Riboregulation in Nitrogen-Fixing Endosymbiotic Bacteria


Abstract
:1. Introduction
2. Deciphering the Rhizobial Non-Coding Transcriptome: From Comparative Genomics to RNA-Seq
3. Conservation of Rhizobial sRNAs: α-Proteobacterial sRNA Families
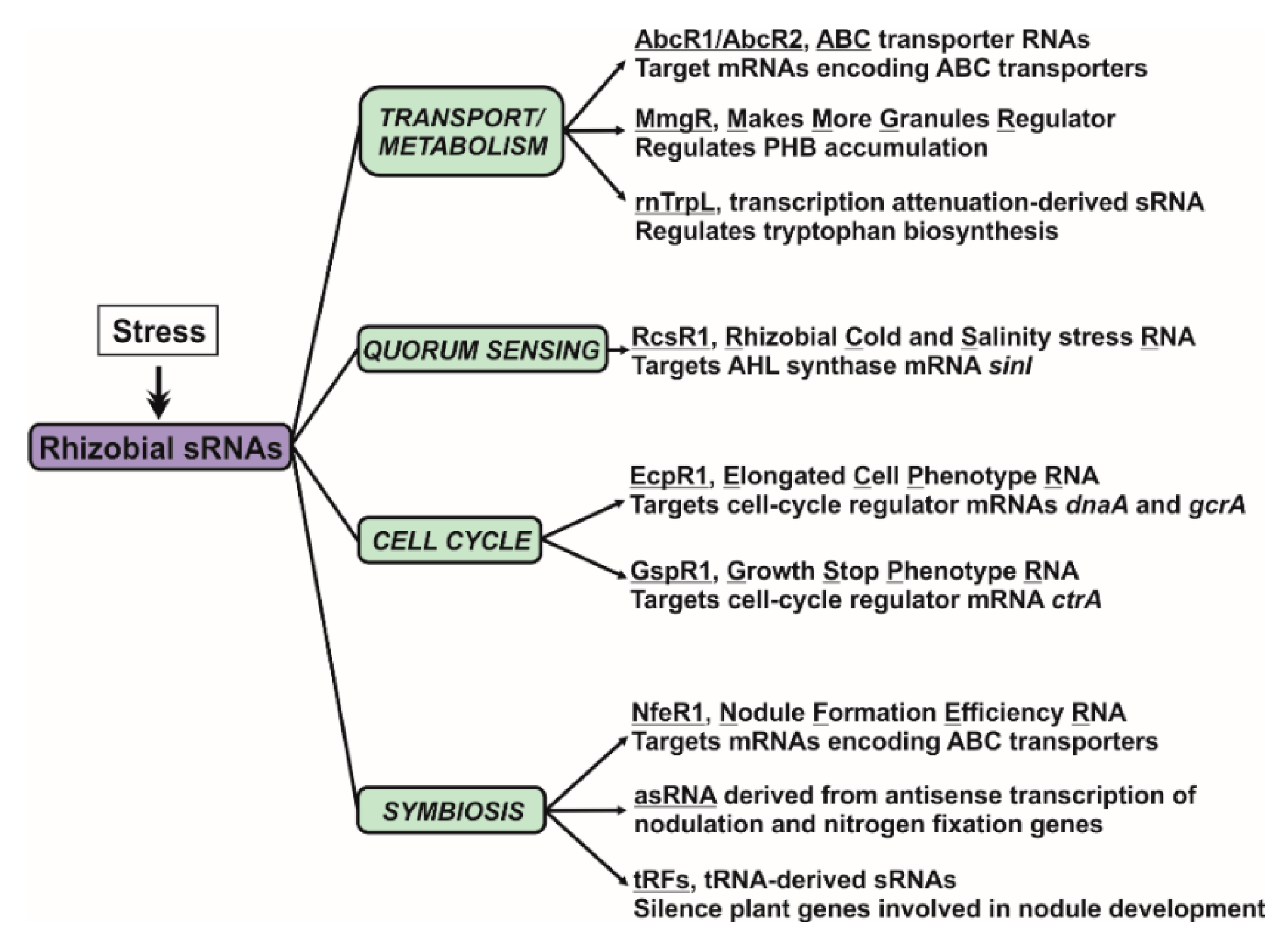
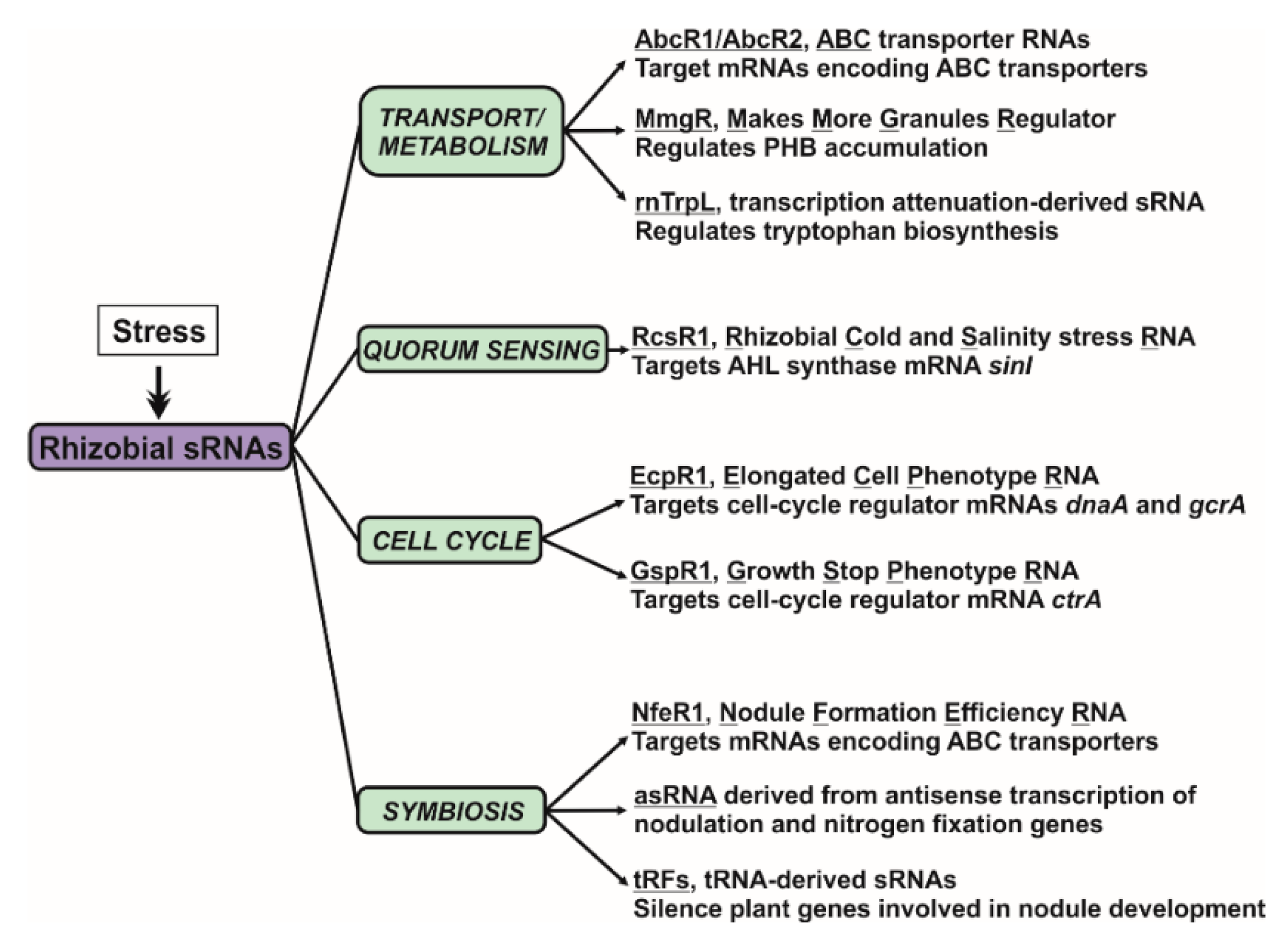
4. Assigning Functions to Rhizobial Trans-sRNAs
4.1. Nutrient Uptake and Metabolism
4.2. Quorum Sensing
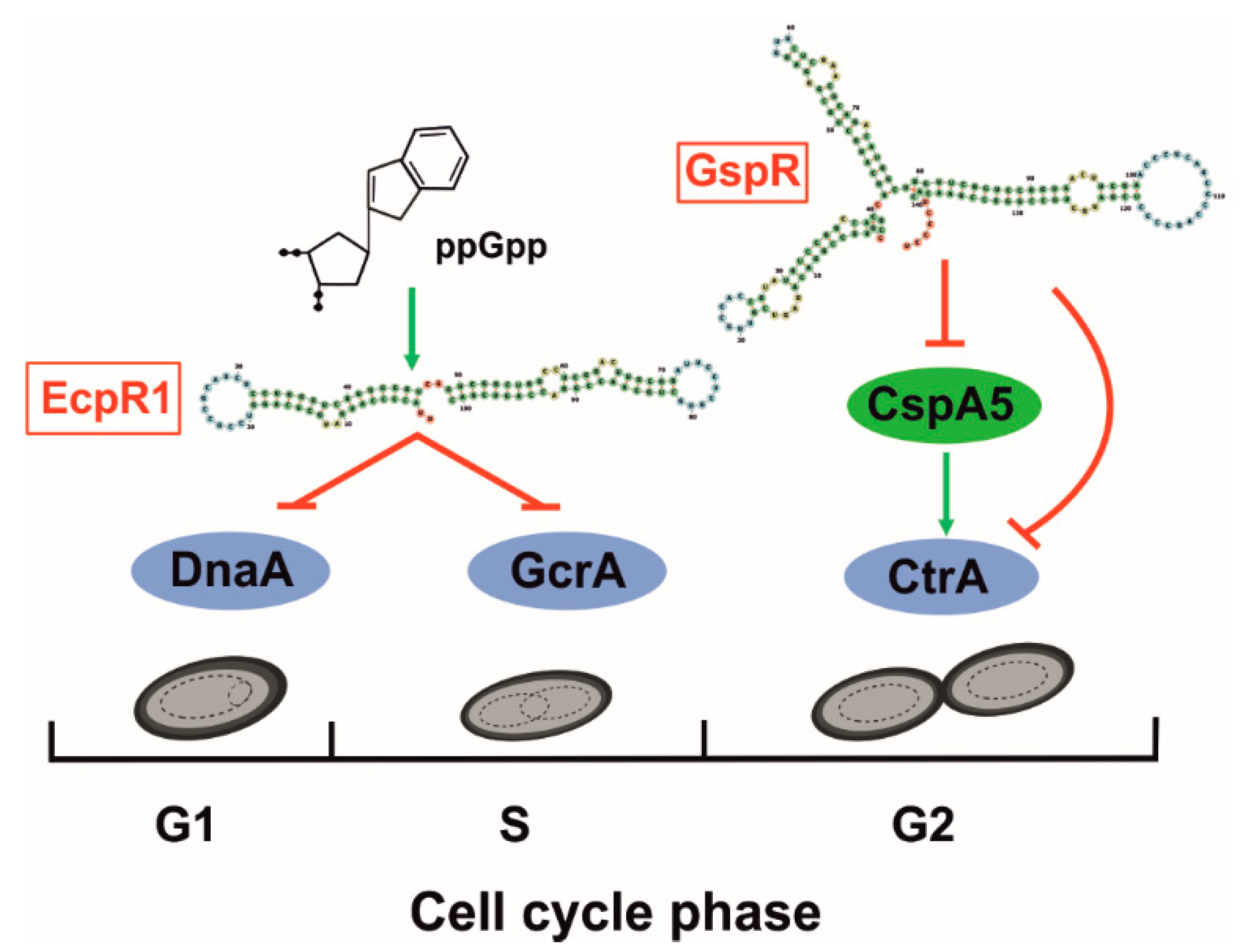
4.3. Cell Cycle
4.4. Nodule Development and Functioning
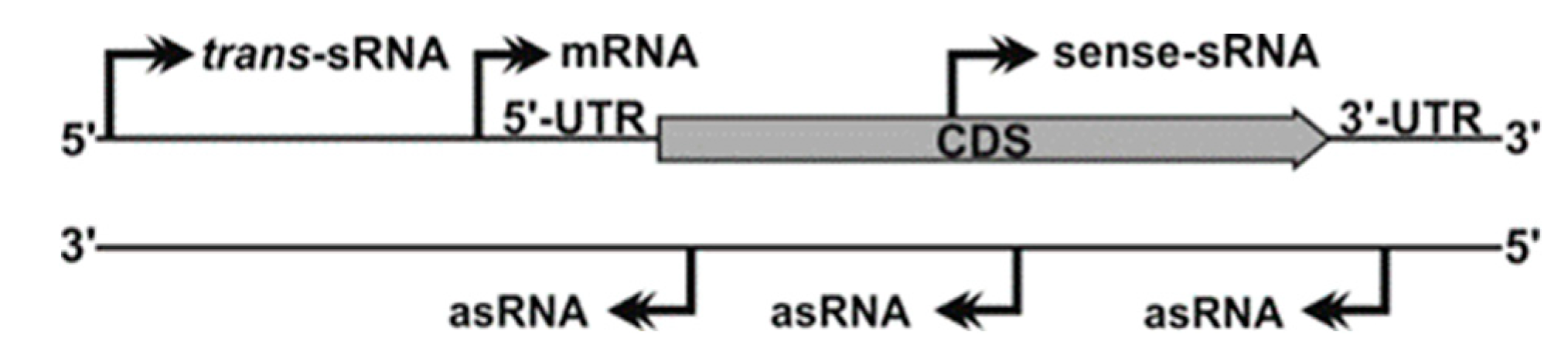
5. Antisense Transcription in S. meliloti
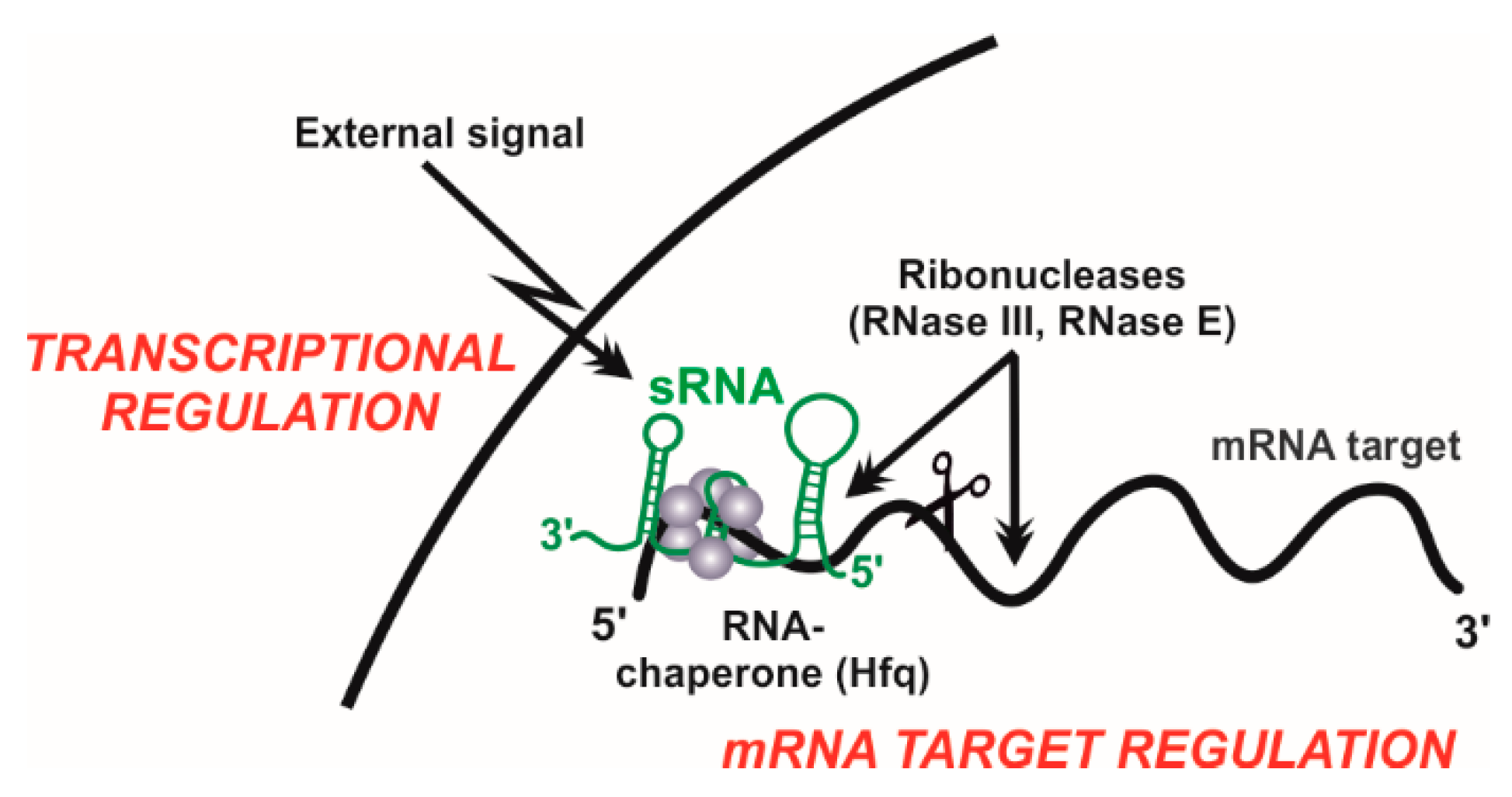
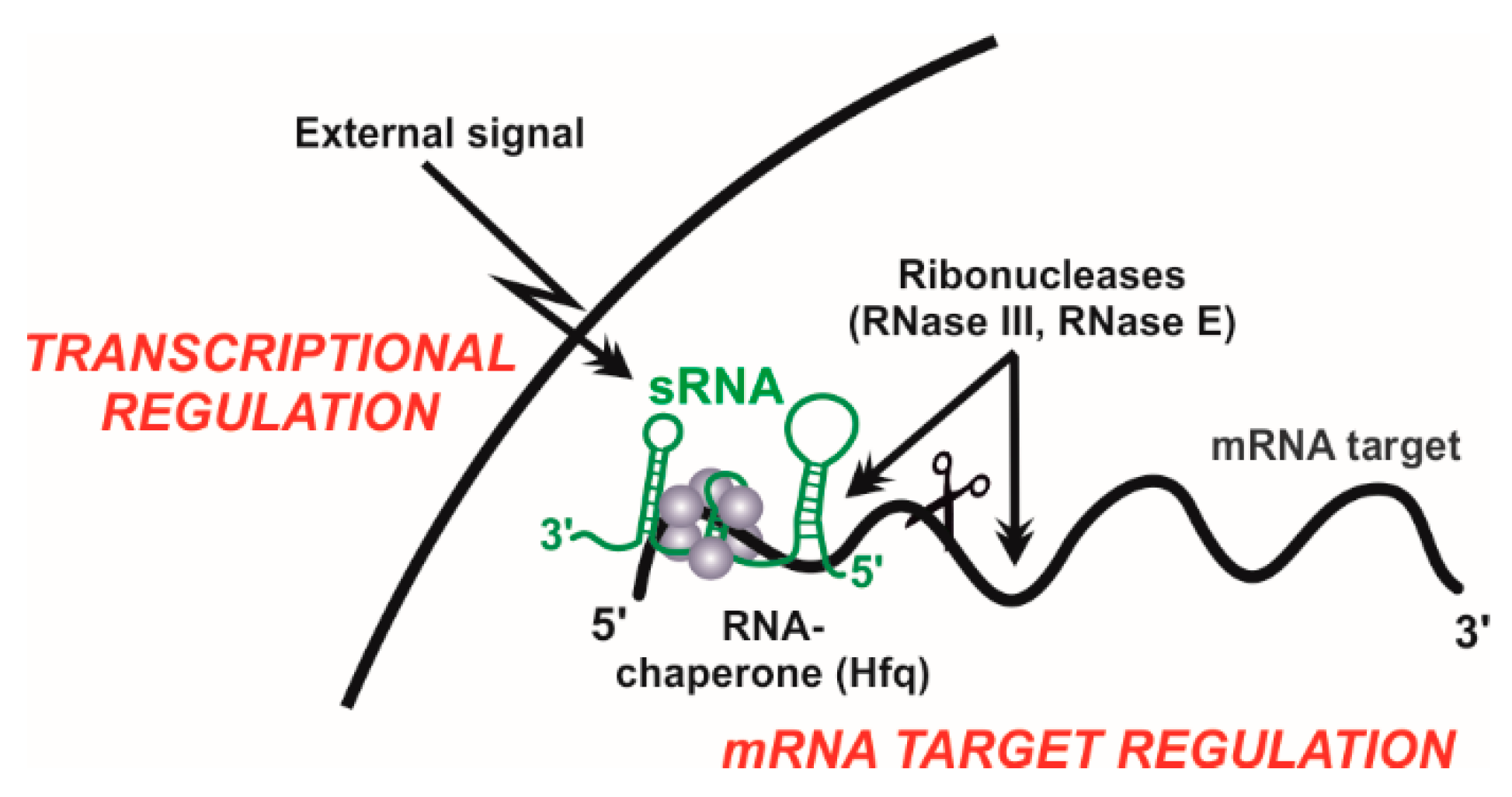
6. Proteins Assisting sRNA Activity
6.1. The Rhizobial RNA Chaperone Hfq
6.2. Rhizobial Ribonucleases
7. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Poole, P.; Ramachandran, V.; Terpolilli, J. Rhizobia: From saprophytes to endosymbionts. Nat. Rev. Microbiol. 2018, 16, 291. [Google Scholar] [CrossRef] [PubMed]
- Gage, D. Infection and invasion of roots by symbiotic, nitrogen-fixing rhizobia during nodulation of temperate legumes. Microbiol. Mol. Biol. Rev. 2004, 68, 280–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, K.E.; Kobayashi, H.; Walker, G.C. Molecular determinants of a symbiotic chronic infection. Annu. Rev. Genet. 2008, 42, 413–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahran, H.H. Rhizobium-legume symbiosis and nitrogen fixation under severe conditions and in an arid climate. Microbiol. Mol. Biol. Rev. 1999, 63, 968–989. [Google Scholar] [CrossRef] [Green Version]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; Van Der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef]
- Masson-Boivin, C.; Giraud, E.; Perret, X.; Batut, J. Establishing nitrogen-fixing symbiosis with legumes: How many rhizobium recipes? Trends Microbiol. 2009, 17, 458–466. [Google Scholar] [CrossRef]
- Soto, M.J.; Domínguez-Ferreras, A.; Pérez-Mendoza, D.; Sanjuán, J.; Olivares, J. Mutualism versus pathogenesis: The give-and-take in plant-bacteria interactions. Cell. Microbiol. 2009, 11, 381–388. [Google Scholar] [CrossRef]
- Van De Velde, W.; Zehirov, G.; Szatmari, A.; Debreczeny, M.; Ishihara, H.; Kevei, Z.; Farkas, A.; Mikulass, K.; Nagy, A.; Tiricz, H.; et al. Plant Peptides Govern Terminal Differentiation of Bacteria in Symbiosis. Science 2010, 327, 1122–1126. [Google Scholar] [CrossRef]
- Galibert, F.; Finan, T.M.; Long, S.R.; Puhler, A.; Abola, P.; Ampe, F.; Barloy-Hubler, F.; Barnett, M.J.; Becker, A.; Boistard, P.; et al. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 2001, 293, 668–672. [Google Scholar] [CrossRef] [Green Version]
- Brilli, M.; Fondi, M.; Fani, R.; Mengoni, A.; Ferri, L.; Bazzicalupo, M.; Biondi, E.G. The diversity and evolution of cell cycle regulation in alpha-proteobacteria: A comparative genomic analysis. BMC Syst. Biol. 2010, 4, 52. [Google Scholar] [CrossRef] [Green Version]
- Krol, E.; Blom, J.; Winnebald, J.; Berhörster, A.; Barnett, M.J.; Goesmann, A.; Baumbach, J.; Becker, A. RhizoRegNet—A database of rhizobial transcription factors and regulatory networks. J. Biotechnol. 2011, 155, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Cossart, P. Prokaryotic transcriptomics: A new view on regulation, physiology and pathogenicity. Nat. Rev. Genet. 2010, 11, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.G.H.; Romby, P. Small RNAs in bacteria and archaea: Who they are, what they do, and how they do it. In Advances in Genetics; Theodore Friedmann, J.C.D., Stephen, F.G., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 90, pp. 133–208. [Google Scholar]
- Sobrero, P.; Valverde, C. The bacterial protein Hfq: Much more than a mere RNA-binding factor. Crit. Rev. Microbiol. 2012, 38, 276–299. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Zurdo, J.I.; Valverde, C.; Becker, A. Insights into the noncoding RNome of nitrogen-fixing endosymbiotic α-proteobacteria. Mol. Plant-Microbe Interact. 2013, 26, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Zurdo, J.I.; Robledo, M. Unraveling the universe of small RNA regulators in the legume symbiont Sinorhizobium Meliloti. Symbiosis 2015, 67, 43–54. [Google Scholar] [CrossRef]
- Becker, A.; Overlöper, A.; Schlüter, J.-P.; Reinkensmeier, J.; Robledo, M.; Giegerich, R.; Narberhaus, F.; Evguenieva-Hackenberg, E. Riboregulation in plant-associated α-proteobacteria. RNA Biol. 2014, 11, 550–562. [Google Scholar] [CrossRef] [Green Version]
- Ulve, V.M.; Sevin, E.W.; Cheron, A.; Barloy-Hubler, F. Identification of chromosomal alpha-proteobacterial small RNAs by comparative genome analysis and detection in Sinorhizobium meliloti strain 1021. BMC Genom. 2007, 8, 467. [Google Scholar] [CrossRef] [Green Version]
- Valverde, C.; Livny, J.; Schluter, J.P.; Reinkensmeier, J.; Becker, A.; Parisi, G. Prediction of Sinorhizobium meliloti sRNA genes and experimental detection in strain 2011. BMC Genom. 2008, 9, 416. [Google Scholar] [CrossRef] [Green Version]
- del Val, C.; Rivas, E.; Torres-Quesada, O.; Toro, N.; Jiménez-Zurdo, J.I. Identification of differentially expressed small non-coding RNAs in the legume endosymbiont Sinorhizobium meliloti by comparative genomics. Mol. Microbiol. 2007, 66, 1080–1091. [Google Scholar] [CrossRef] [Green Version]
- Vercruysse, M.; Fauvart, M.; Cloots, L.; Engelen, K.; Thijs, I.M.; Marchal, K.; Michiels, J. Genome-wide detection of predicted non-coding RNAs in Rhizobium etli expressed during free-living and host-associated growth using a high-resolution tiling array. BMC Genom. 2010, 11, 53. [Google Scholar] [CrossRef] [Green Version]
- Madhugiri, R.; Pessi, G.; Voss, B.; Hahn, J.; Sharma, C.M.; Reinhardt, R.; Vogel, J.; Hess, W.R.; Fischer, H.-M.; Evguenieva-Hackenberg, E. Small RNAs of the Bradyrhizobium/Rhodopseudomonas lineage and their analysis. RNA Biol. 2012, 9, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuli, X.; Wenlong, Z.; Xiao, W.; Jing, Z.; Baohai, H.; Zhengzheng, Z.; Bin-Guang, M.; Youguo, L. A Genome-Wide Prediction and Identification of Intergenic Small RNAs by Comparative Analysis in Mesorhizobium huakuii 7653R. Front. Microbiol. 2017, 8, 1730. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.M.; Vogel, J. Differential RNA-seq: The approach behind and the biological insight gained. Curr. Opin. Microbiol. 2014, 19, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Ettwiller, L.; Buswell, J.; Yigit, E.; Schildkraut, I. A novel enrichment strategy reveals unprecedented number of novel transcription start sites at single base resolution in a model prokaryote and the gut microbiome. BMC Genom. 2016, 17, 199. [Google Scholar] [CrossRef] [Green Version]
- Schlüter, J.P.; Reinkensmeier, J.; Daschkey, S.; Evguenieva-Hackenberg, E.; Janssen, S.; Janicke, S.; Becker, J.D.; Giegerich, R.; Becker, A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genom. 2010, 11, 245. [Google Scholar] [CrossRef] [Green Version]
- Schlüter, J.P.; Reinkensmeier, J.; Barnett, M.J.; Lang, C.; Krol, E.; Giegerich, R.; Long, S.R.; Becker, A. Global mapping of transcription start sites and promoter motifs in the symbiotic alpha-proteobacterium Sinorhizobium meliloti 1021. BMC Genom. 2013, 14, 156. [Google Scholar] [CrossRef] [Green Version]
- Čuklina, J.; Hahn, J.; Imakaev, M.; Omasits, U.; Förstner, K.U.; Ljubimov, N.; Goebel, M.; Pessi, G.; Fischer, H.-M.; Ahrens, C.H.; et al. Genome-wide transcription start site mapping of Bradyrhizobium japonicum grown free-living or in symbiosis—A rich resource to identify new transcripts, proteins and to study gene regulation. BMC Genom. 2016, 17, 302. [Google Scholar] [CrossRef] [Green Version]
- Sallet, E.; Roux, B.; Sauviac, L.; Jardinaud, M.-F.o.; Carrère, S.; Faraut, T.; de Carvalho-Niebel, F.; Gouzy, J.; Gamas, P.; Capela, D.; et al. Next-generation annotation of prokaryotic genomes with EuGene-P: Application to Sinorhizobium meliloti 2011. DNA Res. 2013, 20, 339–354. [Google Scholar] [CrossRef]
- Sittka, A.; Lucchini, S.; Papenfort, K.; Sharma, C.M.; Rolle, K.; Binnewies, T.T.; Hinton, J.C.D.; Vogel, J. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 2008, 4, e1000163. [Google Scholar] [CrossRef] [Green Version]
- Sittka, A.; Sharma, C.M.; Rolle, K.; Vogel, J. Deep sequencing of Salmonella RNA associated with heterologous Hfq proteins in vivo reveals small RNAs as a major target class and identifies RNA processing phenotypes. RNA Biol. 2009, 6, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Torres-Quesada, O.; Reinkensmeier, J.; Schlüter, J.-P.; Robledo, M.; Peregrina, A.; Giegerich, R.; Toro, N.; Becker, A.; Jimenez-Zurdo, J.I. Genome-wide profiling of Hfq-binding RNAs uncovers extensive post-transcriptional rewiring of major stress response and symbiotic regulons in Sinorhizobium meliloti. RNA Biol. 2014, 11, 563–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, P.R.; Georg, J. Workflow for a Computational Analysis of an sRNA Candidate in Bacteria; Springer: New York, NY, USA, 2018; pp. 3–30. [Google Scholar] [CrossRef]
- Roux, B.; Rodde, N.; Jardinaud, M.-F.; Timmers, T.; Sauviac, L.; Cottret, L.; Carrère, S.; Sallet, E.; Courcelle, E.; Moreau, S.; et al. An integrated analysis of plant and bacterial gene expression in symbiotic root nodules using laser-capture microdissection coupled to RNA sequencing. Plant J. 2014, 77, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2017, 46, D335–D342. [Google Scholar] [CrossRef] [PubMed]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Keiler, K.C. Biology of trans-Translation. Annu. Rev. Microbiol. 2008, 62, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, S.; Kündig, C.; Hennecke, H. Discovery of a rhizobial RNA that is essential for symbiotic root nodule development. J. Bacteriol. 1991, 173, 6373–6382. [Google Scholar] [CrossRef] [Green Version]
- Ulve, V.M.; Cheron, A.; Trautwetter, A.; Fontenelle, C.; Barloy-Hubler, F. Characterization and expression patterns of Sinorhizobium meliloti tmRNA (ssrA). FEMS Microbiol. Lett. 2007, 269, 117–123. [Google Scholar] [CrossRef]
- Keenan, R.J.; Freymann, D.M.; Stroud, R.M.; Walter, P. The signal recognition particle. Annu. Rev. Biochem. 2001, 70, 755–775. [Google Scholar] [CrossRef] [Green Version]
- Wassarman, K.M. 6S RNA, a Global Regulator of Transcription. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Faucher, S.P.; Friedlander, G.; Livny, J.; Margalit, H.; Shuman, H.A. Legionella pneumophila 6S RNA optimizes intracellular multiplication. Proc. Natl. Acad. Sci. USA 2010, 107, 7533–7538. [Google Scholar] [CrossRef] [Green Version]
- Reinkensmeier, J.; Schlüter, J.-P.; Giegerich, R.; Becker, A. Conservation and Occurrence of Trans-Encoded sRNAs in the Rhizobiales. Genes 2011, 2, 925–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinkensmeier, J.; Giegerich, R. Thermodynamic matchers for the construction of the cuckoo RNA family. RNA Biol. 2015, 12, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagares, A., Jr.; Roux, I.; Valverde, C. Phylogenetic distribution and evolutionary pattern of an α-proteobacterial small RNA gene that controls polyhydroxybutyrate accumulation in Sinorhizobium meliloti. Mol. Phylogenetics Evol. 2016, 99, 182–193. [Google Scholar] [CrossRef] [PubMed]
- del Val, C.; Romero-Zaliz, R.; Torres-Quesada, O.; Peregrina, A.; Toro, N.; Jiménez-Zurdo, J.I. A survey of sRNA families in alpha-proteobacteria. RNA Biol. 2012, 9, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Robledo, M.; García-Tomsig, N.I.; Jiménez-Zurdo, J.I. Primary Characterization of Small RNAs in Symbiotic Nitrogen-Fixing Bacteria. In Host-Pathogen Interactions: Methods and Protocols; Medina, C., López-Baena, F.J., Eds.; Springer: New York, NY, USA, 2018; pp. 277–295. [Google Scholar] [CrossRef]
- Robledo, M.; Matia-Gonzalez, A.M.; Garcia-Tomsig, N.I.; Jimenez-Zurdo, J.I. Identification of Small RNA-Protein Partners in Plant Symbiotic Bacteria. Methods Mol. Biol. 2018, 1737, 351–370. [Google Scholar] [CrossRef]
- Wright, P.R.; Georg, J.; Mann, M.; Sorescu, D.A.; Richter, A.S.; Lott, S.; Kleinkauf, R.; Hess, W.R.; Backofen, R. CopraRNA and IntaRNA: Predicting small RNA targets, networks and interaction domains. Nucleic Acids Res. 2014, 42, W119–W123. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.R.; Richter, A.S.; Papenfort, K.; Mann, M.; Vogel, J.; Hess, W.R.; Backofen, R.; Georg, J. Comparative genomics boosts target prediction for bacterial small RNAs. Proc. Natl. Acad. Sci. USA 2013, 110, E3487–E3496. [Google Scholar] [CrossRef] [Green Version]
- Lalaouna, D.; Massé, E. Identification of sRNA interacting with a transcript of interest using MS2-affinity purification coupled with RNA sequencing (MAPS) technology. Genom. Data 2015, 5, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.M.; Vogel, J. Experimental approaches for the discovery and characterization of regulatory small RNA. Curr. Opin. Microbiol. 2009, 12, 536–546. [Google Scholar] [CrossRef]
- Mauchline, T.H.; Fowler, J.E.; East, A.K.; Sartor, A.L.; Zaheer, R.; Hosie, A.H.F.; Poole, P.S.; Finan, T.M. Mapping the Sinorhizobium meliloti 1021 solute-binding protein-dependent transportome. Proc. Natl. Acad. Sci. USA 2006, 103, 17933–17938. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.F. ABC transporters: Physiology, structure and mechanism—An overview. Res. Microbiol. 2001, 152, 205–210. [Google Scholar] [CrossRef]
- Nogales, J.; Munoz, S.; Olivares, J.; Sanjuan, J. Genetic characterization of oligopeptide uptake systems in Sinorhizobium meliloti. FEMS Microbiol. Lett. 2009, 293, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djordjevic, M.A. Sinorhizobium meliloti metabolism in the root nodule: A proteomic perspective. Proteomics 2004, 4, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Melior, H.; Li, S.; Madhugiri, R.; Stotzel, M.; Azarderakhsh, S.; Barth-Weber, S.; Baumgardt, K.; Ziebuhr, J.; Evguenieva-Hackenberg, E. Transcription attenuation-derived small RNA rnTrpL regulates tryptophan biosynthesis gene expression in trans. Nucleic Acids Res. 2019, 47, 6396–6410. [Google Scholar] [CrossRef]
- Miyakoshi, M.; Chao, Y.; Vogel, J. Cross talk between ABC transporter mRNAs via a target mRNA-derived sponge of the GcvB small RNA. EMBO J. 2015, 34, 1478–1492. [Google Scholar] [CrossRef]
- Wilms, I.; Voss, B.; Hess, W.R.; Leichert, L.I.; Narberhaus, F. Small RNA-mediated control of the Agrobacterium tumefaciens GABA binding protein: Small RNA-controlled GABA uptake. Mol. Microbiol. 2011, 80, 492–506. [Google Scholar] [CrossRef]
- Overloper, A.; Kraus, A.; Gurski, R.; Wright, P.R.; Georg, J.; Hess, W.R.; Narberhaus, F. Two separate modules of the conserved regulatory RNA AbcR1 address multiple target mRNAs in and outside of the translation initiation region. RNA Biol. 2014, 11, 624–640. [Google Scholar] [CrossRef] [Green Version]
- Caswell, C.C.; Gaines, J.M.; Roop, R.M., 2nd. The RNA chaperone Hfq independently coordinates expression of the VirB type IV secretion system and the LuxR-type regulator BabR in Brucella abortus 2308. J. Bacteriol. 2012, 194, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Torres-Quesada, O.; Millán, V.; Nisa-Martínez, R.; Bardou, F.; Crespi, M.; Toro, N.; Jiménez-Zurdo, J.I. Independent activity of the homologous small regulatory RNAs AbcR1 and AbcR2 in the legume symbiont Sinorhizobium meliloti. PLoS ONE 2013, 8, e68147. [Google Scholar] [CrossRef]
- Sheehan, L.M.; Caswell, C.C. An account of evolutionary specialization: The AbcR small RNAs in the Rhizobiales. Mol. Microbiol. 2018, 107, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, L.M.; Budnick, J.A.; Blanchard, C.; Dunman, P.M.; Caswell, C.C. A LysR-family transcriptional regulator required for virulence in Brucella abortus is highly conserved among the alpha-proteobacteria. Mol. Microbiol. 2015, 98, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Yao, S.-Y.; Becker, A.; Rüberg, S.; Yu, G.-Q.; Zhu, J.-B.; Cheng, H.-P. Two New Sinorhizobium meliloti LysR-Type Transcriptional Regulators Required for Nodulation. J. Bacteriol. 2005, 187, 4562–4572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Lu, D.; Wang, D.; Luo, L. Sinorhizobium meliloti lsrB is involved in alfalfa root nodule development and nitrogen-fixing bacteroid differentiation. Chin. Sci. Bull. 2013, 58, 4077–4083. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.; Wang, Y.; Luo, L. Transcriptional regulator LsrB of Sinorhizobium meliloti positively regulates the expression of genes involved in lipopolysaccharide biosynthesis. Appl. Environ. Microbiol. 2014, 80, 5265–5273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodwig, E.M.; Hosie, A.H.F.; Bourdès, A.; Findlay, K.; Allaway, D.; Karunakaran, R.; Downie, J.A.; Poole, P.S. Amino-acid cycling drives nitrogen fixation in the legume–Rhizobium symbiosis. Nature 2003, 422, 722–726. [Google Scholar] [CrossRef]
- Prell, J.; White, J.P.; Bourdes, A.; Bunnewell, S.; Bongaerts, R.J.; Poole, P.S. Legumes regulate Rhizobium bacteroid development and persistence by the supply of branched-chain amino acids. Proc. Natl. Acad. Sci. USA 2009, 106, 12477–12482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udvardi, M.; Poole, P.S. Transport and metabolism in legume-rhizobia symbioses. Annu. Rev. Plant Biol. 2013, 64, 781–805. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.M.; Kobayashi, H.; Davies, B.W.; Taga, M.E.; Walker, G.C. How rhizobial symbionts invade plants: The Sinorhizobium–Medicago model. Nat. Rev. Microbiol. 2007, 5, 619–633. [Google Scholar] [CrossRef] [Green Version]
- Lagares, A., Jr.; Borella, G.C.; Linne, U.; Becker, A.; Valverde, C. Regulation of Polyhydroxybutyrate Accumulation in Sinorhizobium meliloti by the Trans-Encoded Small RNA MmgR. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef] [Green Version]
- Ceizel Borella, G.; Lagares, A., Jr.; Valverde, C. Expression of the small regulatory RNA gene mmgR is regulated negatively by AniA and positively by NtrC in Sinorhizobium meliloti 2011. Microbiology 2018, 164, 88–98. [Google Scholar] [CrossRef]
- Wang, C.; Saldanha, M.; Sheng, X.; Shelswell, K.J.; Walsh, K.T.; Sobral, B.W.; Charles, T.C. Roles of poly-3-hydroxybutyrate (PHB) and glycogen in symbiosis of Sinorhizobium meliloti with Medicago sp. Microbiology 2007, 153, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cevallos, M.A.; Encarnación, S.; Leija, A.; Mora, Y.; Mora, J. Genetic and physiological characterization of a Rhizobium etli mutant strain unable to synthesize poly-beta-hydroxybutyrate. J. Bacteriol. 1996, 178, 1646–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodwig, E.M.; Leonard, M.; Marroqui, S.; Wheeler, T.R.; Findlay, K.; Downie, J.A.; Poole, P.S. Role of polyhydroxybutyrate and glycogen as carbon storage compounds in pea and bean bacteroids. Mol. Plant-Microbe Interact. 2005, 18, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Rutherford Steven, T.; Papenfort, K.; Bagert John, D.; van Kessel Julia, C.; Tirrell David, A.; Wingreen Ned, S.; Bassler Bonnie, L. A Qrr Noncoding RNA Deploys Four Different Regulatory Mechanisms to Optimize Quorum-Sensing Dynamics. Cell 2015, 160, 228–240. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, M.; Meyer, S.; Becker, A. Novel Sinorhizobium meliloti quorum sensing positive and negative regulatory feedback mechanisms respond to phosphate availability. Mol. Microbiol. 2009, 74, 1238–1256. [Google Scholar] [CrossRef]
- Gao, M.; Barnett, M.J.; Long, S.R.; Teplitski, M. Role of the Sinorhizobium meliloti global regulator Hfq in gene regulation and symbiosis. Mol. Plant-Microbe Interact. 2010, 23, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Baumgardt, K.; Charoenpanich, P.; McIntosh, M.; Schikora, A.; Stein, E.; Thalmann, S.; Kogel, K.-H.; Klug, G.; Becker, A.; Evguenieva-Hackenberg, E. RNase E Affects the Expression of the Acyl-Homoserine Lactone Synthase Gene sinI in Sinorhizobium meliloti. J. Bacteriol. 2014, 196, 1435–1447. [Google Scholar] [CrossRef] [Green Version]
- Baumgardt, K.; Šmídová, K.; Rahn, H.; Lochnit, G.; Robledo, M.; Evguenieva-Hackenberg, E. The stress-related, rhizobial small RNA RcsR1 destabilizes the autoinducer synthase encoding mRNA sinI in Sinorhizobium meliloti. RNA Biol. 2016, 13, 486–499. [Google Scholar] [CrossRef] [Green Version]
- Jonas, K. To divide or not to divide: Control of the bacterial cell cycle by environmental cues. Curr. Opin. Microbiol. 2014, 18, 54–60. [Google Scholar] [CrossRef]
- Penterman, J.; Abo, R.P.; De Nisco, N.J.; Arnold, M.F.F.; Longhi, R.; Zanda, M.; Walker, G.C. Host plant peptides elicit a transcriptional response to control the Sinorhizobium meliloti cell cycle during symbiosis. Proc. Natl. Acad. Sci. USA 2014, 111, 3561–3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frage, B.; Döhlemann, J.; Robledo, M.; Lucena, D.; Sobetzko, P.; Graumann, P.L.; Becker, A. Spatiotemporal choreography of chromosome and megaplasmids in the Sinorhizobium meliloti cell cycle. Mol. Microbiol. 2016, 100, 808–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtzendorff, J.; Hung, D.; Brende, P.; Reisenauer, A.; Viollier, P.H.; McAdams, H.H.; Shapiro, L. Oscillating Global Regulators Control the Genetic Circuit Driving a Bacterial Cell Cycle. Science 2004, 304, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Robledo, M.; Frage, B.; Wright, P.R.; Becker, A. A stress-induced small RNA modulates alpha-rhizobial cell cycle progression. PLoS Genet. 2015, 11, e1005153. [Google Scholar] [CrossRef] [Green Version]
- Pini, F.; De Nisco, N.J.; Ferri, L.; Penterman, J.; Fioravanti, A.; Brilli, M.; Mengoni, A.; Bazzicalupo, M.; Viollier, P.H.; Walker, G.C.; et al. Cell Cycle Control by the Master Regulator CtrA in Sinorhizobium meliloti. PLoS Genet. 2015, 11, e1005232. [Google Scholar] [CrossRef] [Green Version]
- Robledo, M.; Schlüter, J.-P.; Loehr, L.O.; Linne, U.; Albaum, S.P.; Jiménez-Zurdo, J.I.; Becker, A. An sRNA and Cold Shock Protein Homolog-Based Feedforward Loop Post-transcriptionally Controls Cell Cycle Master Regulator CtrA. Front. Microbiol. 2018, 9, 763. [Google Scholar] [CrossRef]
- Barnett, M.J.; Toman, C.J.; Fisher, R.F.; Long, S.R. A dual-genome Symbiosis Chip for coordinate study of signal exchange and development in a prokaryote-host interaction. Proc. Natl. Acad. Sci. USA 2004, 101, 16636–16641. [Google Scholar] [CrossRef] [Green Version]
- Lang, C.; Long, S.R. Transcriptomic analysis of Sinorhizobium meliloti and Medicago truncatula symbiosis using nitrogen fixation–deficient nodules. Mol. Plant-Microbe Interact. 2015, 28, 856–868. [Google Scholar] [CrossRef] [Green Version]
- Robledo, M.; Peregrina, A.; Millán, V.; García-Tomsig, N.I.; Torres-Quesada, O.; Mateos, P.F.; Becker, A.; Jiménez-Zurdo, J.I. A conserved α-proteobacterial small RNA contributes to osmoadaptation and symbiotic efficiency of rhizobia on legume roots. Environ. Microbiol. 2017, 19, 2661–2680. [Google Scholar] [CrossRef]
- Starker, C.G.; Parra-Colmenares, A.L.; Smith, L.; Mitra, R.M.; Long, S.R. Nitrogen fixation mutants of Medicago truncatula fail to support plant and bacterial symbiotic gene expression. Plant Physiol. 2006, 140, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Patriarca, E.J.; Tatè, R.; Iaccarino, M. Key Role of Bacterial NH(4)(+) Metabolism in Rhizobium-Plant Symbiosis. Microbiol. Mol. Biol. Rev. 2002, 66, 203–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, B.J.; Mens, C.; Hastwell, A.H.; Zhang, M.; Su, H.; Jones, C.H.; Chu, X.; Gresshoff, P.M. Legume nodulation: The host controls the party. Plant Cell Environ. 2019, 42, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, B.; Wang, X.; Duan, J.; Ma, J. Rhizobial tRNA-derived small RNAs are signal molecules regulating plant nodulation. Science 2019, 365, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Robledo, M.; Jiménez-Zurdo, J.I.; Becker, A. Antisense transcription of symbiotic genes in Sinorhizobium meliloti. Symbiosis 2015, 67, 55–67. [Google Scholar] [CrossRef]
- Georg, J.; Hess, W.R. Widespread Antisense Transcription in Prokaryotes. Microbiol. Spectr. 2018, 6, 191–210. [Google Scholar] [CrossRef]
- MacLellan, S.R.; Smallbone, L.A.; Sibley, C.D.; Finan, T.M. The expression of a novel antisense gene mediates incompatibility within the large repABC family of α-proteobacterial plasmids. Mol. Microbiol. 2005, 55, 611–623. [Google Scholar] [CrossRef]
- Raghavan, R.; Sloan, D.B.; Ochman, H. Antisense Transcription Is Pervasive but Rarely Conserved in Enteric Bacteria. mBio 2012, 3, e00156-12. [Google Scholar] [CrossRef] [Green Version]
- Lybecker, M.; Bilusic, I.; Raghavan, R. Pervasive transcription: Detecting functional RNAs in bacteria. Transcription 2014, 5, e944039. [Google Scholar] [CrossRef] [Green Version]
- Holmqvist, E.; Vogel, J. RNA-binding proteins in bacteria. Nat. Rev. Microbiol. 2018. [Google Scholar] [CrossRef]
- Jiménez-Zurdo, J.I.; Robledo, M. RNA silencing in plant symbiotic bacteria: Insights from a protein-centric view. RNA Biol. 2017, 14, 1672–1677. [Google Scholar] [CrossRef]
- De Lay, N.; Schu, D.J.; Gottesman, S. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J. Biol. Chem. 2013, 288, 7996–8003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakulskas, C.A.; Potts, A.H.; Babitzke, P.; Ahmer, B.M.M.; Romeo, T. Regulation of Bacterial Virulence by Csr (Rsm) Systems. Microbiol. Mol. Biol. Rev. 2015, 79, 193–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, C.J.; Menendez-Gil, P.; Catalan-Moreno, A.; Vergara-Irigaray, M.; García, B.; Segura, V.; Irurzun, N.; Villanueva, M.; Ruiz de los Mozos, I.; Solano, C.; et al. The regulon of the RNA chaperone CspA and its auto-regulation in Staphylococcus aureus. Nucleic Acids Res. 2018, 46, 1345–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, E.G. Cycling of RNAs on Hfq. RNA Biol. 2013, 10, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnov, A.; Förstner, K.U.; Holmqvist, E.; Otto, A.; Günster, R.; Becher, D.; Reinhardt, R.; Vogel, J. Grad-seq guides the discovery of ProQ as a major small RNA-binding protein. Proc. Natl. Acad. Sci. USA 2016, 113, 11591–11596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, I.J.; Saramago, M.; Dressaire, C.; Domingues, S.; Viegas, S.C.; Arraiano, C.M. Importance and key events of prokaryotic RNA decay: The ultimate fate of an RNA molecule. Wiley Interdiscip. Rev. RNA 2011, 2, 818–836. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Zurdo, J.I.; Torres-Quesada, O.; Valverde, C.; Sobrero, P. Contribution of the RNA Chaperone Hfq to Environmental Fitness and Symbiosis in Sinorhizobium meliloti; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2015; pp. 737–745. [Google Scholar]
- Vogel, J.; Luisi, B.F. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 2011, 9, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Sobrero, P.; Valverde, C. Evidences of autoregulation of hfq expression in Sinorhizobium meliloti strain 2011. Arch. Microbiol. 2011, 193, 629–639. [Google Scholar] [CrossRef]
- Kaminski, P.A.; Desnoues, N.; Elmerich, C. The expression of nifA in Azorhizobium caulinodans requires a gene product homologous to Escherichia coli HF-I, an RNA-binding protein involved in the replication of phage Q beta RNA. Proc. Natl. Acad. Sci. USA 1994, 91, 4663–4667. [Google Scholar] [CrossRef] [Green Version]
- Barra-Bily, L.; Pandey, S.P.; Trautwetter, A.; Blanco, C.; Walker, G.C. The Sinorhizobium meliloti RNA chaperone Hfq mediates symbiosis of Sinorhizobium meliloti and alfalfa. J. Bacteriol. 2010, 192, 1710–1718. [Google Scholar] [CrossRef] [Green Version]
- Torres-Quesada, O.; Oruezabal, R.I.; Peregrina, A.; Jofre, E.; Lloret, J.; Rivilla, R.; Toro, N.; Jimenez-Zurdo, J.I. The Sinorhizobium meliloti RNA chaperone Hfq influences central carbon metabolism and the symbiotic interaction with alfalfa. BMC Microbiol. 2010, 10, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobrero, P.; Schluter, J.P.; Lanner, U.; Schlosser, A.; Becker, A.; Valverde, C. Quantitative proteomic analysis of the Hfq-regulon in Sinorhizobium meliloti 2011. PLoS ONE 2012, 7, e48494. [Google Scholar] [CrossRef]
- Gao, M.; Nguyen, H.; Salas González, I.; Teplitski, M. Regulation of fixLJ by Hfq controls symbiotically important genes in Sinorhizobium meliloti. Mol Plant-Microbe Interact. 2016, 29, 844–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, P.A.; Elmerich, C. The control of Azorhizobium caulinodans nifA expression by oxygen, ammonia and by the HF-I-like protein, NrfA. Mol. Microbiol. 1998, 28, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Barra-Bily, L.; Fontenelle, C.; Jan, G.; Flechard, M.; Trautwetter, A.; Pandey, S.P.; Walker, G.C.; Blanco, C. Proteomic alterations explain phenotypic changes in Sinorhizobium meliloti lacking the RNA chaperone Hfq. J. Bacteriol. 2010, 192, 1719–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulley, G.; White, J.P.; Karunakaran, R.; Prell, J.; Bourdes, A.; Bunnewell, S.; Hill, L.; Poole, P.S. Mutation of GOGAT prevents pea bacteroid formation and N2 fixation by globally downregulating transport of organic nitrogen sources. Mol. Microbiol. 2011, 80, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Roop, R.M.N.; Robertson, G.T.; Ferguson, G.P.; Milford, L.E.; Winkler, M.E.; Walker, G.C. Seeking a niche: Putative contributions of the hfq and bacA gene products to the successful adaptation of the brucellae to their intracellular home. Vet. Microbiol. 2002, 90, 349–363. [Google Scholar] [CrossRef]
- Wilms, I.; Moller, P.; Stock, A.M.; Gurski, R.; Lai, E.M.; Narberhaus, F. Hfq influences multiple transport systems and virulence in the plant pathogen Agrobacterium tumefaciens. J. Bacteriol. 2012, 194, 5209–5217. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Benge, A.; Mesa, J.M.; Javier, R.; Liu, F.-X. Use of RNA Immunoprecipitation Method for Determining Sinorhizobium meliloti RNA-Hfq Protein Associations In Vivo. Biol. Proced. Online 2018, 20, 8. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, A.; Wang, C.; Drewry, L.L.; Vogel, J. Molecular mechanism of mRNA repression in trans by a ProQ-dependent small RNA. EMBO J. 2017, 36, 1029–1045. [Google Scholar] [CrossRef]
- Olejniczak, M.; Storz, G. ProQ/FinO-domain proteins: Another ubiquitous family of RNA matchmakers? Mol. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arraiano, C.M.; Andrade, J.M.; Domingues, S.; Guinote, I.B.; Malecki, M.; Matos, R.G.; Moreira, R.N.; Pobre, V.; Reis, F.P.; Saramago, M.; et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol. Rev. 2010, 34, 883–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalaouna, D.; Simoneau-Roy, M.; Lafontaine, D.; Massé, E. Regulatory RNAs and target mRNA decay in prokaryotes. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2013, 1829, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Lasa, I.; Toledo-Arana, A.; Dobin, A.; Villanueva, M.; de los Mozos, I.R.; Vergara-Irigaray, M.; Segura, V.; Fagegaltier, D.; Penadés, J.R.; Valle, J.; et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 20172–20177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.; Barnett, M.J.; Capela, D.; Dondrup, M.; Kamp, P.B.; Krol, E.; Linke, B.; Ruberg, S.; Runte, K.; Schroeder, B.K.; et al. A portal for rhizobial genomes: RhizoGATE integrates a Sinorhizobium meliloti genome annotation update with postgenome data. J. Biotechnol. 2009, 140, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, B.; Holscher, M.; Baumgarth, B.; Kalbfleisch, A.; Kaya, C.; Hess, W.R.; Becker, A.; Evguenieva-Hackenberg, E. Expression of small RNAs in Rhizobiales and protection of a small RNA and its degradation products by Hfq in Sinorhizobium meliloti. Biochem. Biophys. Res. Commun. 2009, 390, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hong, G. Post-transcriptional regulation of NifA expression by Hfq and RNase E complex in Rhizobium leguminosarum bv. viciae. Acta Biochim. Biophys. Sin. 2009, 41, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Baumgardt, K.; Melior, H.; Madhugiri, R.; Thalmann, S.; Schikora, A.; McIntosh, M.; Becker, A.; Evguenieva-Hackenberg, E. RNase E and RNase J are needed for S-adenosylmethionine homeostasis in Sinorhizobium meliloti. Microbiology 2017, 163, 570–583. [Google Scholar] [CrossRef]
- Madhugiri, R.; Evguenieva-Hackenberg, E. RNase J is involved in the 5′-end maturation of 16S rRNA and 23S rRNA in Sinorhizobium meliloti. FEBS Lett. 2009, 583, 2339–2342. [Google Scholar] [CrossRef] [Green Version]
- Saramago, M.; Robledo, M.; Matos, R.G.; Jiménez-Zurdo, J.I.; Arraiano, C.M. Sinorhizobium meliloti RNase III: Catalytic Features and Impact on Symbiosis. Front. Genet. 2018, 9. [Google Scholar] [CrossRef]
- Gil, R.; Silva, F.J.; Peretó, J.; Moya, A. Determination of the core of a minimal bacterial gene set. Microbiol. Mol. Biol. Rev. 2004, 68, 518–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, S.P.; Minesinger, B.K.; Kumar, J.; Walker, G.C. A highly conserved protein of unknown function in Sinorhizobium meliloti affects sRNA regulation similar to Hfq. Nucleic Acids Res. 2011, 39, 4691–4708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, B.W.; Walker, G.C. A highly conserved protein of unknown function is required by Sinorhizobium meliloti for symbiosis and environmental stress protection. J. Bacteriol. 2008, 190, 1118–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob Asha, I.; Köhrer, C.; Davies Bryan, W.; RajBhandary Uttam, L.; Walker Graham, C. Conserved bacterial RNase YbeY plays key roles in 70S ribosome quality control and 16S rRNA maturation. Mol. Cell 2013, 49, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saramago, M.; Peregrina, A.; Robledo, M.; Matos, R.G.; Hilker, R.; Serrania, J.; Becker, A.; Arraiano, C.M.; Jiménez-Zurdo, J.I. Sinorhizobium meliloti YbeY is an endoribonuclease with unprecedented catalytic features, acting as silencing enzyme in riboregulation. Nucleic Acids Res. 2017, 45, 1371–1391. [Google Scholar] [CrossRef] [PubMed]
- Babu, V.M.P.; Sankari, S.; Budnick, J.A.; Caswell, C.C.; Walker, G.C. Sinorhizobium meliloti YbeY is a zinc-dependent single-strand specific endoribonuclease that plays an important role in 16S ribosomal RNA processing. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbionts | Pathogens | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name/s in S. meliloti 1021 and (S. meliloti 2011) a | Genome Copies | Coordinates in S. meliloti 1021 Genome | Strand | Predicted Promoter Motifs [27] | Hfq Co-IP RNA [32] | Expression in Nodules b [34] | S. meliloti | S. fredii | S. medicae | R. leguminosarum | R. etli | M. loti | B. japonicum | Agrobacterium sp. | Brucella sp. | Others | |
| Housekeeping | SMc04478/tmRNA/ssrA | 1 | 2291143–2291463 | + | RpoD | ||||||||||||
| SmelC035/sra05/4.5S/SRP–RNA/ffs (SMc06075) | 1 | 259927–260032 | + | RpoD RpoN | ZII/IZ/ZIII | ||||||||||||
| SmelC667/sra56/Smr22C/6S RNA/ssrs (SMc06867) | 1 | 2972090–2972252 | - | RpoD | IZ/ZIII | ||||||||||||
| SmelC809/sra50/SmrC19/RNase P/rnpB (SMc06867) | 1 | 2356793–2357134 | - | RpoD | IZ/ZIII | ||||||||||||
| trans-sRNAs | SmelC411/sm3/sra41/smrC15/AbcR2 (SMc06510) | 3 | 1698731–1698617 | - | RpoD RpoH1/2 | IZ/ZIII | |||||||||||
| SmelC412/sm3’/sra41/smrC16/AbcR1 (SMc06511) | 3 | 1698937–1698817 | - | RpoD | ZII/IZ/ZIII | ||||||||||||
| SmelC397/smrC14/NfeR1 (SMc06496) | 6 | 1667613–1667492 | - | RpoD | IZ/ZIII | ||||||||||||
| SmelC689/MmgR (SMc06891) | 3 | 3046712–3046789 | + | RpoD RpoN | IZ/ZIII | ||||||||||||
| SmelC706/smrC45/SpeF (SMc06909) | 1 | 3105298–3105445 | - | RpoH1/2 | ZIII | ||||||||||||
| SmelA075/smA8 (SMa6506) | 5 | 1220693–1220808 | + | RpoD | ZII/IZ | ||||||||||||
| SmelC023/sra03/smrC7 (SMc06056) | 1 | 201679–201825 | + | RpoD RpoH1/2 | ZII/ZIII | ||||||||||||
| SmelC289/sra32/smrC9 (SMc06412) | 1 | 1398278–1398426 | - | RpoD | ZII | ||||||||||||
| SmelC291/sra33/smrC10/EcpR1 (SMc06416+SMc06418) | 1 | 1411678–1411848 | + | ZII/IZ | |||||||||||||
| SmelC671/sm84 (SMc06875) | 1 | 2986421–2986520 | + | ZII | |||||||||||||
| SmelA099/smrA6 (SMa6570) | 1 | 1328175–1328334 | - | ZII/IZ/ZIII | |||||||||||||
| SmelB053/smrB35 (SMb23147) | 3 | 577730–577873 | + | RpoD RpoH1/2 | ZII | ||||||||||||
| SmelC151 (SMc06261) | 1 | 843451–843524 | + | IZ/ZIII | |||||||||||||
| SmelC587/sm104/RcsR1 (rnTrpL?) | 1 | 2575832–2575947 | - | ||||||||||||||
| SmelC165 | 1 | 910182–910297 | + | RpoN | |||||||||||||
| SmelC416/sm138 (SMc06519+) | 1 | 1718814–1718919 | - | ||||||||||||||
| SmelC500 (SMc06663) | 1 | 2180099–2180274 | - | ZII | |||||||||||||
| SmelC507 (SMc06677) | 1 | 2206348–2206579 | + | RpoD RpoH1/2 | ZII | ||||||||||||
| SmelC549/sm4 (SMc06721) | 1 | 2371597–2371855 | + | RpoD RpoH1/2 | ZII/IZ(ZIII | ||||||||||||
| SmelC601 (SMc06778) | 1 | 2625313–2625439 | - | RpoE2 | ZII/IZ/ZIII | ||||||||||||
| SmelC775/GspR1 (SMc07200) | 1 | 3509658–3509803 | + | ZII | |||||||||||||
| SmelA003 | 4 | 15179–15268 | + | ||||||||||||||
| SmelA033/smA3a/smrA2 | 1 | 512140–512221 | - | RpoD | |||||||||||||
| SmelB003 (SMb23003) | 1 | 30240–30345 | + | RpoH1 | IZ/ZIII | ||||||||||||
| SmelB008 | 1 | 65071–65147 | + | RpoE2 | |||||||||||||
| SmelB009 | 1 | 65186–65266 | - | RpoH1/2 | |||||||||||||
| SmelB033 (SMb23075) | 1 | 379319–379391 | - | RpoN | IZ | ||||||||||||
| SmelB044/smrB3b (SMb23116) | 2 | 541771–541909 | - | ZII/IZ | |||||||||||||
| SmelB075 (SMb23231) | 1 | 800730–800793 | - | RpoH1/2 | ZII | ||||||||||||
| SmelB126/smB9 (SMb23362) | 4 | 1325476–1325586 | + | RpoE2 | ZII/ZIII | ||||||||||||
| SmelC434/sm118 (SMc06565) | 1 | 1821211–1821366 | + | ZII | |||||||||||||
| SmelA001 (SMa6000) | 1 | 1002–1054 | + | RpoE2 | IZ/ZIII | ||||||||||||
| SmelA014 (SMa6078) | 1 | 206669–206772 | + | RpoH1/2 | ZII | ||||||||||||
| SmelA018 (SMa6091) | 1 | 234850–234930 | - | RpoE2 | ZIII | ||||||||||||
| SmelA019 | 1 | 235259–235355 | + | RpoH1/2 | ZII/IZ/ZIII | ||||||||||||
| SmelA020 (SMa6094) | 1 | 235393–235497 | - | RpoH1/2 | ZII/IZ/ZIII | ||||||||||||
| SmelA054 (SMa6367) | 1 | 911299–911374 | + | ZII | |||||||||||||
| SmelA056 (SMa6389) | 1 | 954480–954620 | - | ZII | |||||||||||||
| SmelC032 | 1 | 241172–241247 | + | ||||||||||||||
| SmelC749 (SMc07132) | 1 | 3383055–3383121 | - | ZIII | |||||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robledo, M.; García-Tomsig, N.I.; Jiménez-Zurdo, J.I. Riboregulation in Nitrogen-Fixing Endosymbiotic Bacteria. Microorganisms 2020, 8, 384. https://doi.org/10.3390/microorganisms8030384
Robledo M, García-Tomsig NI, Jiménez-Zurdo JI. Riboregulation in Nitrogen-Fixing Endosymbiotic Bacteria. Microorganisms. 2020; 8(3):384. https://doi.org/10.3390/microorganisms8030384
Chicago/Turabian StyleRobledo, Marta, Natalia I. García-Tomsig, and José I. Jiménez-Zurdo. 2020. "Riboregulation in Nitrogen-Fixing Endosymbiotic Bacteria" Microorganisms 8, no. 3: 384. https://doi.org/10.3390/microorganisms8030384
APA StyleRobledo, M., García-Tomsig, N. I., & Jiménez-Zurdo, J. I. (2020). Riboregulation in Nitrogen-Fixing Endosymbiotic Bacteria. Microorganisms, 8(3), 384. https://doi.org/10.3390/microorganisms8030384