Core Binding Factor Leukemia: Chromatin Remodeling Moves Towards Oncogenic Transcription

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
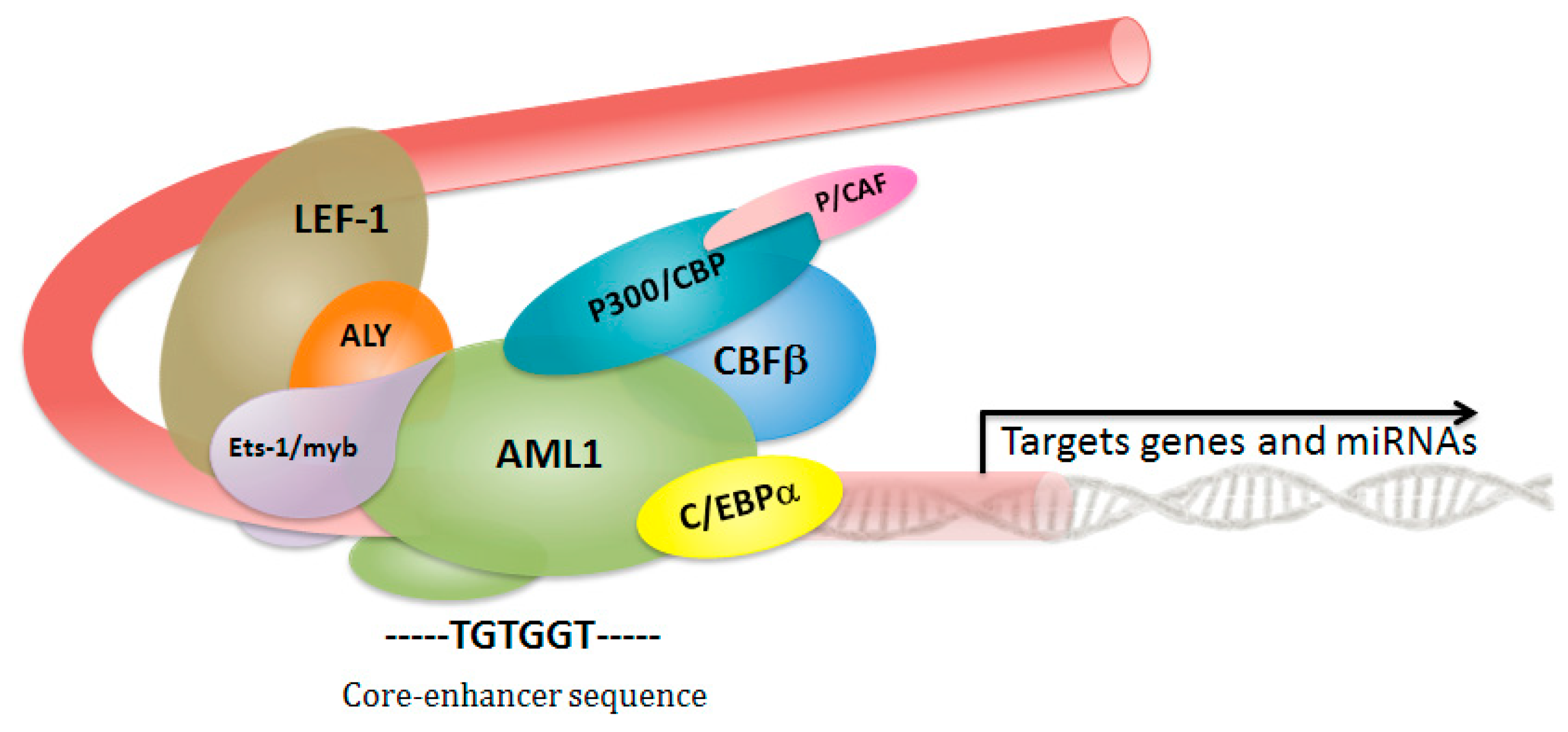
2. Core Binding Factor Complex: A Critical Role in Hematopoietic Stem Cell Fate
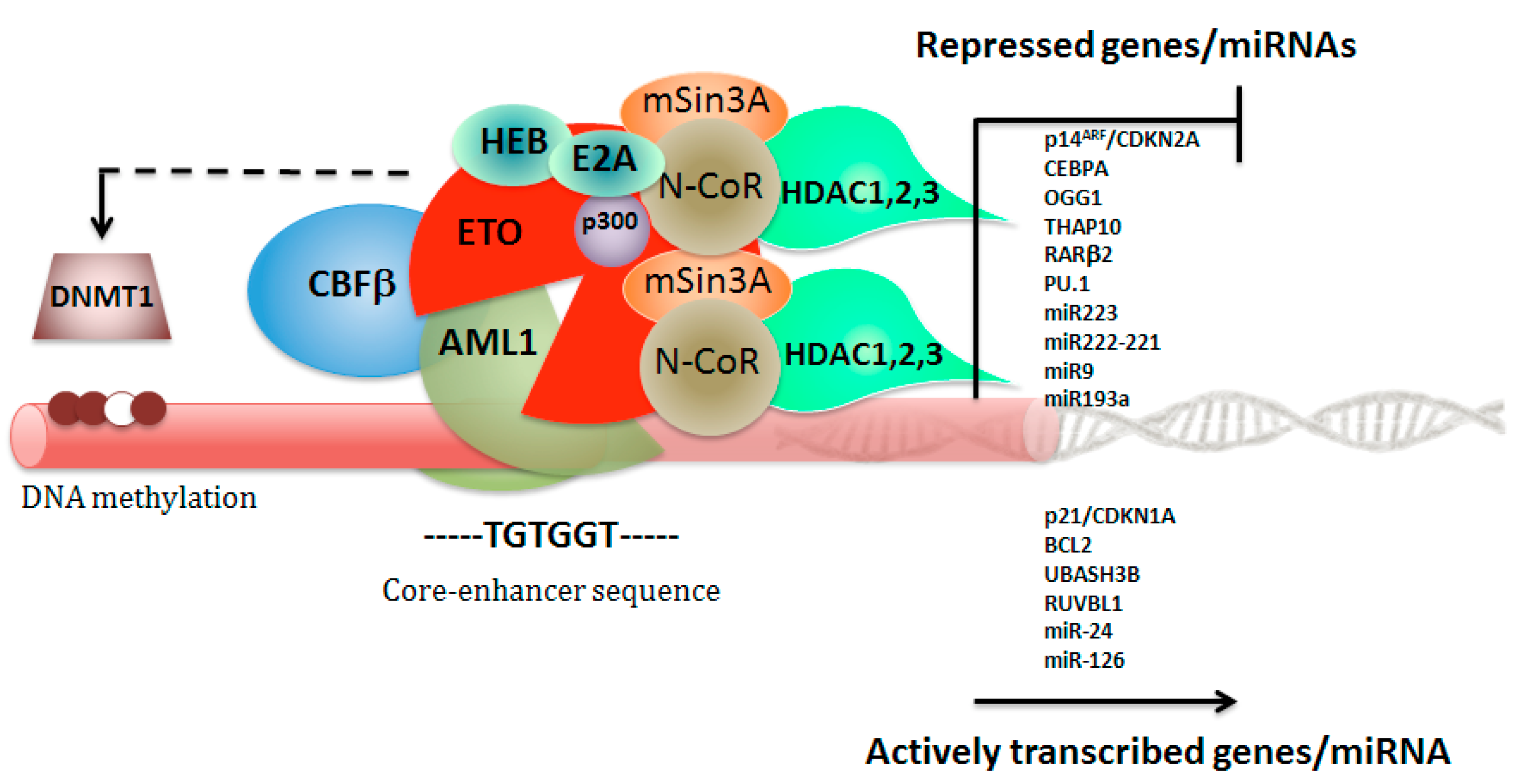
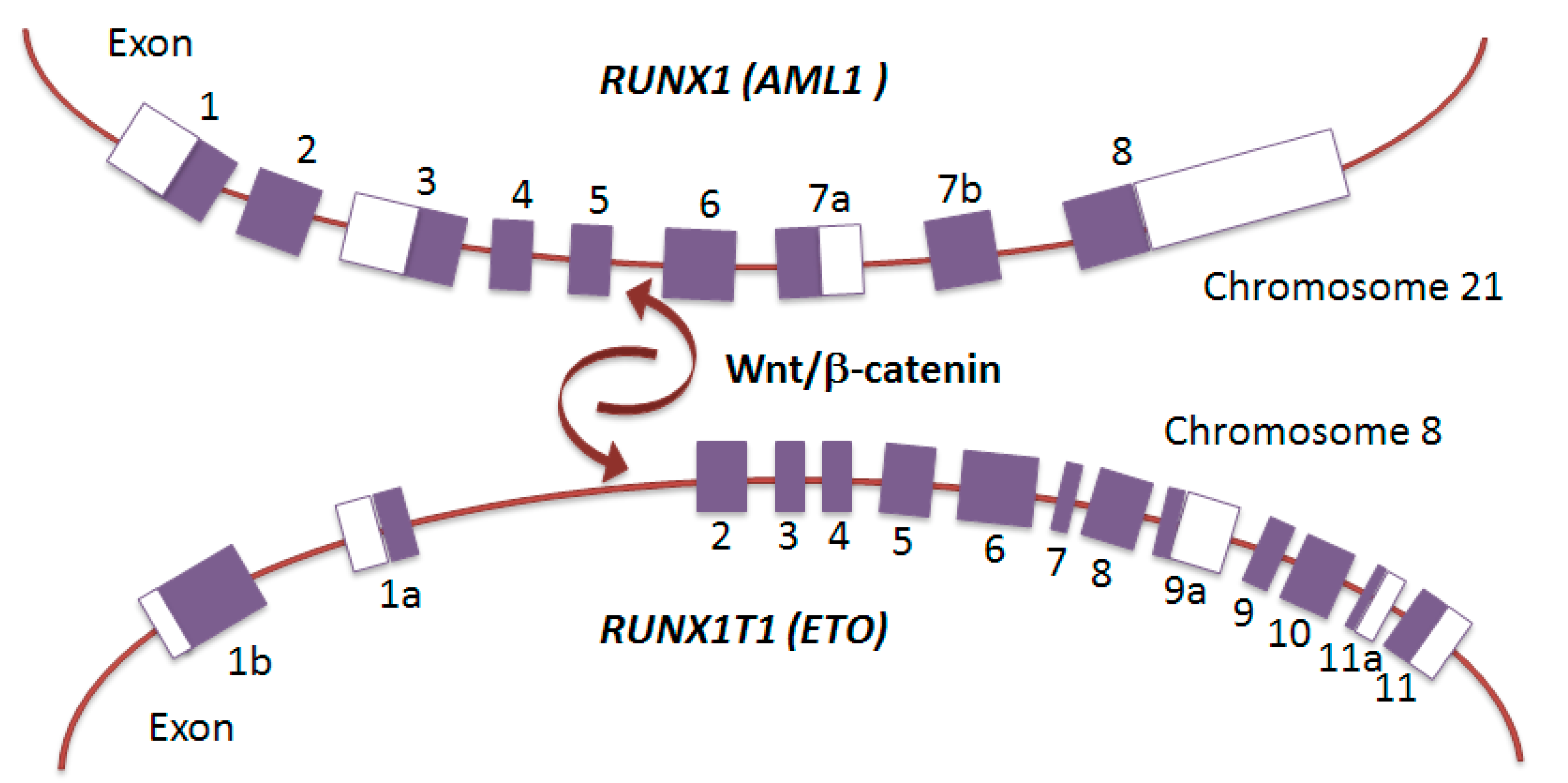
3. Leukemia Triggered by RUNX1–RUNX1T1
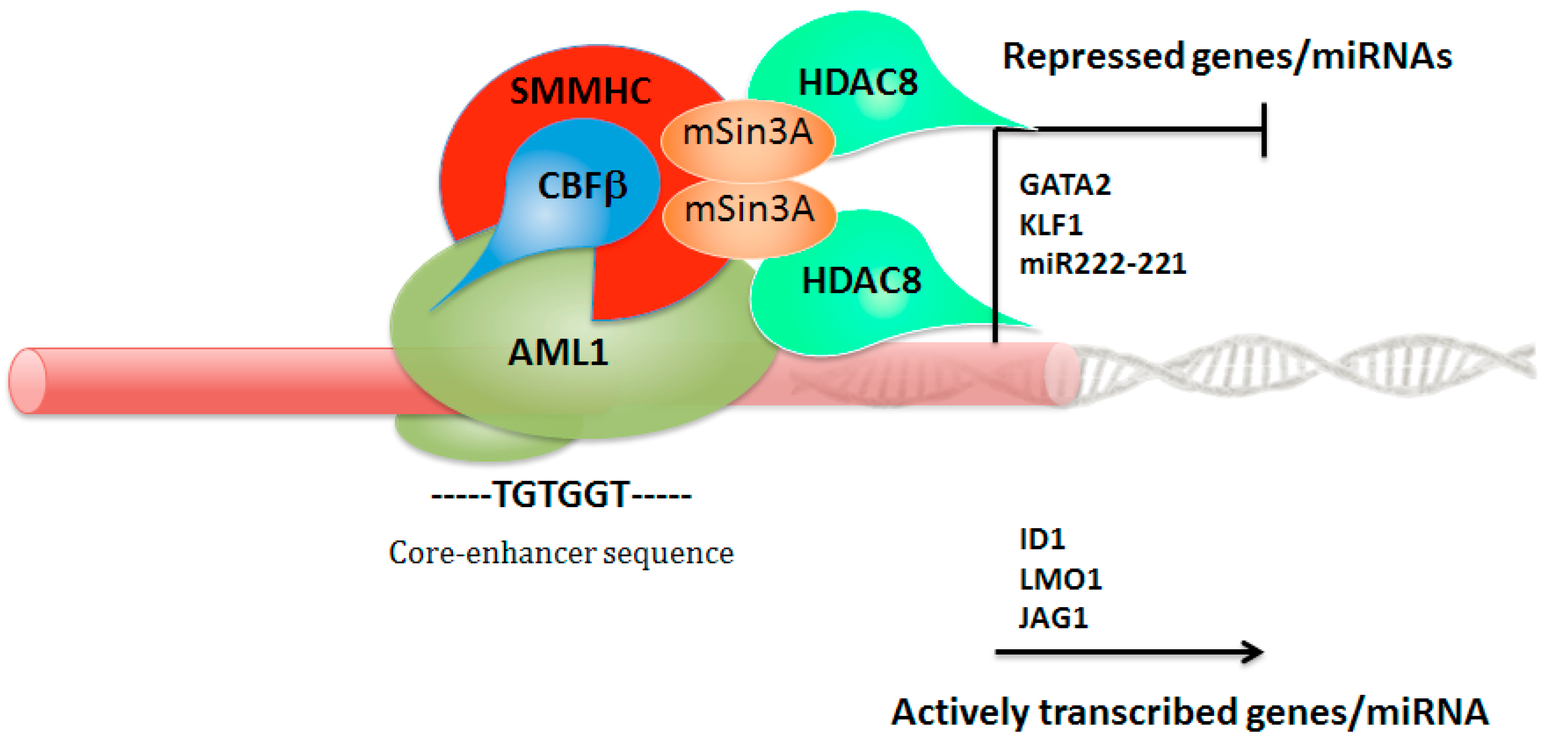
4. Leukemia Triggered by CBFβ-MYH11
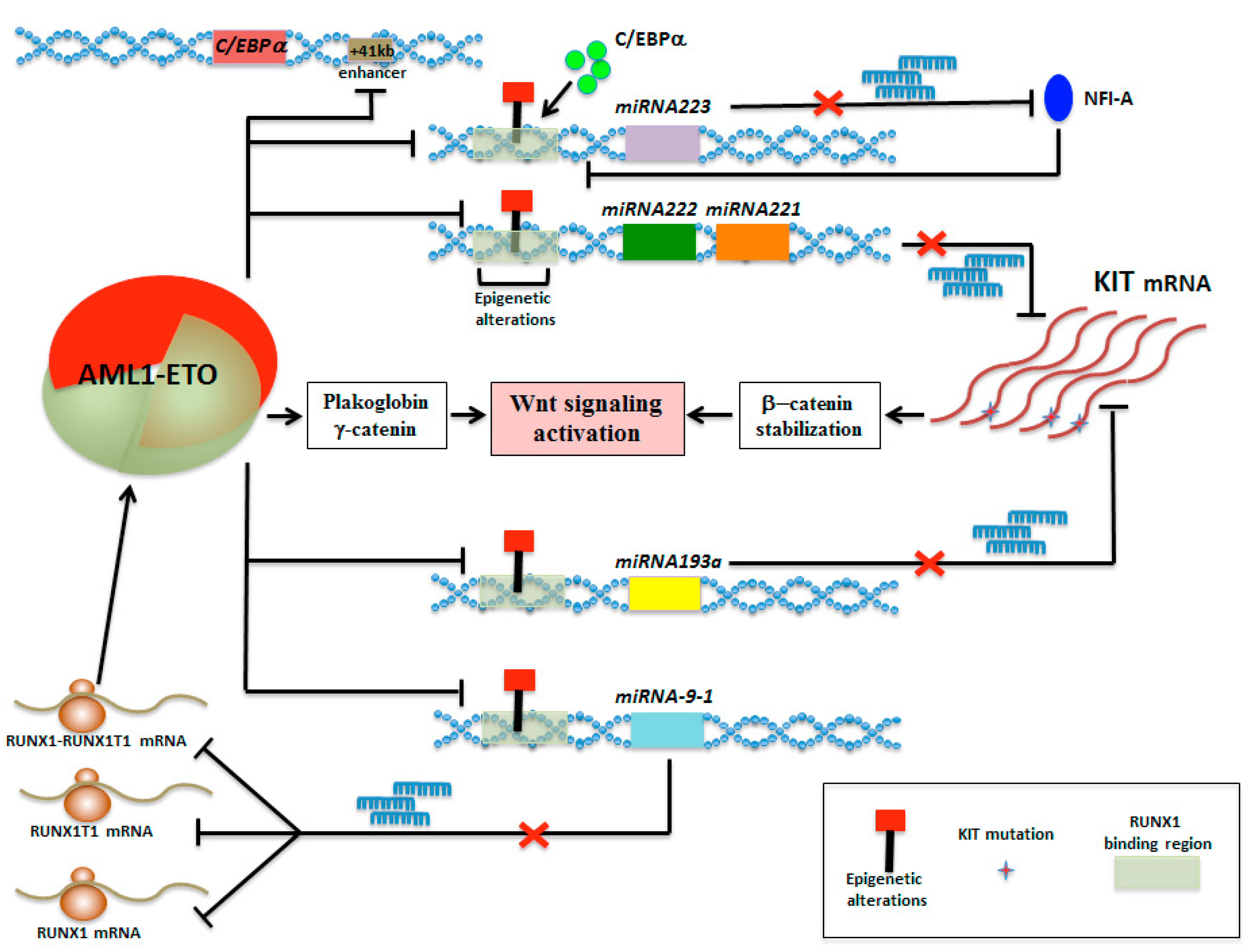
5. MicroRNA Circuitries Contribute to CBF-Mediated Leukemogenesis
5.1. Down-Regulation of miR-222/221 in AML with Deranged Core Binding Factor
5.2. Epigenetic Silencing of miR-193a Contributes to t(8;21)-Mediated AML
5.3. Epigenetic Mini-Circuit AML1-ETO/miR-9-1/miR-383 Contributes to t(8;21) Leukemogenesis
6. The Genomic Landscape of Core-Binding Factor Acute Myeloid Leukemias
7. Mouse Models for Core Binding Factor Leukemia
8. Molecular Targeted Therapy of CBFL: The Progress and Future Prospect
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Miyoshi, H.; Shimizu, K.; Kozu, T.; Maseki, N.; Kaneko, Y.; Ohki, M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc. Natl. Acad. Sci. USA 1991, 88, 10431–10434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, K.A.; Chou, F.-S.; Mulloy, J.C. Core binding factor at the crossroads: Determining the fate of the HSC. J. Cell. Physiol. 2010, 222, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bruijn, M.; Dzierzak, E. Runx transcription factors in the development and function of the definitive hematopoietic system. Blood 2017, 129, 2061–2069. [Google Scholar] [CrossRef] [PubMed]
- Menegatti, S.; de Kruijf, M.; Garcia-Alegria, E.; Lacaud, G.; Kouskoff, V. Transcriptional control of blood cell emergence. FEBS Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Tarlé, S.A.; Hajra, A.; Claxton, D.F.; Marlton, P.; Freedman, M.; Siciliano, M.J.; Collins, F.S. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science 1993, 261, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Goyama, S.; Mulloy, J.C. Molecular pathogenesis of core binding factor leukemia: Current knowledge and future prospects. Int. J. Hematol. 2011, 94, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Sinha, C.; Cunningham, L.C.; Liu, P.P. Core Binding Factor Acute Myeloid Leukemia: New Prognostic Categories and Therapeutic Opportunities. Semin. Hematol. 2015, 52, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Schlenk, R.F.; Benner, A.; Krauter, J.; Büchner, T.; Sauerland, C.; Ehninger, G.; Schaich, M.; Mohr, B.; Niederwieser, D.; Krahl, R.; et al. Individual patient data-based meta-analysis of patients aged 16 to 60 years with core binding factor acute myeloid leukemia: A survey of the German Acute Myeloid Leukemia Intergroup. J. Clin. Oncol. 2004, 22, 3741–3750. [Google Scholar] [CrossRef]
- Jourdan, E.; Boissel, N.; Chevret, S.; Delabesse, E.; Renneville, A.; Cornillet, P.; Blanchet, O.; Cayuela, J.-M.; Recher, C.; Raffoux, E.; et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood 2013, 121, 2213–2223. [Google Scholar] [CrossRef]
- Ghamari, A.; van de Corput, M.P.C.; Thongjuea, S.; van Cappellen, W.A.; van Ijcken, W.; van Haren, J.; Soler, E.; Eick, D.; Lenhard, B.; Grosveld, F.G. In vivo live imaging of RNA polymerase II transcription factories in primary cells. Genes Dev. 2013, 27, 767–777. [Google Scholar] [CrossRef] [Green Version]
- Ugarte, G.D.; Vargas, M.F.; Medina, M.A.; León, P.; Necuñir, D.; Elorza, A.A.; Gutiérrez, S.E.; Moon, R.T.; Loyola, A.; De Ferrari, G.V. Wnt signaling induces transcription, spatial proximity, and translocation of fusion gene partners in human hematopoietic cells. Blood 2015, 126, 1785–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castilla, L.H.; Garrett, L.; Adya, N.; Orlic, D.; Dutra, A.; Anderson, S.; Owens, J.; Eckhaus, M.; Bodine, D.; Liu, P.P. The fusion gene Cbfb-MYH11 blocks myeloid differentiation and predisposes mice to acute myelomonocytic leukaemia. Nat. Genet. 1999, 23, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhou, L.; Miyamoto, T.; Iwasaki, H.; Harakawa, N.; Hetherington, C.J.; Burel, S.A.; Lagasse, E.; Weissman, I.L.; Akashi, K.; et al. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc. Natl. Acad. Sci. USA 2001, 98, 10398–10403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faber, Z.J.; Chen, X.; Gedman, A.L.; Boggs, K.; Cheng, J.; Ma, J.; Radtke, I.; Chao, J.-R.; Walsh, M.P.; Song, G.; et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat. Genet. 2016, 48, 1551–1556. [Google Scholar] [CrossRef]
- Duployez, N.; Marceau-Renault, A.; Boissel, N.; Petit, A.; Bucci, M.; Geffroy, S.; Lapillonne, H.; Renneville, A.; Ragu, C.; Figeac, M.; et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood. 2016, 127, 2451–2459. [Google Scholar] [CrossRef]
- Solh, M.; Yohe, S.; Weisdorf, D.; Ustun, C. Core-binding factor acute myeloid leukemia: Heterogeneity, monitoring, and therapy. Am. J. Hematol. 2014, 89, 1121–1131. [Google Scholar] [CrossRef]
- Corces-Zimmerman, M.R.; Hong, W.-J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [Green Version]
- Shima, T.; Miyamoto, T.; Kikushige, Y.; Yuda, J.; Tochigi, T.; Yoshimoto, G.; Kato, K.; Takenaka, K.; Iwasaki, H.; Mizuno, S.; et al. The ordered acquisition of Class II and Class I mutations directs formation of human t(8;21) acute myelogenous leukemia stem cell. Exp. Hematol. 2014, 42, 955–965. [Google Scholar] [CrossRef]
- Nucifora, G.; Larson, R.A.; Rowley, J.D. Persistence of the 8;21 translocation in patients with acute myeloid leukemia type M2 in long-term remission. Blood 1993, 82, 712–715. [Google Scholar] [CrossRef] [Green Version]
- Jurlander, J.; Caligiuri, M.A.; Ruutu, T.; Baer, M.R.; Strout, M.P.; Oberkircher, A.R.; Hoffmann, L.; Ball, E.D.; Frei-Lahr, D.A.; Christiansen, N.P.; et al. Persistence of the AML1/ETO fusion transcript in patients treated with allogeneic bone marrow transplantation for t(8;21) leukemia. Blood 1996, 88, 2183–2191. [Google Scholar] [CrossRef]
- Miyamoto, T.; Nagafuji, K.; Akashi, K.; Harada, M.; Kyo, T.; Akashi, T.; Takenaka, K.; Mizuno, S.; Gondo, H.; Okamura, T.; et al. Persistence of multipotent progenitors expressing AML1/ETO transcripts in long-term remission patients with t(8;21) acute myelogenous leukemia. Blood 1996, 87, 4789–4796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croce, C.M.; Calin, G.A. miRNAs, cancer, and stem cell division. Cell 2005, 122, 6–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; De Marchis, M.L.; Nervi, C.; Bozzoni, I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005, 123, 819–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, J.; Rossetti, S.; Datta, A.; Eng, K.; Beghini, A.; Sacchi, N.; Datta, A.; Eng, K.; Beghini, A.; Sacchi, N. miR-17 deregulates a core RUNX1-miRNA mechanism of CBF acute myeloid leukemia. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, F.; De Marinis, E.; Grignani, F.; Nervi, C. Epigenetic role of miRNAs in normal and leukemic hematopoiesis. Epigenomics 2013, 5, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Shi, J.; Liu, A.; Zhou, L.; Jiang, M.; Fu, H.; Xu, K.; Li, D.; Deng, A.; Zhang, Q.; et al. A minicircuitry of microRNA-9-1 and RUNX1-RUNX1T1 contributes to leukemogenesis in t(8;21) acute myeloid leukemia. Int. J. Cancer 2017, 140, 653–661. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.A.; O’Connell, R.M. MicroRNAs and acute myeloid leukemia: Therapeutic implications and emerging concepts. Blood 2017, 130, 1290–1301. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ning, Q.; Shi, J.; Chen, Y.; Jiang, M.; Gao, L.; Huang, W.; Jing, Y.; Huang, S.; Liu, A.; et al. A novel epigenetic AML1-ETO/THAP10/miR-383 mini-circuitry contributes to t(8;21) leukaemogenesis. EMBO Mol. Med. 2017, 9, 933–949. [Google Scholar] [CrossRef]
- Okuda, T.; van Deursen, J.; Hiebert, S.W.; Grosveld, G.; Downing, J.R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996, 84, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Stacy, T.; Miller, J.D.; Lewis, A.F.; Gu, T.L.; Huang, X.; Bushweller, J.H.; Bories, J.C.; Alt, F.W.; Ryan, G.; et al. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell 1996, 87, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Niki, M.; Okada, H.; Takano, H.; Kuno, J.; Tani, K.; Hibino, H.; Asano, S.; Ito, Y.; Satake, M.; Noda, T. Hematopoiesis in the fetal liver is impaired by targeted mutagenesis of a gene encoding a non-DNA binding subunit of the transcription factor, polyomavirus enhancer binding protein 2/core binding factor. Proc. Natl. Acad. Sci. USA 1997, 94, 5697–5702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichikawa, M.; Asai, T.; Saito, T.; Seo, S.; Yamazaki, I.; Yamagata, T.; Mitani, K.; Chiba, S.; Ogawa, S.; Kurokawa, M.; et al. AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat. Med. 2004, 10, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Growney, J.D.; Shigematsu, H.; Li, Z.; Lee, B.H.; Adelsperger, J.; Rowan, R.; Curley, D.P.; Kutok, J.L.; Akashi, K.; Williams, I.R.; et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 2005, 106, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Putz, G.; Rosner, A.; Nuesslein, I.; Schmitz, N.; Buchholz, F. AML1 deletion in adult mice causes splenomegaly and lymphomas. Oncogene 2006, 25, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Motoda, L.; Osato, M.; Yamashita, N.; Jacob, B.; Chen, L.Q.; Yanagida, M.; Ida, H.; Wee, H.-J.; Sun, A.X.; Taniuchi, I.; et al. Runx1 protects hematopoietic stem/progenitor cells from oncogenic insult. Stem Cells 2007, 25, 2976–2986. [Google Scholar] [CrossRef]
- Aikawa, Y.; Nguyen, L.A.; Isono, K.; Takakura, N.; Tagata, Y.; Schmitz, M.L.; Koseki, H.; Kitabayashi, I. Roles of HIPK1 and HIPK2 in AML1-and p300-dependent transcription, hematopoiesis and blood vessel formation. EMBO J. 2006, 25, 3955–3965. [Google Scholar] [CrossRef] [Green Version]
- Gu, T.L.; Goetz, T.L.; Graves, B.J.; Speck, N.A. Auto-inhibition and partner proteins, core-binding factor beta (CBFbeta) and Ets-1, modulate DNA binding by CBFalpha2 (AML1). Mol. Cell. Biol. 2000, 20, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cheney, M.D.; Gaudet, J.J.; Chruszcz, M.; Lukasik, S.M.; Sugiyama, D.; Lary, J.; Cole, J.; Dauter, Z.; Minor, W.; et al. The tetramer structure of the Nervy homology two domain, NHR2, is critical for AML1/ETO’s activity. Cancer Cell 2006, 9, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Speck, N.A.; Bushweller, J.H. The role of CBFbeta in AML1-ETO’s activity. Blood 2009, 114, 2849–2850. [Google Scholar] [CrossRef]
- Roudaia, L.; Cheney, M.D.; Manuylova, E.; Chen, W.; Morrow, M.; Park, S.; Lee, C.-T.; Kaur, P.; Williams, O.; Bushweller, J.H.; et al. CBFbeta is critical for AML1-ETO and TEL-AML1 activity. Blood 2009, 113, 3070–3079. [Google Scholar] [CrossRef] [Green Version]
- Corpora, T.; Roudaia, L.; Oo, Z.M.; Chen, W.; Manuylova, E.; Cai, X.; Chen, M.J.; Cierpicki, T.; Speck, N.A.; Bushweller, J.H. Structure of the AML1-ETO NHR3-PKA(RIIα) complex and its contribution to AML1-ETO activity. J. Mol. Biol. 2010, 402, 560–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.-J.; Wang, Z.; Wang, L.; Jiang, Y.; Kost, N.; Soong, T.D.; Chen, W.-Y.; Tang, Z.; Nakadai, T.; Elemento, O.; et al. A stable transcription factor complex nucleated by oligomeric AML1-ETO controls leukaemogenesis. Nature 2013, 500, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hoshino, T.; Redner, R.L.; Kajigaya, S.; Liu, J.M. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc. Natl. Acad. Sci. USA 1998, 95, 10860–10865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Shen, T.; Huynh, L.; Klisovic, M.I.; Rush, L.J.; Ford, J.L.; Yu, J.; Becknell, B.; Li, Y.; Liu, C.; et al. Interplay of RUNX1/MTG8 and DNA methyltransferase 1 in acute myeloid leukemia. Cancer Res. 2005, 65, 1277–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linggi, B.; Müller-Tidow, C.; van de Locht, L.; Hu, M.; Nip, J.; Serve, H.; Berdel, W.E.; van der Reijden, B.; Quelle, D.E.; Rowley, J.D.; et al. The t(8;21) fusion protein, AML1–ETO, specifically represses the transcription of the p14ARF tumor suppressor in acute myeloid leukemia. Nat. Med. 2002, 8, 743–750. [Google Scholar] [CrossRef]
- Friedman, A.D. C/EBPα in normal and malignant myelopoiesis. Int. J. Hematol. 2015, 101, 330–341. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.N.; Yan, F.; Lin, J.; Gao, L.; Lu, X.L.; Wei, S.C.; Shen, N.; Pang, J.X.; Ning, Q.Y.; Komeno, Y.; et al. AML1/ETO cooperates with HIF1α to promote leukemogenesis through DNMT3a transactivation. Leukemia 2015, 29, 1730–1740. [Google Scholar] [CrossRef]
- Ptasinska, A.; Assi, S.A.; Mannari, D.; James, S.R.; Williamson, D.; Dunne, J.; Hoogenkamp, M.; Wu, M.; Care, M.; McNeill, H.; et al. Depletion of RUNX1/ETO in t(8;21) AML cells leads to genome-wide changes in chromatin structure and transcription factor binding. Leukemia 2012, 26, 1829–1841. [Google Scholar] [CrossRef]
- Pabst, T.; Mueller, B.U.; Harakawa, N.; Schoch, C.; Haferlach, T.; Behre, G.; Hiddemann, W.; Zhang, D.E.; Tenen, D.G. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat. Med. 2001, 7, 444–451. [Google Scholar] [CrossRef]
- Vangala, R.K.; Heiss-Neumann, M.S.; Rangatia, J.S.; Singh, S.M.; Schoch, C.; Tenen, D.G.; Hiddemann, W.; Behre, G. The myeloid master regulator transcription factor PU.1 is inactivated by AML1-ETO in t(8;21) myeloid leukemia. Blood 2003, 101, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Lutterbach, B.; Westendorf, J.J.; Linggi, B.; Patten, A.; Moniwa, M.; Davie, J.R.; Huynh, K.D.; Bardwell, V.J.; Lavinsky, R.M.; Rosenfeld, M.G.; et al. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol. Cell. Biol. 1998, 18, 7176–7184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Hug, B.A.; Huang, E.Y.; Chen, C.W.; Gelmetti, V.; Maccarana, M.; Minucci, S.; Pelicci, P.G.; Lazar, M.A. Oligomerization of ETO is obligatory for corepressor interaction. Mol. Cell. Biol. 2001, 21, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutterbach, B.; Westendorf, J.J.; Linggi, B.; Isaac, S.; Seto, E.; Hiebert, S.W. A mechanism of repression by acute myeloid leukemia-1, the target of multiple chromosomal translocations in acute leukemia. J. Biol. Chem. 2000, 275, 651–656. [Google Scholar] [CrossRef] [Green Version]
- Kamikubo, Y.; Zhao, L.; Wunderlich, M.; Corpora, T.; Hyde, R.K.; Paul, T.A.; Kundu, M.; Garrett, L.; Compton, S.; Huang, G.; et al. Accelerated leukemogenesis by truncated CBFβ-SMMHC defective in high-affinity binding with RUNX1. Cancer Cell 2010, 17, 455–468. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, H.; Wang, X.; Jin, W.; Tan, Y.; Fang, H.; Chen, S.; Chen, Z.; Wang, K. Genome-wide studies identify a novel interplay between AML1 and AML1/ETO in t(8;21) acute myeloid leukemia. Blood 2016, 127, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Durst, K.L.; Lutterbach, B.; Kummalue, T.; Friedman, A.D.; Hiebert, S.W. The inv(16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol. Cell. Biol. 2003, 23, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.D.; Stacy, T.; Liu, P.P.; Speck, N.A. Core-binding factor beta (CBFbeta), but not CBFbeta-smooth muscle myosin heavy chain, rescues definitive hematopoiesis in CBFbeta-deficient embryonic stem cells. Blood 2001, 97, 2248–2256. [Google Scholar] [CrossRef]
- Richter, L.E.; Wang, Y.; Becker, M.E.; Coburn, R.A.; Williams, J.T.; Amador, C.; Hyde, R.K. HDAC1 Is a Required Cofactor of CBFβ-SMMHC and a Potential Therapeutic Target in Inversion 16 Acute Myeloid Leukemia. Mol. Cancer Res. 2019, 17, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Mandoli, A.; Singh, A.A.; Jansen, P.W.T.C.; Wierenga, A.T.J.; Riahi, H.; Franci, G.; Prange, K.; Saeed, S.; Vellenga, E.; Vermeulen, M.; et al. CBFB-MYH11/RUNX1 together with a compendium of hematopoietic regulators, chromatin modifiers and basal transcription factors occupies self-renewal genes in inv(16) acute myeloid leukemia. Leukemia 2014, 28, 770–778. [Google Scholar] [CrossRef] [Green Version]
- Hyde, R.K.; Zhao, L.; Alemu, L.; Liu, P.P. Runx1 is required for hematopoietic defects and leukemogenesis in Cbfb-MYH11 knock-in mice. Leukemia 2015, 29, 1771–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, T.; Kwon, E.M.; Zhao, L.; Hsu, J.; Hyde, R.K.; Lu, Y.; Alemu, L.; Speck, N.A.; Liu, P.P. Chd7 deficiency delays leukemogenesis in mice induced by Cbfb-MYH11. Blood 2017, 130, 2431–2442. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.J.; Bravo, J.; Williams, R.L.; Rabbitts, T.H. Structural basis for the heterodimeric interaction between the acute leukaemia-associated transcription factors AML1 and CBFβ. EMBO J. 2000, 19, 3004–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukasik, S.M.; Zhang, L.; Corpora, T.; Tomanicek, S.; Li, Y.; Kundu, M.; Hartman, K.; Liu, P.P.; Laue, T.M.; Biltonen, R.L.; et al. Altered affinity of CBF beta-SMMHC for Runx1 explains its role in leukemogenesis. Nat. Struct. Biol. 2002, 9, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Shigesada, K.; Wee, H.-J.; Liu, P.P.; Osato, M.; Ito, Y. Molecular basis for a dominant inactivation of RUNX1/AML1 by the leukemogenic inversion 16 chimera. Blood 2004, 103, 3200–3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wee, H.-J.; Voon, D.C.-C.; Bae, S.-C.; Ito, Y. PEBP2-beta/CBF-beta-dependent phosphorylation of RUNX1 and p300 by HIPK2: Implications for leukemogenesis. Blood 2008, 112, 3777–3787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, G.; Mandoli, A.; Jussen, L.; Tijchon, E.; van Bergen, M.G.J.M.; Cordonnier, G.; Hansen, M.; Kim, B.; Nguyen, L.N.; Jansen, P.W.T.C.; et al. CBFβ-MYH11 interferes with megakaryocyte differentiation via modulating a gene program that includes GATA2 and KLF1. Blood Cancer J. 2019, 9, e33. [Google Scholar] [CrossRef] [Green Version]
- Saida, S.; Zhen, T.; Kim, E.; Yu, K.; Lopez, G.; McReynolds, L.J.; Liu, P.P. Gata2 deficiency delays leukemogenesis while contributing to aggressive leukemia phenotype in Cbfb-MYH11 knockin mice. Leukemia 2019. [Google Scholar] [CrossRef]
- Sood, R.; Hansen, N.F.; Donovan, F.X.; Carrington, B.; Bucci, D.; Maskeri, B.; Young, A.; Trivedi, N.S.; Kohlschmidt, J.; Stone, R.M.; et al. Somatic mutational landscape of AML with inv(16) or t(8;21) identifies patterns of clonal evolution in relapse leukemia. Leukemia 2016, 30, 501–504. [Google Scholar] [CrossRef]
- Visconte, V.; Nakashima, M.O.; Rogers, H.J. Mutations in splicing factor genes in myeloid malignancies: Significance and impact on clinical features. Cancers 2019, 11, 1844. [Google Scholar] [CrossRef] [Green Version]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nature 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Felli, N.; Fontana, L.; Pelosi, E.; Botta, R.; Bonci, D.; Facchiano, F.; Liuzzi, F.; Lulli, V.; Morsilli, O.; Santoro, S.; et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc. Natl. Acad. Sci. USA 2005, 102, 18081–18086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garzon, R.; Pichiorri, F.; Palumbo, T.; Iuliano, R.; Cimmino, A.; Aqeilan, R.; Volinia, S.; Bhatt, D.; Alder, H.; Marcucci, G.; et al. MicroRNA fingerprints during human megakaryocytopoiesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5078–5083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnnidis, J.B.; Harris, M.H.; Wheeler, R.T.; Stehling-Sun, S.; Lam, M.H.; Kirak, O.; Brummelkamp, T.R.; Fleming, M.D.; Camargo, F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008, 451, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Petriv, O.I.; Kuchenbauer, F.; Delaney, A.D.; Lecault, V.; White, A.; Kent, D.; Marmolejo, L.; Heuser, M.; Berg, T.; Copley, M.; et al. Comprehensive microRNA expression profiling of the hematopoietic hierarchy. Proc. Natl. Acad. Sci. USA 2010, 107, 15443–15448. [Google Scholar] [CrossRef] [Green Version]
- Georgantas, R.W.; Hildreth, R.; Morisot, S.; Alder, J.; Liu, C.; Heimfeld, S.; Calin, G.A.; Croce, C.M.; Civin, C.I. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: A circuit diagram of differentiation control. Proc. Natl. Acad. Sci. USA 2007, 104, 2750–2755. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, A.; Emmrich, S.; Schmidt, F.; Beck, D.; Ng, M.; Reimer, C.; Adams, F.F.; Grasedieck, S.; Witte, D.; Käbler, S.; et al. The non-coding RNA landscape of human hematopoiesis and leukemia. Nat. Commun. 2017, 8, e218. [Google Scholar] [CrossRef]
- Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. Available online: https://www.nejm.org/doi/full/10.1056/nejmoa1301689 (accessed on 1 May 2013).
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Junge, A.; Zandi, R.; Havgaard, J.H.; Gorodkin, J.; Cowland, J.B. Assessing the miRNA sponge potential of RUNX1T1 in t(8;21) acute myeloid leukemia. Gene 2017, 615, 35–40. [Google Scholar] [CrossRef]
- Fazi, F.; Racanicchi, S.; Zardo, G.; Starnes, L.M.; Mancini, M.; Travaglini, L.; Diverio, D.; Ammatuna, E.; Cimino, G.; Lo-Coco, F.; et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 2007, 12, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Y.; Zhou, G.-B.; Yin, T.; Chen, B.; Shi, J.-Y.; Liang, W.-X.; Jin, X.-L.; You, J.-H.; Yang, G.; Shen, Z.-X.; et al. AML1-ETO and C-KIT mutation/overexpression in t(8;21) leukemia: Implication in stepwise leukemogenesis and response to Gleevec. Proc. Natl. Acad. Sci. USA 2005, 102, 1104–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brioschi, M.; Fischer, J.; Cairoli, R.; Rossetti, S.; Pezzetti, L.; Nichelatti, M.; Turrini, M.; Corlazzoli, F.; Scarpati, B.; Morra, E.; et al. Down-regulation of microRNAs 222/221 in acute myelogenous leukemia with deranged core-binding factor subunits. Neoplasia 2010, 12, 866–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beghini, A.; Cairoli, R.; Morra, E.; Lariza, L. In vivo differentiation of mast cells from acute myeloid leukemia blasts carrying a novel activating ligand-independent C-kit mutation. Blood Cells Mol. Dis. 1998, 24, 262–270. [Google Scholar] [CrossRef] [Green Version]
- Beghini, A.; Peterlongo, P.; Ripamonti, C.B.; Larizza, L.; Cairoli, R.; Morra, E.; Mecucci, C. C-kit mutations in core binding factor leukemias. Blood 2000, 95, 726–727. [Google Scholar] [CrossRef]
- Cairoli, R.; Beghini, A.; Grillo, G.; Nadali, G.; Elice, F.; Ripamonti, C.B.; Colapietro, P.; Nichelatti, M.; Pezzetti, L.; Lunghi, M.; et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: An Italian retrospective study. Blood 2006, 107, 3463–3468. [Google Scholar] [CrossRef]
- Paschka, P.; Marcucci, G.; Ruppert, A.S.; Mrózek, K.; Chen, H.; Kittles, R.A.; Vukosavljevic, T.; Perrotti, D.; Vardiman, J.W.; Carroll, A.J.; et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): A Cancer and Leukemia Group B Study. J. Clin. Oncol. 2006, 24, 3904–3911. [Google Scholar] [CrossRef]
- Liu, S.; Wu, L.-C.; Pang, J.; Santhanam, R.; Schwind, S.; Wu, Y.-Z.; Hickey, C.J.; Yu, J.; Becker, H.; Maharry, K.; et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell 2010, 17, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gao, L.; Luo, X.; Wang, L.; Gao, X.; Wang, W.; Sun, J.; Dou, L.; Li, J.; Xu, C.; et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood 2013, 121, 499–509. [Google Scholar] [CrossRef]
- Tickenbrock, L.; Hehn, S.; Sargin, B.; Evers, G.; Ng, P.R.; Choudhary, C.; Berdel, W.E.; Müller-Tidow, C.; Serve, H. Activation of Wnt signaling in cKit-ITD mediated transformation and imatinib sensitivity in acute myeloid leukemia. Int. J. Hematol. 2008, 88, 174–180. [Google Scholar] [CrossRef]
- Medina, M.A.; Ugarte, G.D.; Vargas, M.F.; Avila, M.E.; Necuñir, D.; Elorza, A.A.; Gutiérrez, S.E.; De Ferrari, G.V. Alternative RUNX1 Promoter Regulation by Wnt/β-Catenin Signaling in Leukemia Cells and Human Hematopoietic Progenitors. J. Cell. Physiol. 2016, 231, 1460–1467. [Google Scholar] [CrossRef]
- Mathas, S.; Kreher, S.; Meaburn, K.J.; Jöhrens, K.; Lamprecht, B.; Assaf, C.; Sterry, W.; Kadin, M.E.; Daibata, M.; Joos, S.; et al. Gene deregulation and spatial genome reorganization near breakpoints prior to formation of translocations in anaplastic large cell lymphoma. Proc. Natl. Acad. Sci. USA 2009, 106, 5831–5836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzaroni, F.; Del Giacco, L.; Biasci, D.; Turrini, M.; Prosperi, L.; Brusamolino, R.; Cairoli, R.; Beghini, A. Intronless WNT10B-short variant underlies new recurrent allele-specific rearrangement in acute myeloid leukaemia. Sci. Rep. 2016, 6, e37201. [Google Scholar] [CrossRef] [PubMed]
- Müller-Tidow, C.; Steffen, B.; Cauvet, T.; Tickenbrock, L.; Ji, P.; Diederichs, S.; Sargin, B.; Köhler, G.; Stelljes, M.; Puccetti, E.; et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol. Cell. Biol. 2004, 24, 2890–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.; Kohlmann, A.; Schnittger, S.; Brors, B.; Dugas, M.; Mergenthaler, S.; Kern, W.; Hiddemann, W.; Eils, R.; Haferlach, T. Acute myeloid leukemias with reciprocal rearrangements can be distinguished by specific gene expression profiles. Proc. Natl. Acad. Sci. USA 2002, 99, 10008–10013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.-H.; Nguyen, C.; Yan, C.; Ries, R.E.; Chen, Q.-R.; Hu, Y.; Ostronoff, F.; Stirewalt, D.L.; Komatsoulis, G.; Levy, S.; et al. Transcriptome Profiling of Pediatric Core Binding Factor AML. PLoS ONE 2015, 10, e0138782. [Google Scholar] [CrossRef] [Green Version]
- Care, R.S.; Valk, P.J.M.; Goodeve, A.C.; Abu-Duhier, F.M.; Geertsma-Kleinekoort, W.M.C.; Wilson, G.A.; Gari, M.A.; Peake, I.R.; Löwenberg, B.; Reilly, J.T. Incidence and prognosis of c-KIT and FLT3 mutations in core binding factor (CBF) acute myeloid leukaemias. Br. J. Haematol. 2003, 121, 775–777. [Google Scholar] [CrossRef] [Green Version]
- Beghini, A.; Ripamonti, C.B.; Cairoli, R.; Cazzaniga, G.; Colapietro, P.; Elice, F.; Nadali, G.; Grillo, G.; Haas, O.A.; Biondi, A.; et al. KIT activating mutations: Incidence in adult and pediatric acute myeloid leukemia, and identification of an internal tandem duplication. Haematologica 2004, 89, 920–925. [Google Scholar]
- Schnittger, S.; Kohl, T.M.; Haferlach, T.; Kern, W.; Hiddemann, W.; Spiekermann, K.; Schoch, C. KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood 2006, 107, 1791–1799. [Google Scholar] [CrossRef] [Green Version]
- Cairoli, R.; Grillo, G.; Beghini, A.; Tedeschi, A.; Ripamonti, C.B.; Larizza, L.; Morra, E. C-Kit point mutations in core binding factor leukemias: Correlation with white blood cell count and the white blood cell index. Leukemia 2003, 17, 471–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ustun, C.; Morgan, E.; Moodie, E.E.M.; Pullarkat, S.; Yeung, C.; Broesby-Olsen, S.; Ohgami, R.; Kim, Y.; Sperr, W.; Vestergaard, H.; et al. Core-binding factor acute myeloid leukemia with t(8;21): Risk factors and a novel scoring system (I-CBFit). Cancer Med. 2018, 7, 4447–4455. [Google Scholar] [CrossRef] [PubMed]
- . Madan, V.; Han, L.; Hattori, N.; Teoh, W.W.; Mayakonda, A.; Sun, Q.Y.; Ding, L.W.; Nordin, H.B.M.; Lim, S.L.; Shyamsunder, P.; et al. ASXL2 regulates hematopoiesis in mice and its deficiency promotes myeloid expansion. Haematologica. 2018, 103, 1980–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Soria, N.; McKenzie, L.; Draper, J.; Ptasinska, A.; Issa, H.; Poltluri, S.; Blair, H.J.; Pickin, A.; Isa, A.; Chin, P.S.; et al. The Oncogenic Transcription Factor RUNX1/ETO Corrupts Cell Cycle Regulation to Drive Leukemic Transformation. Cancer Cell 2018, 34, 626–642. [Google Scholar] [CrossRef] [Green Version]
- Mazumdar, C.; Shen, Y.; Xavy, S.; Zhao, F.; Reinisch, A.; Li, R.; Corces, M.R.; Flynn, R.A.; Buenrostro, J.D.; Chan, S.M.; et al. Leukemia-Associated Cohesin Mutants Dominantly Enforce Stem Cell Programs and Impair Human Hematopoietic Progenitor Differentiation. Cell Stem Cell 2015, 17, 675–688. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, N.; Akashi, A.; Nagata, Y.; Kihara, R.; Ishikawa, Y.; Asou, N.; Ohtake, S.; Miyawaki, S.; Sakura, T.; Ozawa, Y.; et al. Clinical significance of ASXL2 and ZBTB7A mutations and C-terminally truncated RUNX1-RUNX1T1 expression in AML patients with t(8;21) enrolled in the JALSG AML201 study. Ann. Hematol. 2019, 98, 83–91. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Gao, J.; Adli, M.; Dey, A.; Trimarchi, T.; Chung, Y.R.; Kuscu, C.; Hricik, T.; Ndiaye-Lobry, D.; Lafave, L.M.; et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013, 210, 2641–2659. [Google Scholar] [CrossRef] [Green Version]
- Inoue, D.; Kitaura, J.; Togami, K.; Nishimura, K.; Enomoto, Y.; Uchida, T.; Kagiyama, Y.; Kawabata, K.C.; Nakahara, F.; Izawa, K.; et al. Myelodysplastic syndromes are induced by histone methylation–altering ASXL1 mutations. J. Clin. Investig. 2013, 123, 4627–4640. [Google Scholar] [CrossRef]
- Micol, J.-B.; Duployez, N.; Boissel, N.; Petit, A.; Geffroy, S.; Nibourel, O.; Lacombe, C.; Lapillonne, H.; Etancelin, P.; Figeac, M.; et al. Frequent ASXL2 mutations in acute myeloid leukemia patients with t(8;21)/RUNX1-RUNX1T1 chromosomal translocations. Blood 2014, 124, 1445–1449. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.; Yao, H.; Romans, A.; Dando, C.; Pierce, S.; Borthakur, G.; Hamilton, A.; Bueso-Ramos, C.; Ravandi, F.; Garcia-Manero, G.; et al. Modeling interactions between leukemia-specific chromosomal changes, somatic mutations, and gene expression patterns during progression of core-binding factor leukemias. Genes Chromosomes Cancer 2010, 49, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Chou, W.-C.; Huang, H.-H.; Hou, H.-A.; Chen, C.-Y.; Tang, J.-L.; Yao, M.; Tsay, W.; Ko, B.-S.; Wu, S.-J.; Huang, S.-Y.; et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood 2010, 116, 4086–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thota, S.; Viny, A.D.; Makishima, H.; Spitzer, B.; Radivoyevitch, T.; Przychodzen, B.; Sekeres, M.A.; Levine, R.L.; Maciejewski, J.P. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 2014, 124, 1790–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, B.; Mertens, F.; Mitelman, F. Secondary chromosomal abnormalities in acute leukemias. Leukemia 1994, 8, 953–962. [Google Scholar] [PubMed]
- Nishii, K.; Usui, E.; Katayama, N.; Lorenzo, F.; Nakase, K.; Kobayashi, T.; Miwa, H.; Mizutani, M.; Tanaka, I.; Nasu, K.; et al. Characteristics of t(8;21) acute myeloid leukemia (AML) with additional chromosomal abnormality: Concomitant trisomy 4 may constitute a distinctive subtype of t(8;21) AML. Leukemia 2003, 17, 731–737. [Google Scholar] [CrossRef] [Green Version]
- Kuchenbauer, F.; Schnittger, S.; Look, T.; Gilliland, G.; Tenen, D.; Haferlach, T.; Hiddemann, W.; Buske, C.; Schoch, C. Identification of additional cytogenetic and molecular genetic abnormalities in acute myeloid leukaemia with t(8;21)/AML1-ETO. Br. J. Haematol. 2006, 134, 616–619. [Google Scholar] [CrossRef]
- Beghini, A.; Magnani, I.; Ripamonti, C.B.; Larizza, L. Amplification of a novel c-Kit activating mutation Asn(822)-Lys in the Kasumi-1 cell line: A t(8;21)-Kit mutant model for acute myeloid leukemia. Hematol. J. 2002, 3, 157–163. [Google Scholar] [CrossRef]
- Matsuura, S.; Yan, M.; Lo, M.-C.; Ahn, E.-Y.; Weng, S.; Dangoor, D.; Matin, M.; Higashi, T.; Feng, G.-S.; Zhang, D.-E. Negative effects of GM-CSF signaling in a murine model of t(8;21)-induced leukemia. Blood 2012, 119, 3155–3163. [Google Scholar] [CrossRef]
- Klug, C.A. GM-CSFRα: The sex-chromosome link to t(8;21)(+) AML? Blood 2012, 119, 2976–2977. [Google Scholar] [CrossRef] [Green Version]
- Krauth, M.-T.; Eder, C.; Alpermann, T.; Bacher, U.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T.; Schnittger, S. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1T1: Frequency and impact on clinical outcome. Leukemia 2014, 28, 1449–1458. [Google Scholar] [CrossRef]
- Chen, M.J.; Yokomizo, T.; Zeigler, B.M.; Dzierzak, E.; Speck, N.A. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 2009, 457, 887–891. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Stacy, T.; Binder, M.; Marin-Padilla, M.; Sharpe, A.H.; Speck, N.A. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc. Natl. Acad. Sci. USA 1996, 93, 3444–3449. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Yagi, H.; Bronson, R.T.; Tominaga, K.; Matsunashi, T.; Deguchi, K.; Tani, Y.; Kishimoto, T.; Komori, T. Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proc. Natl. Acad. Sci. USA 1996, 93, 12359–12363. [Google Scholar] [CrossRef] [Green Version]
- Yergeau, D.A.; Hetherington, C.J.; Wang, Q.; Zhang, P.; Sharpe, A.H.; Binder, M.; Marín-Padilla, M.; Tenen, D.G.; Speck, N.A.; Zhang, D.E. Embryonic lethality and impairment of haematopoiesis in mice heterozygous for an AML1-ETO fusion gene. Nat. Genet. 1997, 15, 303–306. [Google Scholar] [CrossRef]
- Okuda, T.; Cai, Z.; Yang, S.; Lenny, N.; Lyu, C.J.; van Deursen, J.M.; Harada, H.; Downing, J.R. Expression of a knocked-in AML1-ETO leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood 1998, 91, 3134–3143. [Google Scholar] [CrossRef] [Green Version]
- Castilla, L.H.; Perrat, P.; Martinez, N.J.; Landrette, S.F.; Keys, R.; Oikemus, S.; Flanegan, J.; Heilman, S.; Garrett, L.; Dutra, A.; et al. Identification of genes that synergize with Cbfb-MYH11 in the pathogenesis of acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 4924–4929. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, M.; O’Brien, D.; Kumaravelu, P.; Lenny, N.; Yeoh, E.-J.; Downing, J.R. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell 2002, 1, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Valk, P.J.M.; Bowen, D.T.; Frew, M.E.; Goodeve, A.C.; Löwenberg, B.; Reilly, J.T. Second hit mutations in the RTK/RAS signaling pathway in acute myeloid leukemia with inv(16). Haematologica 2004, 89, e106. [Google Scholar]
- Bowen, D.T.; Frew, M.E.; Hills, R.; Gale, R.E.; Wheatley, K.; Groves, M.J.; Langabeer, S.E.; Kottaridis, P.D.; Moorman, A.V.; Burnett, A.K.; et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood 2005, 106, 2113–2119. [Google Scholar] [CrossRef] [Green Version]
- Goemans, B.F.; Zwaan, C.M.; Miller, M.; Zimmermann, M.; Harlow, A.; Meshinchi, S.; Loonen, A.H.; Hählen, K.; Reinhardt, D.; Creutzig, U.; et al. Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia 2005, 19, 1536–1542. [Google Scholar] [CrossRef] [Green Version]
- Boissel, N.; Leroy, H.; Brethon, B.; Philippe, N.; de Botton, S.; Auvrignon, A.; Raffoux, E.; Leblanc, T.; Thomas, X.; Hermine, O.; et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia 2006, 20, 965–970. [Google Scholar] [CrossRef] [Green Version]
- Kühn, M.W.M.; Radtke, I.; Bullinger, L.; Goorha, S.; Cheng, J.; Edelmann, J.; Gohlke, J.; Su, X.; Paschka, P.; Pounds, S.; et al. High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood 2012, 119, 67–75. [Google Scholar] [CrossRef]
- Speck, N.A.; Gilliland, D.G. Core-binding factors in haematopoiesis and leukaemia. Nat. Rev. Cancer 2002, 2, 502–513. [Google Scholar] [CrossRef]
- Castilla, L.H.; Wijmenga, C.; Wang, Q.; Stacy, T.; Speck, N.A.; Eckhaus, M.; Marín-Padilla, M.; Collins, F.S.; Wynshaw-Boris, A.; Liu, P.P. Failure of embryonic hematopoiesis and lethal hemorrhages in mouse embryos heterozygous for a knocked-in leukemia gene CBFB-MYH11. Cell 1996, 87, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Mulloy, J.C.; Jankovic, V.; Wunderlich, M.; Delwel, R.; Cammenga, J.; Krejci, O.; Zhao, H.; Valk, P.J.M.; Lowenberg, B.; Nimer, S.D. AML1-ETO fusion protein up-regulates TRKA mRNA expression in human CD34+ cells, allowing nerve growth factor-induced expansion. Proc. Natl. Acad. Sci. USA 2005, 102, 4016–4021. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhang, W.; Ramdas, L.; Stivers, D.N.; Jones, D.M.; Kantarjian, H.M.; Estey, E.H.; Vadhan-Raj, S.; Medeiros, L.J.; Bueso-Ramos, C.E. Comparative analysis of genes regulated in acute myelomonocytic leukemia with and without inv(16)(p13q22) using microarray techniques, real-time PCR, immunohistochemistry, and flow cytometry immunophenotyping. Mod. Pathol. 2007, 20, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Görgens, A.; Radtke, S.; Mollmann, M.; Durig, J.; Horn, P.A.; Giebel, B. Revision of the Human Hematopoietic Tree: Granulocyte Subtypes Derive from Distinct Hematopoietic Lineages. Cell Rep. 2013, 3, 1539–1552. [Google Scholar] [CrossRef] [Green Version]
- Ben-Ami, O.; Friedman, D.; Leshkowitz, D.; Goldenberg, D.; Orlovsky, K.; Pencovich, N.; Lotem, J.; Tanay, A.; Groner, Y. Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell Rep. 2013, 4, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Goyama, S.; Schibler, J.; Cunningham, L.; Zhang, Y.; Rao, Y.; Nishimoto, N.; Nakagawa, M.; Olsson, A.; Wunderlich, M.; Link, K.A.; et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J. Clin. Investig. 2013, 123, 3876–3888. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Kanbe, E.; Peterson, L.F.; Boyapati, A.; Miao, Y.; Wang, Y.; Chen, I.-M.; Chen, Z.; Rowley, J.D.; Willman, C.L.; et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat. Med. 2006, 12, 945–949. [Google Scholar] [CrossRef]
- Peterson, L.F.; Boyapati, A.; Ahn, E.-Y.; Biggs, J.R.; Okumura, A.J.; Lo, M.-C.; Yan, M.; Zhang, D.-E. Acute myeloid leukemia with the 8q22;21q22 translocation: Secondary mutational events and alternative t(8;21) transcripts. Blood 2007, 110, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Burel, S.A.; Peterson, L.F.; Kanbe, E.; Iwasaki, H.; Boyapati, A.; Hines, R.; Akashi, K.; Zhang, D.-E. Deletion of an AML1-ETO C-terminal NcoR/SMRT-interacting region strongly induces leukemia development. Proc. Natl. Acad. Sci. USA 2004, 101, 17186–17191. [Google Scholar] [CrossRef] [Green Version]
- Ihle, J.N. Cytokine receptor signalling. Nature 1995, 377, 591–594. [Google Scholar] [CrossRef]
- Marcucci, G.; Geyer, S.; Zhao, W.; Caroll, A.J.; Bucci, D.; Uy, G.L.; Blum, W.; Pardee, T.; Wetzler, M.; Stock, W.; et al. Adding KIT Inhibitor Dasatinib (DAS) to Chemotherapy Overcomes the Negative Impact of KIT Mutation/over-Expression in Core Binding Factor (CBF) Acute Myeloid Leukemia (AML): Results from CALGB 10801 (Alliance). Blood 2014, 124, e8. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Weber, D.; Benner, A.; Bullinger, L.; Heuser, M.; Gaidzik, V.I.; Thol, F.; Agrawal, M.; Teleanu, V.; et al. Adding dasatinib to intensive treatment in core-binding factor acute myeloid leukemia-results of the AMLSG 11-08 trial. Leukemia 2018, 32, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Arber, D.A., Hasserjian, R.P., Le Beau, M.M., et al., Eds.; IARC: Lyon, France, 2017.
- Tarlock, K.; Alonzo, T.A.; Wang, Y.C.; Gerbing, R.B.; Ries, R.; Loken, M.R.; Pardo, L.; Hylkema, T.; Joaquin, J.; Sarukkai, L.; et al. Functional Properties of KIT mitations are associated with differential clinical outcomes and response to targeted therapeutics in CBF acute myeloid leukemia. Clin. Cancer Res. 2019, 25, 5038–5048. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhao, L.J.; Wu, C.F.; Liu, P.; Shi, L.; Liang, Y.; Xiong, S.M.; Mi, J.Q.; Chen, Z.; Ren, R.; et al. C-KIT mutation cooperates with full-length AML1-ETO to induce acute myeloid leukemia in mice. Proc. Natl. Acad. Sci. USA. 2011, 108, 2450–2455. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.; Lee, S.C. Mutations in spliceosoma genes and therapeutic opportunities in myeloid malignancies. Genes Chromosomes Cancer 2019, 58, 889–902. [Google Scholar] [CrossRef]




© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beghini, A. Core Binding Factor Leukemia: Chromatin Remodeling Moves Towards Oncogenic Transcription. Cancers 2019, 11, 1973. https://doi.org/10.3390/cancers11121973
Beghini A. Core Binding Factor Leukemia: Chromatin Remodeling Moves Towards Oncogenic Transcription. Cancers. 2019; 11(12):1973. https://doi.org/10.3390/cancers11121973
Chicago/Turabian StyleBeghini, Alessandro. 2019. "Core Binding Factor Leukemia: Chromatin Remodeling Moves Towards Oncogenic Transcription" Cancers 11, no. 12: 1973. https://doi.org/10.3390/cancers11121973
APA StyleBeghini, A. (2019). Core Binding Factor Leukemia: Chromatin Remodeling Moves Towards Oncogenic Transcription. Cancers, 11(12), 1973. https://doi.org/10.3390/cancers11121973