1. Introduction
There are more than trillions of microorganisms inhabiting the human gut, and they live in homeostatic balance [
1,
2]. However, a microbial imbalance called dysbiosis causes metabolic disorders and affects the interactions of microorganisms in the host, which can eventually lead to inflammatory bowel disease (IBD) and ulcerative colitis (UC) [
1,
3]. The increasing global incidence of UC is growing, as is the concern for treating the disease [
4,
5].
A novel therapy named fecal microbiota transplantation (FMT) has recently shown promise in treating gut dysbiosis in UC patients [
6,
7,
8]. However, it is difficult to determine the safety and efficacy of FMT in UC patients, and it is also difficult to determine the factors that affect the success of FMT [
9,
10]. The success of FMT may be associated with microbial shift [
11,
12]. Previous studies have characterized gut dysbiosis as an alteration in the relative abundance of the two dominant phyla in the human gut, Bacteroidetes and Firmicutes [
13]. Thus, for the FMT technique to be successful, it is crucial to characterize and monitor the gut microbiota of UC patients [
6,
9].
For decades, next-generation sequencing (NGS) has been used to analyze changes in gut microbiota composition, and it has played a critical role in advancing gut microbiome research [
14,
15,
16]. Despite the popularity of NGS, the cost and time constraints make it difficult to use for emergency services. A rapid, sensitive, and cost-effective method is required. Conventional and quantitative PCR methods have been useful to specifically detect organisms at the phylum level [
17]. However, these methods are not able to simultaneously quantify different phyla in a single tube. TaqMan PCR is a powerful tool for the simultaneous detection and quantification of microbes in samples from different sources [
18,
19,
20]. The relative composition of the three major phyla in the human gut, Bacteroidetes, Firmicutes, and Proteobacteria, has been proposed as a potential diagnostic tool for UC [
9,
21]. This study aimed to develop an MTq-PCR assay using novel TaqMan probes for profiling the dominant gut microbiota phyla. Our study confirmed that MTq-PCR is a viable alternative to NGS when characterizing the three dominant phyla (Bacteroidetes, Firmicutes, and Proteobacteria) in fecal samples from healthy and UC patients.
3. Discussion
Ulcerative colitis (UC) is a chronic inflammatory bowel disease (IBD) that adversely affects the quality of a patient’s life [
27,
28]. Recent reports have indicated that UC is characterized by a low diversity of intestinal bacterial flora [
29,
30]. The relative composition of the three major gut flora phyla (Bacteroidetes, Firmicutes, and Proteobacteria) could be a potential diagnostic tool for UC [
9,
21]. Fecal microbiota transplantation (FMT) is a novel therapy for UC that restores the composition and function of the bacterial flora in the gut [
31,
32]. For FMT to be successful, it is crucial to characterize the presence and absence of individual taxa in the samples of potential fecal donors and UC patients [
33]. For decades, next-generation sequencing (NGS) has been used to analyze gut microbiota [
34]. Despite the popularity of NGS, the cost and time constraints make it difficult for emergency services to employ it [
33,
34,
35]. In addition, and of concern because the number of UC patients is rapidly increasing [
36], it is more costly to monitor the clinical efficacy of FMT (i.e., analyzing the gut microbiota) of numerous people with NGS. A rapid, sensitive, and cost-effective method is required [
20].
TaqMan qPCR is a powerful tool used in the microbial diagnosis of samples from different sources [
19]. Hence, in this study, we developed a multiplex TaqMan qPCR (MTq-PCR) with three novel phylum-specific TaqMan probes to determine the proportions of the three dominant phyla in the gut microbiota of UC patients (
n = 6) and healthy subjects (
n = 6), namely Bacteroidetes, Firmicutes, and Proteobacteria. The effectiveness of our MTq-PCR assay in profiling the three phyla was compared with NGS.
The phylum-specific probes and primers were designed to target 16S rRNA and were evaluated in silico using the SILVA database [
37]. Our probes were comparatively more sensitive than the previously reported probes in discriminating non-target phyla, and they were more specific to gut microbiota [
24,
25,
26]. This indicates that our probes detected more gut microbiota than the previously reported probes. Our probes exhibited low cross-hybridization to non-target phyla. In addition, phylum-specific primers from previous reports were designed for a singleplex PCR and were less sensitive at discriminating non-target taxa [
17,
24]. In our conventional PCR assay, none of the designed primers and probes interfered with each other during the multiplex PCR reaction, and the reaction was confirmed to be reproducible and sensitive. The multiplex assay showed that the probes were specific, and it was possible to discriminate the three phyla in a single tube. Similar studies reported that real-time PCR assays based on TaqMan hydrolysis probes were specific, sensitive, and rapid compared to conventional PCR [
20,
37,
38]. It is important to note that our qPCR assay was based on the proportion of amplicons; therefore, it cannot directly measure the number of individual cells. This is due to the fact that multiple 16S rRNA copies are found in different bacteria [
39]. In connection with multiple copies of 16S rRNA, previous studies similarly reported a biased determination of microbial composition using an NGS approach [
40]. To the best of our knowledge, this is the first study to profile human gut microbiota with three different phylum-specific TaqMan probes in a single assay without cross-reactivity.
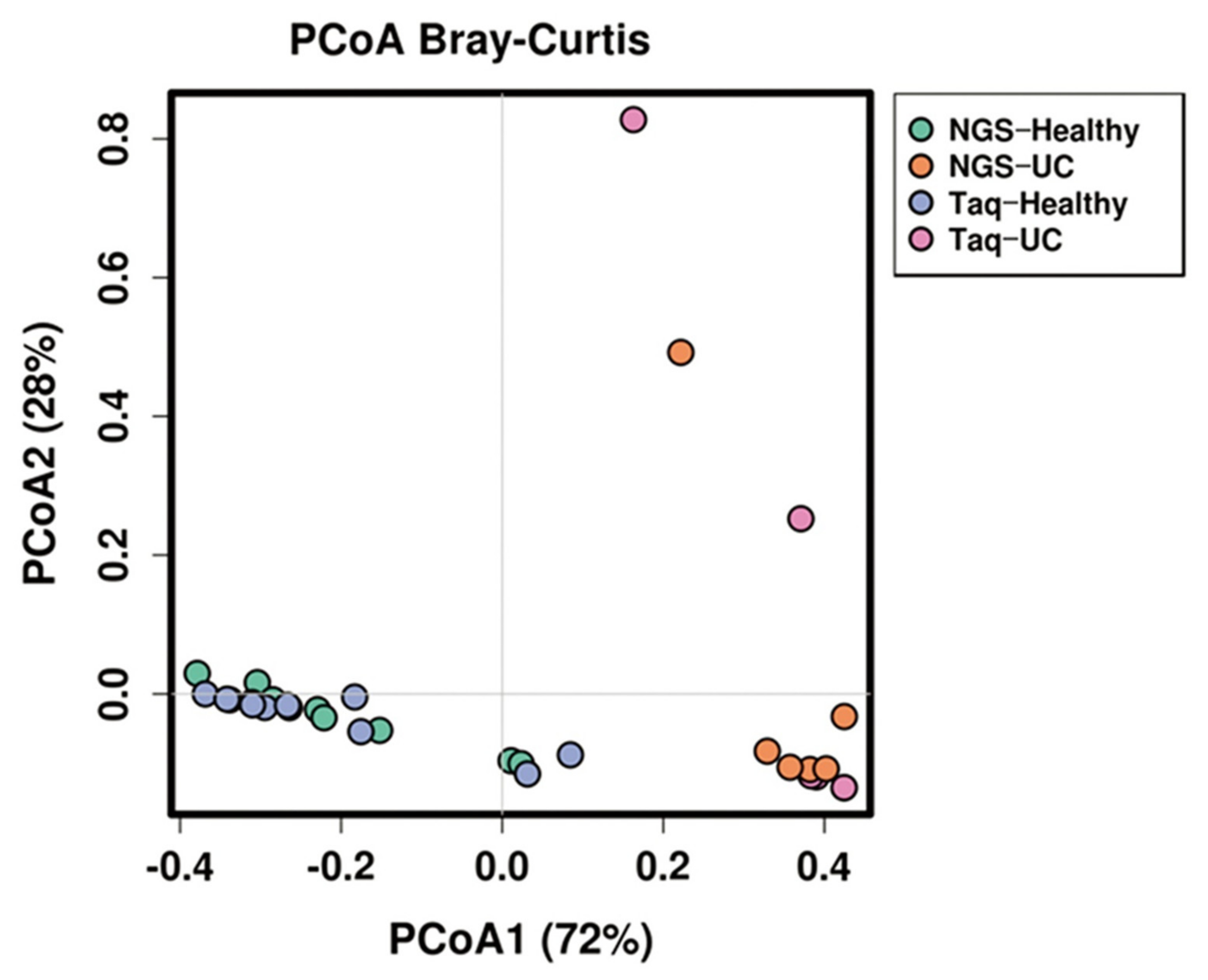
In our study, both NGS and multiplex PCR methods showed a similar relative abundance between the three major phyla in fecal samples of either UC patients or healthy subjects. Both profiling methods also confirmed that the proportion of Bacteroidetes was greatly reduced in all fecal samples of UC patients when compared to healthy subjects. On the other hand, both methods proved that the relative abundance of Proteobacteria was very high in UC patients when compared to healthy subjects. The results of our study were consistent with a previous report showing that Proteobacteria are integral in the formation of UC, and the load of this phylum in the gut could be an effective diagnostic criterion for UC [
21]. Bacteroidetes and Firmicutes are the two dominant phyla in healthy gut microbiota, representing about 90% of the bacterial population [
13,
40,
41]. However, dysbiosis (a microbial imbalance) leads to ulcerative colitis (UC) [
3,
9]. In our study, both NGS and MTq-PCR showed similar results in determining dysbiosis. PCA and an Anosim and Adonis analysis revealed that NGS and MTq-PCR were not significantly different (
p > 0.05) in profiling the abundance of the three phyla from fecal samples of either healthy volunteers or UC patients. Both NGS and MTq-PCR indicated significant differences (
p < 0.001) in the phylum composition between healthy and UC patients. This indicates that our MTq-PCR assay could be used as an alternative method in profiling the three dominant phyla of the gut microbiota since the assay is less costly, rapid, and more accessible than NGS. NGS techniques are costly, time-consuming, and complex for routine applications in resource-limited health care organizations with limited genomic facilities and trained personnel [
42].
In summary, we developed a rapid, sensitive, and cost-effective MTq-PCR assay that reliably profiled the three major phyla in fecal samples of healthy subjects and UC patients: Bacteroidetes, Firmicutes, and Proteobacteria. The results revealed that the proportion of the three phyla in either healthy subjects or UC patients were similar among NGS and MTq-PCR. This suggests that our assay could be a practical microbiota profiling alternative that can characterize the three phyla involved with gut dysbiosis in UC patients during emergency cases of pre-FMT, and it could also help monitor the clinical efficacy and safety of FMT in UC patients at a low cost.
4. Materials and Methods
4.1. Bacterial Strains
Three bacterial strains (B. fragilis GUT-04, C. butyricum TO-A, and S. sonnei KCCM41282) representing the three dominant gut flora phyla (Bacteroidetes, Firmicutes, and Proteobacteria, respectively) were obtained from Kyungpook National University culture collections. The strains were grown in fastidious anaerobe broth medium (FAB: peptone, 23 g; soluble starch, 1 g; sodium bicarbonate, 0.4 g; sodium chloride, 5 g; glucose, 1 g; sodium pyruvate, 1 g; L-arginine, 1 g; L-cysteine HCL 0.5 g; sodium pyrophosphate, 0.25 g; sodium succinate, 0.5 g; Hemin, 0.01 g; and Vitamin K, 0.001 g; Seoul, South Korea) at 37 °C for 48 h.
4.2. Fecal Sample Collection
Fresh fecal samples from healthy volunteers (n = 10) were collected at Kyungpook National University (KNU), Daegu, South Korea. In addition, fecal samples from six UC patients (n = 6) who had been diagnosed with dysbiosis at Yonsei University, Seoul, South Korea were collected. Collection of human fecal samples was performed with the approval of the Institutional Review Board (IRB) at Yonsei University and Kyungpook National University (permit numbers: YSU4-2018-0438 and KNU-2019-0129, respectively). All fecal samples were collected in Transwab® (Medical Wire, UK) following the manufacturer’s protocol. Within 24 h, all collected samples were transported to our laboratory in ice packs and stored at −70 °C until processing.
4.3. DNA Extraction
Genomic DNA from pure cultures of the three bacterial strains was extracted using the Wizard® Genomic Purification Kit (Promega Corporation, USA). DNA from fecal samples was extracted using the QIAamp Powerfecal DNA kit (QIAGEN, Germany) according to the manufacturer’s protocol. The DNA concentration was measured using a Qubit® Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and stored at −20 °C until used.
4.4. NGS Method
The V4–V5 variable region of the 16S rRNA gene was PCR amplified using a universal primer pair: forward (515F 5′-GTGCCAGCMGCCGCGG-3′) and reverse (907R 5′-CCGTCAATTCMTTTRAGTTT-3′). For sequencing, an Ion Torrent PGM adapter and barcode tailored to the primer pair was used. The library preparation reactions consisted of 50 μL and were composed of 25 μL of EmeraldAmp
® Max PCR Master Mix (Takara Korea Biomedical Inc., Seoul, Korea), 1 μL of each bacterial primer, 1 μL of extracted DNA, and 22 μL of ultra-pure water. The first amplification reaction was performed using the following thermocycling program: pre-denaturation at 95 °C for 3 min, followed by five cycles of 95 °C for 30 s, 57 °C for 30 s, and 72 °C for 30 s. The second amplification reaction was 30 cycles of denaturation at 95 °C for 30 s and annealing–extension at 72 °C for 1 min. The final extension was performed at 72 °C for 5 min [
43].
In this study, NGS was employed, and the quality of the amplified DNA library was assessed using an Agilent 2100 Bioanalyzer High-Sensitivity DNA Assay kit (Agilent Technology, Santa Clara, CA, USA). The pre-amplified DNA library was further diluted to 6 pM to perform emulsion PCR with Ion Sphere™ Particles (ISPs) using the Ion OneTouch System II (Thermo Fisher Scientific Korea Inc., Seoul, Korea) followed by enrichment of template-positive ISPs with Dynabeads™ MyOne™ streptavidin C1 beads (Thermo Fisher Scientific, Waltham, MA, USA). Each sample was loaded on an Ion 316 Chip Kit v2 bar-coded chip. Sequencing was performed on the Ion Torrent PGM for 1200 flows with an Ion PGM™ Hi Q Sequencing Kit (Thermo Fisher Scientific Korea Inc., Seoul, Korea). The Torrent Suite™ and Ion Torrent PGM-specific pipeline software was employed to generate sequence reads, trim adapter sequences, filter, and remove low-quality signal-profile reads. Quality filtering of generated sequences and taxonomic classification were performed using QIIME software based on the Greengenes database [
44].
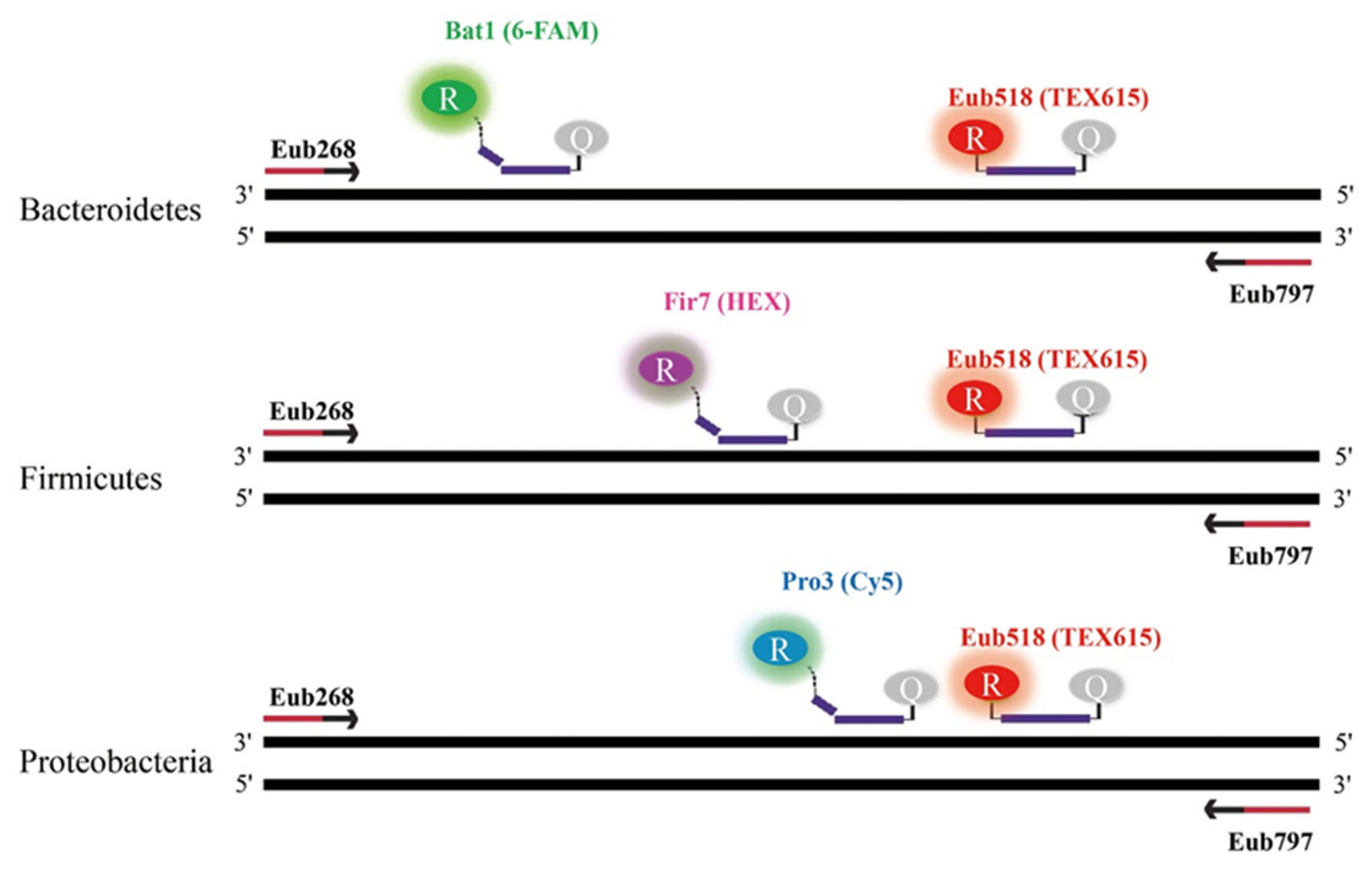
4.5. TaqMan Probe and Primer Design
Five bacterial species from the three dominant phyla in the human gut (Bacteroidetes, Firmicutes, and Proteobacteria) were used for probe design (
Table 9.). Complete 16S rRNA gene sequences from 15 strains representing the three phyla were downloaded from the National Center for Biotechnology Information (NCBI) GeneBank database. Multiple sequence alignments of the 16S rRNA gene sequences from the 15 strains were performed using CLC Main Workbench 8.1 software (Qiagen, Hilden, Germany). Following alignment, a primer pair targeting the 16S rRNA of Eubacteria was designed from a conserved region of all strains, and phylum-specific TaqMan probes were selected from the consensus sequence of each phylum. All probes were designed to be within the binding site of the eubacteria primer pair. A universal probe for eubacteria that targets the opposite strand of the conserved region of the three phyla was used [
22]. The specificity of the primer pair and TaqMan probes were confirmed by in silico analysis using the SILVA database [
37].
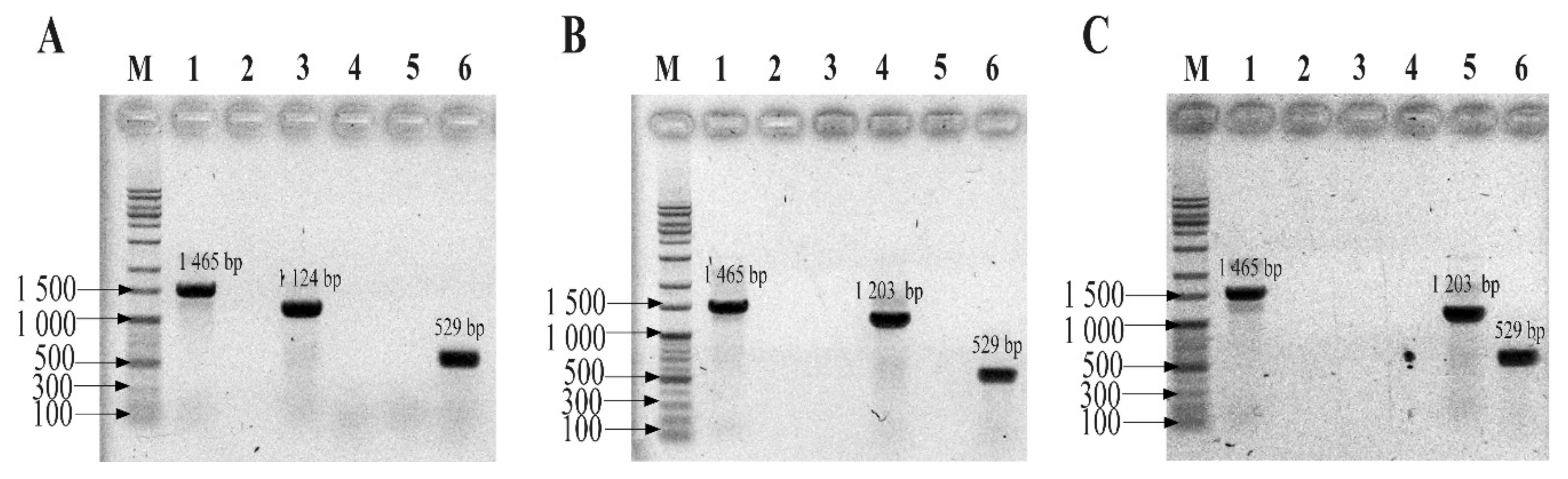
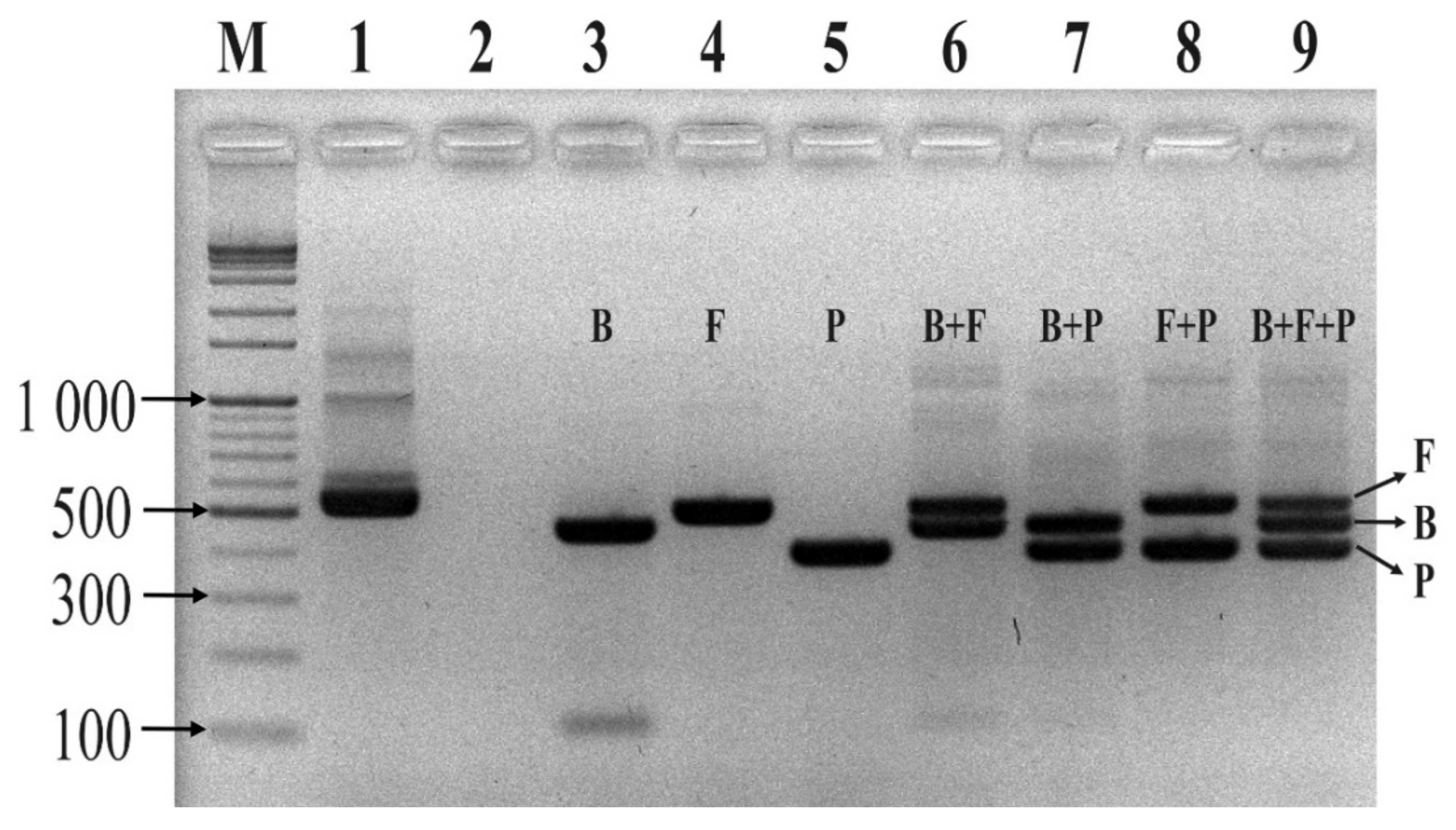
4.6. Conventional PCR Assay
Before conducting the multiplex TaqMan qPCR (mTqPCR) assay, conventional PCR assays were performed to determine the specificity of the designed primer/probe sets using a Mastercycler Nexus PCR machine (Eppendorf, Hamburg, Germany). The primer pairs and TaqMan probes were purchased from Integrated DNA Technologies (IDT, Coralvile, IA, USA). Conventional PCR reactions were 50 μL and were composed of 1 μL of each forward primer (Eub268), 1 μL of universal primer (1492R), 25 μL of EmeraldAmp Max PCR Master Mix (Takara, Japan), and 1 μL of genomic DNA from one of the three strains from the three phyla. Sterilized ultra-pure water was used to make up a total reaction volume of 50 μL. Genomic DNA from the three strains (B. fragilis GUT-04, C. butyricum TO-A, and S. sonnei KCCM 41282) was added separately. After confirming the specificity of each primer and probe set for their respective phyla, the sets were further evaluated by introducing a mock community (i.e., a mixture of the template DNA from the above-mentioned strains in equal proportions) into the MTq-PCR reaction. All PCR assays were performed with the following thermocycling program: 95 °C for 5 min; 35 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s; and a final extension at 72 °C for 5 min. PCR mixtures using universal primers (both forward and reverse) served as positive controls. PCR mixtures without a template DNA served as negative controls.
4.7. Multiplex TaqMan qPCR
In order to quantify the relative proportions of the three phyla (Bacteroidetes, Firmicutes, and Proteobacteria) in fecal samples from UC patients and healthy subjects, a multiplex, real-time TaqMan qPCR was carried out using a CFX96 (Bio-Rad, Hercules, USA). All qPCR reactions for both healthy and UC samples were performed in duplicate, and the average cycle threshold (Ct) values were calculated. Each qPCR reaction was 20 μL and was comprised of 10 μL of 2 X qPCR master mix (MGmed, Seoul, Korea), 1 μL of each primer, 0.5 μL of each probe, 1 μL of sample DNA, and sterilized ultra-pure water to make up a total reaction volume of 20 μL. Based on the conventional results, simultaneous quantification of the three phyla was performed with three TaqMan probes labeled with three distinct fluorophores. Bacteriodetes, Firmicutes, Proteobacteria, and Eubacteria probes were labeled with 6-FAM/BHQ1, HEX/BHQ1, Cy5/BHQ2, and TEX615/BHQ2, respectively (
Figure 6). All qPCR reactions were performed using the following thermocycling program: initial denaturation at 95 °C for 5 min and 40 cycles of 30 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C. The relative proportions of Bacteroidetes, Firmicutes, and Proteobacteria were calculated as follows. The Ct values of each phylum were normalized by subtracting their respective Ct values from 40, and the resulting Ct values were summed to give a total Ct value. The relative abundance of each phylum was calculated by dividing their respective normalized Ct value with the total Ct value, and was expressed as a percentage by multiplying the resulting ratio by 100.
4.8. Statistical Analysis
A comparison of the relative abundances from NGS and TaqMan qPCR assays was conducted using Calypso version 8.84 [
45]. The statistical similarity between NGS and TaqMan qPCR was performed using the analysis of similarities (ANOSIM) test. A dissimilarity analysis was performed using permutational manova (PERMANOVA, Adonis function) [
46]. The similarity and dissimilarity analyses were computed based on Bray–Curtis distance.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}