, P.; Sylla, B.; Scoazec, J.Y.; Tost, J.; et al. Aberrant DNA methylation distinguishes hepatocellular carcinoma associated with HBV and HCV infection and alcohol intake. J. Hepatol. 2011, 54, 705–715. [Google Scholar] [CrossRef]
- Hosel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; Langenkamp, A.; et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, X.; Harry, B.L.; Zhou, Q.; Skeen-Gaar, R.R.; Ge, X.; Lee, E.S.; Mitani, S.; Xue, D. Hepatitis B virus X protein targets the Bcl-2 protein CED-9 to induce intracellular Ca2+ increase and cell death in caenorhabditid elegans. Proc. Natl. Acad. Sci. USA 2012, 109, 18465–18470. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Bouchard, M.J. Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J. Virol. 2008, 82, 6798–6811. [Google Scholar] [CrossRef] [PubMed]
- Tordjmann, T.; Soulie, A.; Guettier, C.; Schmidt, M.; Berthou, C.; Beaugrand, M.; Sasportes, M. Perforin and granzyme B lytic protein expression during chronic viral and autoimmune hepatitis. Liver 1998, 18, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, A.; Reis, H.M.; Feitelson, M.A. Pathogenic mechanisms in HBV- and HCV associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alix-Panabieres, C.; Schwarzenbach, H.; Pantel, K. Circulating tumor cells and circulating tumor DNA. Annu. Rev. Med. 2012, 63, 199–215. [Google Scholar] [CrossRef]
- Ng, C.K.Y.; Di Costanzo, G.G.; Terracciano, L.M.; Piscuoglio, S. Circulating cell-free DNA in hepatocellular carcinoma: Current insights and outlook. Front. Med. 2018, 26, 78. [Google Scholar] [CrossRef]
- Gailhouste, L.; Gomez-Santos, L.; Ochiya, T. Potential applications of miRNAs as diagnostic and prognostic markers in liver cancer. Front. Biosci. 2013, 18, 199–223. [Google Scholar]
- Cardin, R.; Picocchi, M.; Sinigaglia, A.; Lavezzo, E.; Bortolami, M.; Kotsafti, A.; Cillo, U.; Zanus, G.; Mescoli, C.; Rugge, M.; et al. Oxidative DNA damage correlates with cell immortalization and mir-92 expression in hepatocellular carcinoma. BMC Cancer 2012, 12, 177. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. USA 2004, 27, 6379–6384. [Google Scholar] [CrossRef] [PubMed]
- Plafker, K.S.; Nguyen, L.; Barneche, M.; Mirza, S.; Crawford, D.; Plafker, S.M. The ubiquitin-conjugating enzyme UbcM2 can regulate the stability and activity of the antioxidant transcription factor Nrf2. J. Biol. Chem. 2010, 285, 23064–23074. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef]
- Kim, J.H.; Yu, S.; Chen, J.D.; Kong, A.N. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 2013, 32, 514–527. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobon-Velasco, J.C.; Devijver, H.; Garcia-Mayoral, M.F.; van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef]
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen ynthase kinase 3/β-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, V.J.; Treisman, R. The POZ domain: A conserved protein-protein interaction motif. Genes Dev. 1994, 8, 1664–1677. [Google Scholar] [CrossRef] [PubMed]
- Zipper, L.M.; Mulcahy, R.T. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohstuji, J.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Cooper, C.D.; Kroler, T.; Murray, J.W.; Pike, A.C.; Chaikuad, A.; Keates, T.; Thangaratnarajah, C.; Hoizan, V.; Ayinampudi, V.; et al. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J. Biol. Chem. 2013, 288, 7803–7814. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Hannink, M.; Beamer, L.J. Crystal structure of the Kelch domain of human Keap1. J. Biol. Chem. 2004, 279, 54750–54758. [Google Scholar] [CrossRef]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohstuji, M.; Kang, M.I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell. 2006, 21, 689–700. [Google Scholar] [CrossRef]
- Adams, J.; Kelso, R.; Cooley, L. The kelch repeat superfamily of proteins: Propellers of cell function. Trends Cell Biol. 2000, 10, 17–24. [Google Scholar] [CrossRef]
- Tong, K.I.; Kobayahi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem. 2006, 387, 1311–1320. [Google Scholar] [CrossRef]
- Lo, S.C.; Li, X.; Henzi, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef] [Green Version]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- Hast, B.E.; Cloer, E.W.; Goldfarb, D.; Li, H.; Siesser, P.F.; Yan, F.; Walter, V.; Zheng, N.; Hayes, D.N.; Major, M.B. Cancer-derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res. 2014, 74, 808–817. [Google Scholar] [CrossRef]
- Eades, G.; Yang, M.; Yao, Y.; Zhang, Y.; Zhou, Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J. Biol. Chem. 2011, 286, 40725–40733. [Google Scholar] [CrossRef]
- Van Jaarsveld, M.T.; Helleman, J.; Boersma, A.W.; van Kujik, P.F.; van Ijcken, W.F.; Despierre, E.; Vergote, I.; Mathijssen, R.H.; Berns, E.M.; Verweij, J.; et al. miR-141 regulates KEAP1 and modulates cisplatin sensitivity in ovarian cancer cells. Oncogene 2013, 32, 4284–4293. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Wu, B.; Yan, J.; Li, X.; Sun, H.; Zhou, D. A possible gene silencing mechanism: Hypermethylation of the Keap1 promoter abrogates binding of the transcription factor Sp1 in lung cancer cells. Biochem. Biophys. Res. Commun. 2012, 428, 80–85. [Google Scholar] [CrossRef]
- Jeong, Y.; Hoang, N.T.; Lovejoy, A.; Stehr, H.; Newman, A.M.; Gentles, A.J.; Kong, W.; Truong, D.; Martin, S.; Chaudhuri, A.; et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamus cell carcinoma development and radiation resistance. Cancer Discov. 2017, 7, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Itoch, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Iqarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifiying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The dual roles of NRF2 in cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaki, H.; Kawakami, Y.; Ueno, M.; Gotoh, K.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yu, Y.; Ji, T.; Ma, R.; Chen, M.; Li, G.; Li, F.; Ding, Q.; Kang, Q.; Huang, D.; et al. Clinical implication of Keap1 and phosphorylated Nrf2 expression in hepatocellular carcinoma. Cancer Med. 2016, 5, 2678–2687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavattari, P.; Perra, A.; Menegon, S.; Kowalik, M.A.; Petrelli, A.; Angioni, M.M.; Follenzi, A.; Quagliata, L.; Ledda-Columbano, G.M.; Terracciano, L.; et al. Nrf2, but not β-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology 2015, 62, 851–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 system in cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, J.C.; Zucman-Rossi, J. Genetics of hepatocellular carcinoma: The next generation. J. Hepatol. 2014, 60, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.K.C.; Kim, D.H.; Cha, Y.N.; Na, H.K.; Surh, Y.J. Nrf2 mutagenic activation drives hepatocarcinogenesis. Cancer Res. 2017, 77, 4797–4808. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Dhar, D. Liver carcinogenesis: From naughty chemicals to soothing fat and the surprising role of NRF2. Carcinogenesis 2016, 37, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Jaiswal, A.K. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic. Biol. Med. 2013, 57, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma-On, C.; Sanpavat, A.; Whongsiri, P.; Suwannasin, S.; Hirankarn, N.; Tangkijvanich, P.; Boonla, C. Oxidative stress indicated by elevated expression of Nrf2 and 8-OHdG promotes hepatocellular carcinoma progression. Med. Oncol. 2017, 34, 57. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, M.; Kuroiwa, Y.; Inoue, T.; Hibi, D.; Matsushita, K.; Kijima, A.; Maruyama, S.; Nishikawa, A.; Umemura, T. Lack of nrf2 results in progression of proliferative lesions to neoplasms induced by long-term exposure to non-genotoxic hepatocarcinogens involving oxidative stress. Exp. Toxicol. Pathol. 2014, 66, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Nishi, S.; Sakai, M. Transcription factor Nrf2/MafK regulates rat placental glutathione S-transferase gene during hepatocarcinogenesis. Biochem. J. 2004, 380, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Inoue, K.; Hachiva, H.; Shibuya, N.; Aoki, T.; Kubota, K. Accumulation of phosphorylated p62 is associated with NF-E2-related factor 2 activation in hepatocellular carcinoma. J. Hepatobiliary Pancreat Sci. 2016, 23, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantely, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Umemura, A.; He, F.; Taniquchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Dodson, M.; Chapman, E.; Zhang, D.D. NRF2-targeted therapeutics: New targets and modes of NRF2 regulation. Curr. Opin. Toxicol. 2016, 1, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolini, D.; Dallaglio, K.; Torguato, P.; Piroddi, M.; Galli, F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. 2018, 193, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, H.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.; Lu, Z.N.; Guo, X.L. Regulation and role of nuclear factor-E2-related factor 2 (Nrf2) in multidrug resistance of hepatocellular carcinoma. Chem. Biol. Interact. 2018, 280, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Kato, S.; Saeki, M.; DeMartino, G.N.; Slaughter, C.A.; Takeuchi, J.; Toh-e, A.; Tanaka, K. cDNA cloning and functional analysis of p28 (Nas6p) and p40.5 (Nas7p), two novel regulatory subunits of the 26S proteasome. Gene 1998, 216, 113–122. [Google Scholar] [CrossRef]
- Higasthitsuji, H.; Iton, K.; Nagao, T.; Dawson, S.; Nonoguchi, K.; Kido, T.; Maver, R.J.; Arii, S.; Fujita, J. Reduced stability of retinoblastoma protein by gankyrin, an oncogenic ankyrin-repeat protein overexpressed in hepatomas. Nat. Med. 2000, 6, 96–99. [Google Scholar] [CrossRef]
- Fu, X.Y.; Wang, H.Y.; Tan, L.; Liu, S.Q.; Cao, H.F.; Wu, M.C. Overexpression of p28/gankyrin in human hepatocellular carcinoma and its clinical significance. World J. Gastroenterol. 2002, 8, 638–643. [Google Scholar] [CrossRef]
- Yang, C.; Tan, Y.X.; Yang, G.Z.; Zhang, J.; Pan, Y.F.; Liu, C.; Fu, J.; Chen, Y.; Ding, Z.W.; Dong, L.W.; et al. Gankyrin has an antioxidative role through the feedback regulation of Nrf2 in hepatocellular carcinoma. J. Exp. Med. 2016, 213, 859–875. [Google Scholar] [CrossRef] [Green Version]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef]
- Sid, B.; Glorieux, C.; Valenzuela, M.; Rommelaere, G.; Najimi, M.; Dejeans, N.; Renard, P.; Verrax, J.; Calderon, P.B. AICAR induces Nrf2 activation by an AMPK-independent mechanism in hepatocarcinoma cells. Biochem. Pharmacol. 2014, 91, 168–180. [Google Scholar] [CrossRef]
- Yokomizo, T.; Izumi, T.; Takahashi, T.; Kasama, T.; Kobayashi, Y.; Sato, F.; Taketni, Y.; Shimizu, T. Enzymatic inactivation of leukotriene B4 by a novel enzyme found in the porcine kidney. Purification and properties of leukotriene B4 12-hydroxydehydrogenase. J. Biol. Chem. 1993, 268, 18128–18135. [Google Scholar] [PubMed]
- Sanchez-Rodriguez, R.; Torres-Mena, J.E.; De-la-Luz-Crus, M.; Bernal-Ramos, G.A.; Villa-Trevino, S.; Chagoya-Hazas, L.; Landero-Lopez, L.; Garcia-Roman, R.; Rouimi, P.; Perez-Carreon, J.I.; et al. Increased expression of prostaglandin reductase 1 in hepatocellular carcinomas from clinical cases and experimental tumors in rats. Int. J. Biochem. Cell Biol. 2014, 53, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rodriguez, R.; Torres-Mena, J.E.; Quintanar-Jurado, V.; Chagoya-Hazas, V.; Rojas Del Castillo, E.; Del Pozo Yauner, L.; Villa-Trevino, S.; Perez-Carreon, J.I. Ptgr1 expression is regulated by NRF2 in rat hepatocarcinogenesis and promotes cell proliferation and resistance to oxidative stress. Free Radic. Biol. Med. 2017, 102, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J. More than spice: Capsaicin in hot chili peppers makes tumor cells commit suicide. J. Natl. Cancer Inst. 2002, 94, 1263–1265. [Google Scholar] [CrossRef]
- Joung, E.J.; Li, M.H.; Lee, H.G.; Somparn, N.; Jung, Y.S.; Na, H.K.; Kim, S.H.; Cha, Y.N.; Surh, Y.J. Capsaicin induces heme oxygenase-1 expression in HepG2 cells via activation of PI3K-Nrf2 signaling: NAD(P)H: Quinone oxidoreductase as a potential target. Antioxid. Redox Signal. 2007, 9, 2087–2098. [Google Scholar] [CrossRef] [PubMed]
- Feron, V.J.; Til, H.P.; de Vrijer, F.; Woutersen, R.A.; Cassee, F.R.; van Bladeren, P.J. Aldehydes: Occurrence, carcinogenic potential, mechanism of action and risk assessment. Mutat. Res. 1991, 259, 363–385. [Google Scholar] [CrossRef]
- Lee, S.E.; Yang, H.; Jeong, S.I.; Jin, Y.H.; Park, C.S.; Park, Y.S. Induction of heme oxygenase-1 inhibits cell death in crotonaldehyde-stimulated HepG2 cells via the PKC-δ-p38-Nrf2 pathway. PLoS ONE. 2012, 7, e41676. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, C.Y.; Lee, H.J.; Yun, J.H.; Nho, C.W. Induction of the phase II detoxification enzyme NQO1 in hepatocarcinoma cells by lignans from the fruit of Schisandra chinensis through nuclear accumulation of Nrf2. Planta Med. 2009, 75, 1314–1318. [Google Scholar] [CrossRef]
- Puri, M.; Sharma, D.; Tiwari, A.K. Downstream processing of stevioside and its potential applications. Biotechnol. Adv. 2011, 29, 781–791. [Google Scholar] [CrossRef]
- Ceunen, S.; Geuns, J.M. Steviol glycosides: Chemical diversity, metabolism, and function. J. Nat. Prod. 2013, 76, 1201–1228. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Esmaeili, S.A.; Abdollah, E.; Sahebkar, A. A review on the pharmacology and toxicology of steviol glycosides extracted from Stevia rebaudiana. Curr. Pharm. Des. 2017, 23, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Wang, Y.; Zhu, X.; Jiang, M.; Song, E.; Song, Y. New application of the commercial sweetener rebaudioside a as a hepatoprotective candidate: Induction of the Nrf2 signaling pathway. Eur. J. Pharmacol. 2018, 822, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Blanc, P.J.; Laussac, J.P.; Le Bars, J.; Le Bars, P.; Loret, M.O.; Pareilleux, A.; Prome, D.; Prome, J.C.; Santerre, A.L.; Goma, G. Characterization of monascidin A from Monascus as citrinin. Int. J. Food Microbiol. 1995, 27, 201–213. [Google Scholar] [CrossRef]
- Sharath Babu, G.R.; Anand, T.; Ilaiyaraja, N.; Khanum, F.; Gopalan, N. Peargonidin modulates Keap1/Nrf2 parhway gene expression and ameliorates citrinin induced oxidative stress in HepG2 cells. Front. Pharmacol. 2017, 8, 868. [Google Scholar] [CrossRef] [PubMed]
- Giusti, M.M.; Wrolstad, R.E. Characterization of red radish anthocyanins. J. Food Sci. 1996, 61, 322–326. [Google Scholar] [CrossRef]
- Gil, M.I.; Tomas-Barberan, F.A.; Hess-Pierce, B.; Holcroft, D.M.; Kader, A.A. Antioxidant activity of pomegranate juice and its relationship with phenolic composition and processing. J. Agric. Food. Chem. 2000, 48, 4581–4589. [Google Scholar] [PubMed]
- Faria, A.; Monteiro, R.; Mateus, N.; Azevedo, I.; Calhau, C. Effect of pomegranate (Punica granatum) juice intake on hepatic oxidative stress. Eur. J. Nutr. 2007, 46, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Bhatia, D.; Thoppil, R.T.; Darvesh, A.S.; Nevo, E.; Lansky, E.P. Pomegranate-mediated chemoprevention of experimental hepatocarcinogenesis involves Nrf2 regulated antioxidant mechanisms. Carcinogenesis 2011, 32, 888–896. [Google Scholar] [PubMed]
- Krajka-Kuzniak, V.; Paluszczak, J.; Szaefer, H.; Baer-Dubowska, W. The activation of the Nrf2/ARE pathway in HepG2 hepatoma cells by phytochemicals and subsequent modulation of phase II and antioxidant enzyme expression. J. Physiol. Biochem. 2015, 71, 227–238. [Google Scholar] [CrossRef]
- Wall, M.E.; Wani, M.C. Camptothecin and taxol: Discovery to clinic—Thirteenth Bruce F. Cain Memorial Award Lecture. Cancer Res. 1995, 55, 753–760. [Google Scholar] [PubMed]
- Chen, F.; Wang, H.; Zhu, J.; Zhao, R.; Xue, P.; Zhang, Q.; Bud Nelson, M.; Qu, W.; Feng, B.; Pi, J. Camptothecin suppresses NRF2-ARE activity and sensitises hepatocellular carcinoma cells to anticancer drugs. Br. J. Cancer 2017, 117, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Yin, S.; Huo, Y.; Liang, M.; Fan, L.; Ye, M.; Hu, H. Glycycoumarin ameliorates alcohol-induced hepatotoxicity via activation of Nrf2 and autophagy. Free Radic. Biol. Med. 2015, 89, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shen, T.; Yang, H.; Gu, W. Ruthenium complexes induce HepG2 human hepatocellular carcinoma cell apoptosis and inhibit cell migration and invasion through regulation of the Nrf2 pathway. Int. J. Mol. Sci. 2016, 17, 775. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.M.; Ke, Z.P.; Wang, J.N.; Yang, J.Y.; Chen, S.Y.; Chen, H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis 2013, 34, 1806–1814. [Google Scholar] [CrossRef] [Green Version]
- Gao, A.M.; Ke, Z.P.; Shi, F.; Sun, G.C.; Chen, H. Chrysin enhances sensitivity of BEL-7402/ADM cells to doxorubicin by suppressing PI3K/Akt/Nrf2 and ERK/Nrf2 pathway. Chem. Biol. Interact. 2013, 206, 100–108. [Google Scholar] [CrossRef]
- Ye, C.L.; Lu, Y.H.; Wei, D.Z. Flavonoids from Cleistocalyx operculatus. Phytochemistry 2004, 65, 445–447. [Google Scholar] [CrossRef]
- Yu, W.G.; Qian, J.; Lu, Y.H. Hepatoprotective effects of 2′,4′-dihydroxy-6′-methoxy-3′,5′-dimethylchalcone on CCl4-induced acute liver injury in mice. J. Agric. Food Chem. 2011, 59, 12821–12829. [Google Scholar] [CrossRef]
- Ye, C.L.; Liu, J.W.; Wei, D.Z.; Lu, Y.H.; Qian, F. In vivo antitumor activity by 2′,4′-dihydroxy-6′-methoxy-3′,5′-dimethylchalcone in a solid human carcinoma xenograft model. Cancer Chemother. Pharmacol. 2005, 56, 70–74. [Google Scholar] [CrossRef]
- Wei, X.; Mo, X.; An, F.; Ji, X.; Lu, Y. 2′,4′-Dihydroxy-6′-methoxy-3′,5′-dimethylchalcone, a potent Nrf2/ARE pathway inhibitor, reverses drug resistance by decreasing glutathione synthesis and drug efflux in BEL-7402/5-FU cells. Food Chem. Toxicol. 2018, 119, 252–259. [Google Scholar] [CrossRef]
- Keating, G.M.; Santoro, A. Sorafenib: A review of its use in advanced hepatocellular carcinoma. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Ye, W.; Duan, X.; Zhang, M.; Wang, J. The noncytotoxic dose of sorafenib sensitizes Bel-7402/5-FU cells to 5-FU by down regulating 5-FU induced Nrf2 expression. Dig. Dis. Sci. 2013, 58, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Trojan, H.; Kopp, T.; Laszcyzck, M.N.; Scheffler, A. Pentacyclic triterpene distribution in various plants-rich sources for a new group of multi-potent plant extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, T.; Du, J. Urosolic acid sensitizes cisplatin-resistant HepG2/DDP cells to cisplatin via inhibiting Nrf2/ARE pathway. Drug Des. Dev. Ther. 2016, 10, 3471–3481. [Google Scholar] [CrossRef]
- Terbach, N.; Williams, R.S. Structure-function studies for the panacea, valproic acid. Biochem. Soc. Trans. 2009, 37, 1126–1132. [Google Scholar] [PubMed]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.I.; Choi, C.; Shin, S.W.; Son, A.; Lee, G.H.; Kim, S.Y.; Park, H.C. Valproic Acid Sensitizes Hepatocellular Carcinoma Cells to Proton Therapy by Suppressing NRF2 Activation. Sci. Rep. 2018, 8, 7597. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Moenner, M.; Pluguet, O.; Bouchecareilh, M.; Chevet, E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007, 67, 10631–10634. [Google Scholar] [CrossRef]
- Martinon, F. Targeting endoplasmic reticulum signaling pathways in cancer. Acta Oncol. 2012, 51, 822–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Y.; Zhao, H.; Gao, L.; Zhang, W.; Shuli, A.Y.; Shay, C. FGF19 protect hepatocellular carcinoma cells against endoplasmic reticulum stress via activation of FGFR4-GSK3β-Nrf2 Signaling. Cancer Res. 2017, 77, 6215–6225. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Hobert, O. Gene regulation by transcription factors and microRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.M.; Zhang, X.Y.; Ke, Z.P. Apigenin sensitizes BEL-7402/ADM cells to doxorubicin through inhibiting miR-101/Nrf2 pathway. Oncotarget 2017, 8, 82085–82091. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Ye, W.; Zhang, Y.; Yu, D.; Shao, Q.; Liang, J.; Zhang, M. miR-144 reverses chemoresistance of hepatocellular carcinoma cell lines by targeting Nrf2-dependent antioxidant pathway. Am. J. Transl. Res. 2016, 8, 2992–3002. [Google Scholar]
- Shi, L.; Chen, Z.G.; Wu, L.L.; Zheng, J.J.; Yang, J.R.; Chen, X.F.; Chen, X.F.; Chen, Z.Q.; Liu, C.L.; Chi, S.Y.; et al. miR-340 reverses cisplatin resistance of hepatocellular carcinoma cell lines by targeting Nrf2-dependent antioxidant pathway. Asian Pac. J. Cancer Prev. 2014, 15, 10439–10444. [Google Scholar] [CrossRef]
- Petrelli, A.; Perra, A.; Cora, D.; Sulas, P.; Menegon, C.; Manca, C.; Migliore, C.; Kowalik, M.A.; Ledda-Columbano, G.M.; Giordano, S.; et al. MicroRNA/gene profiling unveils early molecular changes and nuclear factor erythroid related factor 2 (NRF2) activation in a rat model recapitulating human hepatocellular carcinoma (HCC). Hepatology 2014, 59, 228–241. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Shi, L.; Wu, L.; Chen, Z.; Yang, J.; Yu, F.; Zheng, F.; Lin, X. MiR-141 activates Nrf2-dependent antioxidant pathway via down-regulating the expression of Keap1 conferring the resistance of hepatocellular carcinoma cells to 5-fluorouracil. Cell Physiol. Biochem. 2015, 35, 2333–2348. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Chen, J.; Liu, X.M.; Zhao, R.; Zhe, H. Nrf2-mediated metabolic reprogramming in cancer. Oxid. Med. Cell Longev. 2018, 2018, 9304091. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R. Transcription factor NRF2 regulates miR-1 and miR-201 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, P.; Li, H.; Cheng, P.; Zhang, L.; Wei, D.; Su, X.; Peng, J.; Gao, H.; Tan, Y.; et al. MicroRNA-1 inhibits proliferation of hepatocarcinoma cells by targeting endothelin-1. Life Sci. 2012, 91, 440–447. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Dose and Duration | Cell Lines/Animal Model | Molecular Targets | Molecular Mechanism | Reference |
|---|---|---|---|---|---|
| Camptothecin | 0.1 and 0.5 µM for 24 h | HepG2 | ↓GCLC, ↓GCLM, ↓NQO1, ↓HMOX-1, ↓AKR1C1, ↓AKR1C2, ↓AKR1C3 | Down regulation of NRF2 suppression of ARE- dependent genes | [143] |
| Capsaicin | 200 µM for 24 h | HepG2 | ↑HO-1, ↓NQO1, ↑p-AKT, ↑p-ERK, ↑NRF2 | Down regulation of NQO1 triggers the production of ROS, leading to phosphorylation of AKT, ERK and ARE binding of NRF2 | [127] |
| Glycycoumarin | 10 mg and 20 mg/kg for once a day for 3 weeks | C57BL/6 mice | ↑NRF2, ↑HO-1, ↑GCLC | Glycycoumarin activates NRF2 and induces autophagy via up regulation of p62 and p38 | [144] |
| Glycycoumarin | 50 µM for 24 h | HepG2 | ↑Nrf2, ↑HO-1, ↑GCLC, ↑p38, ↑p-ERK1/2, ↑p62, ↓KEAP1, ↑LC3-II | Glycycoumarin activates NRF2 | [144] |
| Crotonaldehyde | 50 µM for 24 h | HepG2 | ↑HO-1, ↑p38, ↑p-PKC-δ, ↑NRF2 | Anti-apoptotic effect of crotonaldehyde induced by HO-1 through the PKC-δ-p38 MAPK-NRF2 signalling pathway | [129] |
| Pelargonidin Chloride | 50 and 100 µM for 2 h | HepG2 | ↑NRF2, ↑HO-1, ↑GST, ↑NQO1, ↑CAT, ↑SOD1, ↑GPX1 | Up regulation of detoxification enzymes genes through the KEAP1/NRF2 signalling pathway | [136] |
| Pomegranate emulsion | 1 g and 10 g/kg for three times a week | Male Sprague-Dawley rats | ↑GSTA2, ↑GSTA5, ↑GSTM1, ↑GSTM7, ↑GSTT1, ↑NQO1, ↑UGT1A1, ↑UGT2B17, ↑NRF2 | Induction of antioxidant and phase 2 xenobiotic enzymes leading to up regulation of NRF2 | [140] |
| Ruthenium complex | 2 and 4 µM for 24 h | HepG2 | ↓NRF2, ↓NQO1, ↓HO-1 | Suppression of NQO1 and HO-1 expression through down regulation of the Nrf2 signalling pathway | [145] |
| Compounds | Drug Sensitized | Dose and Duration | Cell Lines | Mode of Nrf2 Inhibition | Molecular Targets | Reference |
|---|---|---|---|---|---|---|
| Apigenin | Doxorubicin | Apigenin-20 μM for 24 h Doxorubicin-2 μM for 24 h | BEL-7402/ADM | NRF2 expression was inhibited by down regulation of the PI3K/AKT pathway | ↓NRF2, ↓HO-1, ↓AKR1B10, ↓MRP5 | [147] |
| Chrysin | Doxorubicin | Chrysin-20 μM for 24 h | BEL-7402/ADM | Chrysin suppressed the activation of NRF2 and its downstream genes through inhibition of the PI3K/AKT and ERK signalling pathway | ↓NRF2, ↓HO-1, ↓AKR1B10, ↓MRP5, ↓p-Akt, ↓p-ERK1/2 | [148] |
| DMC | 5-FU | DMD-5,10 and 20 μM for 24 h | BEL-7402/5-FU | NRF2 suppression, prevented NRF2 translocation and inhibited the ARE binding | ↓NRF2, ↓GCLC, ↓GCLM, ↓GST, ↓GSH | [151] |
| Sorafenib | 5-FU | Sorafenib-2 µM for 24 h 5-flurouracil-1000 μg/mL for 24 h | Bel-7402/5-FU | Sorafenib inhibited the expression of NRF2 induced by 5-flurouracil | ↓NRF2, ↓MRP1, ↓MRP2, ↓MRP3 | [153] |
| Ursolic acid | Cisplatin | Ursolic acid-2.25 μg/mL for 48 h | HepG2/DDP | Ursolic acid highly induced ROS and reduced mitochondrial membrane potential, leading to suppression of NRF2 expression and its downstream genes | ↓NRF2, ↓HO-1, ↓NQO1, ↓GST | [155] |
| Valproic acid | Proton therapy | Valproic acid-1 mM for 2 h and 24 h | Hep3B | NRF2 expression was suppressed by NADPH oxidase activation through increased intracellular ROS level | ↑PARP cleavage, ↑caspase-3 cleavage, ↓NRF2, ↓HO-1 | [158] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raghunath, A.; Sundarraj, K.; Arfuso, F.; Sethi, G.; Perumal, E. Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance. Cancers 2018, 10, 481. https://doi.org/10.3390/cancers10120481
Raghunath A, Sundarraj K, Arfuso F, Sethi G, Perumal E. Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance. Cancers. 2018; 10(12):481. https://doi.org/10.3390/cancers10120481
Chicago/Turabian StyleRaghunath, Azhwar, Kiruthika Sundarraj, Frank Arfuso, Gautam Sethi, and Ekambaram Perumal. 2018. "Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance" Cancers 10, no. 12: 481. https://doi.org/10.3390/cancers10120481
APA StyleRaghunath, A., Sundarraj, K., Arfuso, F., Sethi, G., & Perumal, E. (2018). Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance. Cancers, 10(12), 481. https://doi.org/10.3390/cancers10120481