Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions


Abstract
1. Introduction
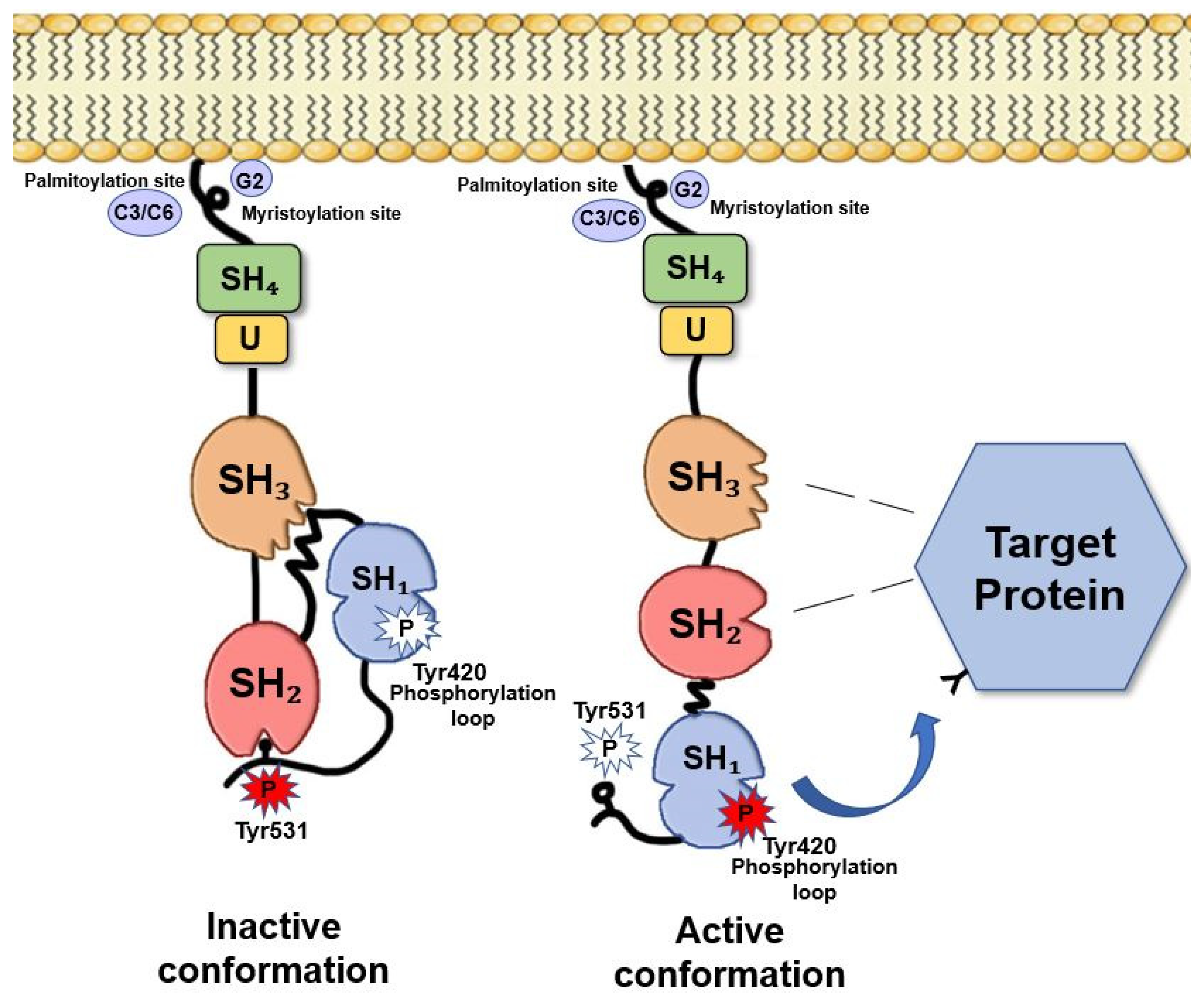
2. Fyn Structure and Activation Mechanisms
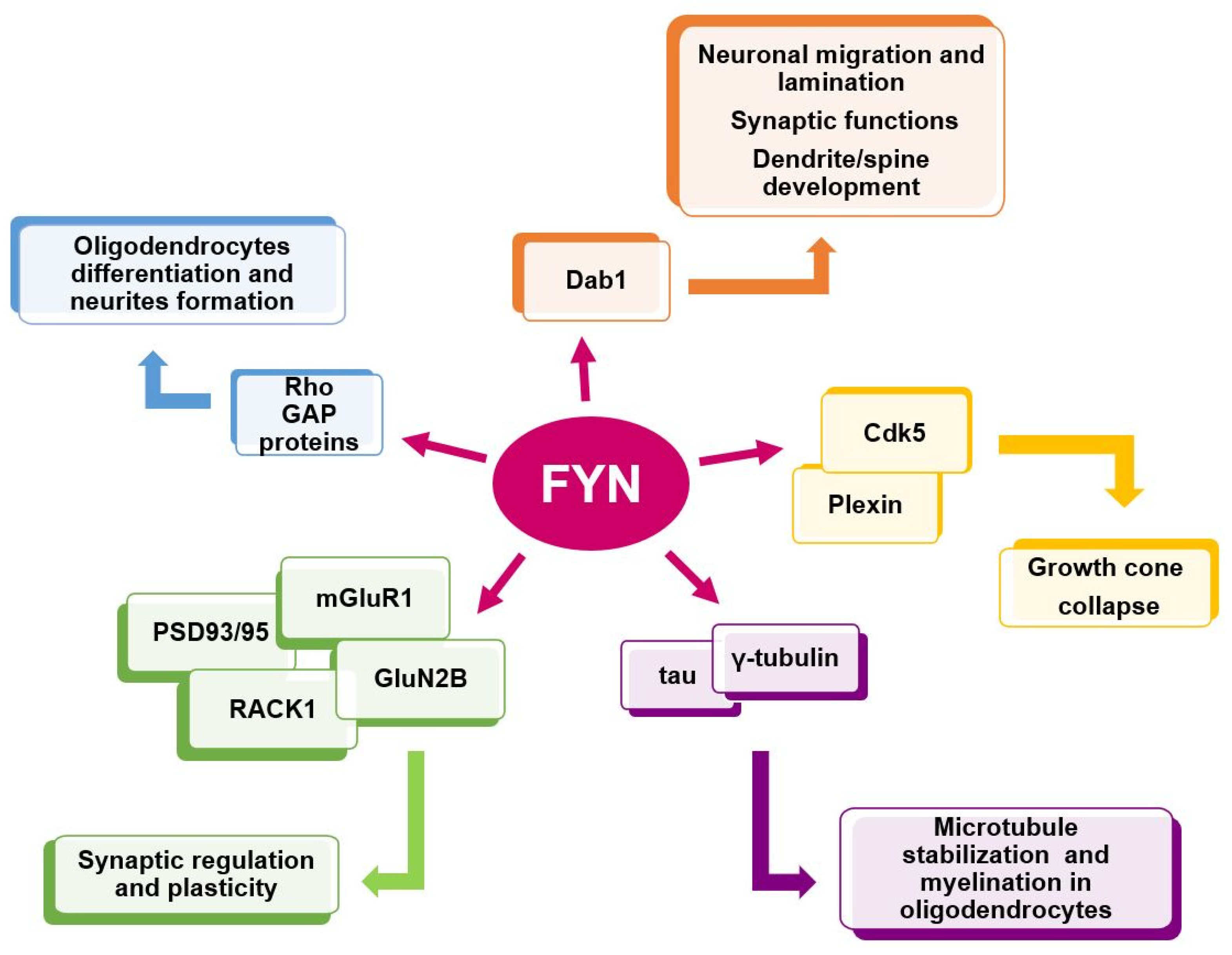
3. Fyn in the Brain: Distribution and Function
3.1. Fyn Promotes Myelination in the CNS
3.2. Fyn Mediates Oligodendrocytes Differentiation and Maturation
3.3. Fyn and Semaphorins in Neurodevelopment and Neurodevelopment-Associated Disorders
3.4. Fyn Controls Neuronal Migration
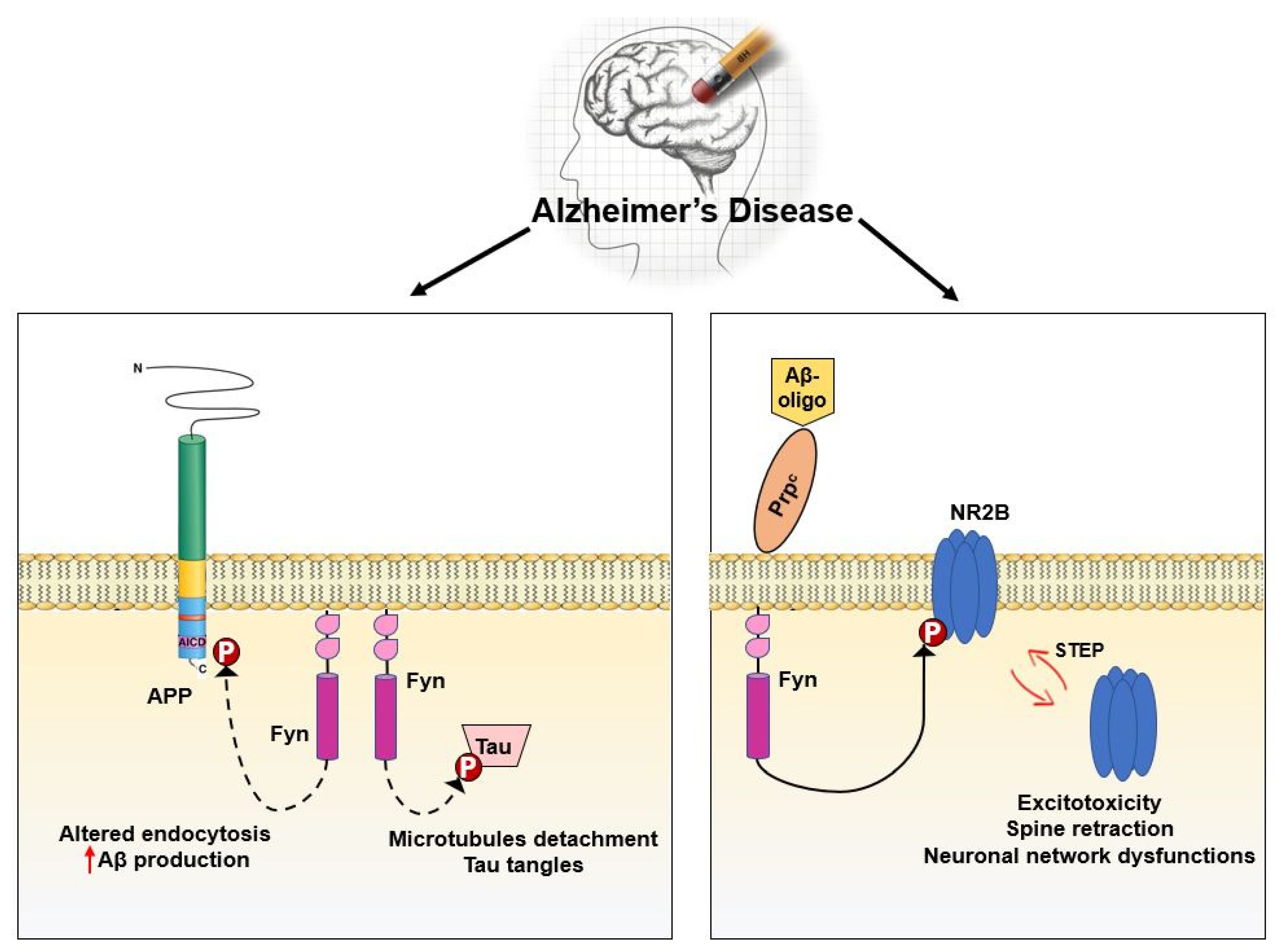
3.5. Fyn in Synaptic Regulation and Dysregulation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPAR | α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor |
| AD | Alzheimer’s Disease |
| APP | Amyloid Precursor Protein |
| ApoER2 | Apolipoprotein E receptor 2 |
| AP2 | Clathrin adaptor protein |
| ASD | Autism Spectrum Disorder |
| A2RE | A2 response element |
| BBB | blood–brain barrier |
| Cdk5 | CiclinA2-dependent kinase 5 |
| CRMP-2 | Collapsin response mediator protein-2 |
| CDT | Carboxy-terminal domains |
| CNS | Central Nervous System |
| DAB1 | Intracellular adaptor protein |
| GAP-domain | GTPase activating domain |
| GRIP1/2 | Glutamate receptor interacting proteins 1 and 2 |
| GSK3β | Glycogen synthase kinase 3 beta |
| HI | Hypoxic–ischemia |
| HnRNP A2 | Heterogeneous nuclear ribonucleoprotein A2 |
| IPT | Immunoglobulin-plexin-transcription domain |
| LTP | Long-term potential |
| L-VGCC | L-type voltage-gated calcium channel |
| MAG | Myelin-associated glycoprotein |
| MAP | Microtubule-associated proteins |
| MBP | Myelin basic protein |
| MGLUR | Metabotropic glutamate receptor |
| MS | Multiple sclerosis |
| NGF | Nerve Growth Factor |
| NMDA | N-methyl-d-aspartate |
| NMDAR | N-methyl-d-aspartate receptor |
| NRTK | Non-Receptor Tyrosine Kinase |
| OL | Oligodendrocytes |
| OTK | Off-track kinases |
| PD | Parkinson Disease |
| PDGF | Platelet-Derived Growth factor |
| PLP | Proteolipid protein |
| PP1,PP2 | Pyrazolopyrimidine |
| PrPC | Prion protein |
| PSD | Postsynaptic density |
| PSI | Plexin-semaphorin-integrin domain |
| PYK2 | Protein tyrosine kinase 2 beta |
| ROS | Reactive oxygen species |
| RPTPα | Receptor-like Protein Tyrosine Phosphatase α |
| RSV | Rous’ virus |
| SFK | Src Family Kinases |
| SNPs | Single nucleotide polymorphisms |
| STEP | Striatal Enriched Phosphatase |
| TK | Tyrosine Kinase |
| VLDL-R | Very-low density lipoprotein receptor |
| Γ-FCR | Γ-chain of immunoglobulin receptor |
References
- Rudd, C.E.; Janssen, O.; Prasad, K.V.; Raab, M.; da Silva, A.; Telfer, J.C.; Yamamoto, M. src-related protein tyrosine kinases and their surface receptors. Biochim. Biophys. Acta 1993, 1155, 239–266. [Google Scholar] [CrossRef]
- Cooper, J.A.; Howell, B. The when and how of Src regulation. Cell 1993, 73, 1051–1054. [Google Scholar] [CrossRef]
- Li, W.; Young, S.L.; King, N.; Miller, W.T. Signaling properties of a non-metazoan Src kinase and the evolutionary history of Src negative regulation. J. Biol. Chem. 2008, 283, 15491–15501. [Google Scholar] [CrossRef] [PubMed]
- Segawa, Y.; Suga, H.; Iwabe, N.; Oneyama, C.; Akagi, T.; Miyata, T.; Okada, M. Functional development of Src tyrosine kinases during evolution from a unicellular ancestor to multicellular animals. Proc. Natl. Acad. Sci. USA 2006, 103, 12021–12026. [Google Scholar] [CrossRef]
- Reinecke, J.; Caplan, S. Endocytosis and the Src family of non-receptor tyrosine kinases. Biomol. Concepts 2014, 5, 143–155. [Google Scholar] [CrossRef]
- Matrone, C.; Iannuzzi, F.; Annunziato, L. The, Y682ENPTY687 motif of APP: Progress and insights toward a targeted therapy for Alzheimer’s disease patients. Ageing. Res. Rev. 2019, 52, 120–128. [Google Scholar] [CrossRef]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef]
- Schenone, S.; Brullo, C.; Musumeci, F.; Biava, M.; Falchi, F.; Botta, M. Fyn kinase in brain diseases and cancer: The search for inhibitors. Curr. Med. Chem. 2011, 18, 2921–2942. [Google Scholar] [CrossRef] [PubMed]
- Resh, M.D. Fyn, a Src family tyrosine kinase. Int. J. Biochem. Cell Biol. 1998, 30, 1159–1162. [Google Scholar] [CrossRef]
- Saito, Y.D.; Jensen, A.R.; Salgia, R.; Posadas, E.M. Fyn: A novel molecular target in cancer. Cancer 2010, 116, 1629–1637. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell. Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed]
- Sicheri, F.; Kuriyan, J. Structures of Src-family tyrosine kinases. Curr. Opin. Struct. Biol. 1997, 7, 777–785. [Google Scholar] [CrossRef]
- Chin, J.; Palop, J.J.; Puoliväli, J.; Massaro, C.; Bien-Ly, N.; Gerstein, H.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2005, 25, 9694–9703. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.T.; Crispino, M.; Tocco, G. The dual response of protein kinase Fyn to neural trauma: Early induction in neurons and delayed induction in reactive astrocytes. Exp. Neurol. 2004, 185, 109–119. [Google Scholar] [CrossRef]
- Cooke, M.P.; Perlmutter, R.M. Expression of a novel form of the fyn proto-oncogene in hematopoietic cells. New. Biol. 1989, 1, 66–74. [Google Scholar]
- Ding, Q.; Stewart, J.; Olman, M.A.; Klobe, M.R.; Gladson, C.L. The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J. Biol. Chem. 2003, 278, 39882–39891. [Google Scholar] [CrossRef]
- Abram, C.L.; Courtneidge, S.A. Src family tyrosine kinases and growth factor signaling. Exp. Cell Res. 2000, 254, 1–13. [Google Scholar] [CrossRef]
- Goldsmith, J.F.; Hall, C.G.; Atkinson, T.P. Identification of an alternatively spliced isoform of the fyn tyrosine kinase. Biochem. Biophys. Res. Commun. 2002, 298, 501–504. [Google Scholar] [CrossRef]
- Knox, R.; Jiang, X. Fyn in Neurodevelopment and Ischemic Brain Injury. Dev. Neurosci. 2015, 37, 311–320. [Google Scholar] [CrossRef]
- Gonfloni, S.; Weijland, A.; Kretzschmar, J.; Superti-Furga, G. Crosstalk between the catalytic and regulatory domains allows bidirectional regulation of Src. Nat. Struct. Biol. 2000, 7, 281–286. [Google Scholar] [CrossRef]
- Kaplan, J.M.; Mardon, G.; Bishop, J.M.; Varmus, H.E. The first seven amino acids encoded by the v-src oncogene act as a myristylation signal: Lysine 7 is a critical determinant. Mol. Cell. Biol. 1988, 8, 2435–2441. [Google Scholar] [CrossRef]
- Resh, M.D. Interaction of tyrosine kinase oncoproteins with cellular membranes. Biochim. Biophys. Acta 1993, 1155, 307–322. [Google Scholar] [CrossRef]
- Rawat, A.; Nagaraj, R. Determinants of membrane association in the SH4 domain of Fyn: Roles of N-terminus myristoylation and side-chain thioacylation. Biochim. Biophys. Acta 2010, 1798, 1854–1863. [Google Scholar] [CrossRef] [PubMed]
- Alland, L.; Peseckis, S.M.; Atherton, R.E.; Berthiaume, L.; Resh, M.D. Dual myristylation and palmitylation of Src family member p59fyn affects subcellular localization. J. Biol. Chem. 1994, 269, 16701–16705. [Google Scholar]
- Resh, M.D. Myristylation and palmitylation of Src family members: The fats of the matter. Cell 1994, 76, 411–413. [Google Scholar] [CrossRef]
- Patwardhan, P.; Resh, M.D. Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol. Cell. Biol. 2010, 30, 4094–4107. [Google Scholar] [CrossRef]
- Koch, C.A.; Anderson, D.; Moran, M.F.; Ellis, C.; Pawson, T. SH2 and SH3 domains: Elements that control interactions of cytoplasmic signaling proteins. Science 1991, 252, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.J.; Baltimore, D. Signalling through SH2 and SH3 domains. Trends Cell Biol. 1993, 3, 8–13. [Google Scholar] [CrossRef]
- Cohen, G.B.; Ren, R.; Baltimore, D. Modular binding domains in signal transduction proteins. Cell 1995, 80, 237–248. [Google Scholar] [CrossRef]
- Songyang, Z.; Shoelson, S.E.; Chaudhuri, M.; Gish, G.; Pawson, T.; Haser, W.G.; King, F.; Roberts, T.; Ratnofsky, S.; Lechleider, R.J.; et al. SH2 domains recognize specific phosphopeptide sequences. Cell 1993, 72, 767–778. [Google Scholar] [CrossRef]
- Pawson, T. Protein modules and signalling networks. Nature 1995, 373, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.T.; Cooper, J.A. Regulation, substrates and functions of src. Biochim. Biophys. Acta 1996, 1287, 121–149. [Google Scholar] [CrossRef]
- Pucheta-Martínez, E.; Saladino, G.; Morando, M.A.; Martinez-Torrecuadrada, J.; Lelli, M.; Sutto, L.; D’Amelio, N.; Gervasio, F.L. An Allosteric Cross-Talk Between the Activation Loop and the ATP Binding Site Regulates the Activation of Src Kinase. Sci. Rep. 2016, 6, 24235. [Google Scholar] [CrossRef] [PubMed]
- Krämer-Albers, E.M.; White, R. From axon-glial signalling to myelination: The integrating role of oligodendroglial Fyn kinase. Cell. Mol. Life Sci. 2011, 68, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, C.A.; Simantov, R.; Kaplan, P.L.; Hunter, T.; Eckhart, W. Alterations in pp60c-src accompany differentiation of neurons from rat embryo striatum. Mol. Cell. Biol. 1987, 7, 1830–1840. [Google Scholar] [CrossRef]
- Reynolds, A.B.; Vila, J.; Lansing, T.J.; Potts, W.M.; Weber, M.J.; Parsons, J.T. Activation of the oncogenic potential of the avian cellular src protein by specific structural alteration of the carboxy terminus. EMBO J. 1987, 6, 2359–2364. [Google Scholar] [CrossRef]
- Okada, M.; Nakagawa, H. A protein tyrosine kinase involved in regulation of pp60c-src function. J. Biol. Chem. 1989, 264, 20886–20893. [Google Scholar]
- Cooper, J.A.; Gould, K.L.; Cartwright, C.A.; Hunter, T. Tyr527 is phosphorylated in pp60c-src: Implications for regulation. Science 1986, 231, 1431–1434. [Google Scholar] [CrossRef]
- Nada, S.; Okada, M.; MacAuley, A.; Cooper, J.A.; Nakagawa, H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature 1991, 351, 69–72. [Google Scholar] [CrossRef]
- Okada, M. Regulation of the SRC family kinases by Csk. Int. J. Biol. Sci. 2012, 8, 1385–1397. [Google Scholar] [CrossRef]
- Nada, S.; Yagi, T.; Takeda, H.; Tokunaga, T.; Nakagawa, H.; Ikawa, Y.; Okada, M.; Aizawa, S. Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell 1993, 73, 1125–1135. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Liu, J.; Lombroso, P.J. Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J. Biol. Chem. 2002, 277, 24274–24279. [Google Scholar] [CrossRef]
- Hansen, K.; Alonso, G.; Courtneidge, S.A.; Rönnstrand, L.; Heldin, C.H. PDGF-induced phosphorylation of Tyr28 in the N-terminus of Fyn affects Fyn activation. Biochem. Biophys. Res. Commun. 1997, 241, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Gartlehner, G.; Hansen, R.A.; Carson, S.S.; Lohr, K.N. Efficacy and safety of inhaled corticosteroids in patients with COPD: A systematic review and meta-analysis of health outcomes. Ann. Fam. Med. 2006, 4, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Twamley-Stein, G.M.; Pepperkok, R.; Ansorge, W.; Courtneidge, S.A. The Src family tyrosine kinases are required for platelet-derived growth factor-mediated signal transduction in NIH 3T3 cells. Proc. Natl. Acad. Sci. USA 1993, 90, 7696–7700. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Rönnstrand, L.; Claesson-Welsh, L.; Heldin, C.H. Phosphorylation of a 72-kDa protein in PDGF-stimulated cells which forms complex with c-Crk, c-Fyn and Eps15. FEBS Lett. 1997, 409, 195–200. [Google Scholar] [CrossRef]
- Zheng, X.M.; Wang, Y.; Pallen, C.J. Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature 1992, 359, 336–339. [Google Scholar] [CrossRef]
- Sap, J.; D’Eustachio, P.; Givol, D.; Schlessinger, J. Cloning and expression of a widely expressed receptor tyrosine phosphatase. Proc. Natl. Acad. Sci. USA 1990, 87, 6112–6116. [Google Scholar] [CrossRef]
- Bhandari, V.; Lim, K.L.; Pallen, C.J. Physical and functional interactions between receptor-like protein-tyrosine phosphatase alpha and p59fyn. J. Biol. Chem. 1998, 273, 8691–8698. [Google Scholar] [CrossRef]
- Matthews, R.J.; Cahir, E.D.; Thomas, M.L. Identification of an additional member of the protein-tyrosine-phosphatase family: Evidence for alternative splicing in the tyrosine phosphatase domain. Proc. Natl. Acad. Sci. USA 1990, 87, 4444–4448. [Google Scholar] [CrossRef]
- Kaplan, R.; Morse, B.; Huebner, K.; Croce, C.; Howk, R.; Ravera, M.; Ricca, G.; Jaye, M.; Schlessinger, J. Cloning of three human tyrosine phosphatases reveals a multigene family of receptor-linked protein-tyrosine-phosphatases expressed in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 7000–7004. [Google Scholar] [CrossRef]
- Krueger, N.X.; Streuli, M.; Saito, H. Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J. 1990, 9, 3241–3252. [Google Scholar] [CrossRef] [PubMed]
- Ponniah, S.; Wang, D.Z.; Lim, K.L.; Pallen, C.J. Targeted disruption of the tyrosine phosphatase PTPalpha leads to constitutive downregulation of the kinases Src and Fyn. Curr. Biol. 1999, 9, 535–538. [Google Scholar] [CrossRef]
- Wang, P.S.; Wang, J.; Xiao, Z.C.; Pallen, C.J. Protein-tyrosine phosphatase alpha acts as an upstream regulator of Fyn signaling to promote oligodendrocyte differentiation and myelination. J. Biol. Chem. 2009, 284, 33692–33702. [Google Scholar] [CrossRef] [PubMed]
- Umemori, H.; Wanaka, A.; Kato, H.; Takeuchi, M.; Tohyama, M.; Yamamoto, T. Specific expressions of Fyn and Lyn, lymphocyte antigen receptor-associated tyrosine kinases, in the central nervous system. Brain Res. Mol. Brain. Res. 1992, 16, 303–310. [Google Scholar] [CrossRef]
- Yagi, T.; Shigetani, Y.; Okado, N.; Tokunaga, T.; Ikawa, Y.; Aizawa, S. Regional localization of Fyn in adult brain; studies with mice in which fyn gene was replaced by lacZ. Oncogene 1993, 8, 3343–3351. [Google Scholar]
- Yagi, T.; Shigetani, Y.; Furuta, Y.; Nada, S.; Okado, N.; Ikawa, Y.; Aizawa, S. Fyn expression during early neurogenesis in mouse embryos. Oncogene 1994, 9, 2433–2440. [Google Scholar] [PubMed]
- Bixby, J.L.; Jhabvala, P. Tyrosine phosphorylation in early embryonic growth cones. J. Neurosci. 1993, 13, 3421–3432. [Google Scholar] [CrossRef]
- Sudol, M.; Hanafusa, H. Cellular proteins homologous to the viral yes gene product. Mol. Cell. Biol. 1986, 6, 2839–2846. [Google Scholar] [CrossRef]
- Yuasa, S.; Hattori, K.; Yagi, T. Defective neocortical development in Fyn-tyrosine-kinase-deficient mice. Neuroreport 2004, 15, 819–822. [Google Scholar] [CrossRef]
- Goto, J.; Tezuka, T.; Nakazawa, T.; Sagara, H.; Yamamoto, T. Loss of Fyn tyrosine kinase on the C57BL/6 genetic background causes hydrocephalus with defects in oligodendrocyte development. Mol. Cell. Neurosci. 2008, 38, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Hu, X.; Song, L.; An, L.; Duan, M.; Chen, S.; Zhao, S. The SH2 domain is crucial for function of Fyn in neuronal migration and cortical lamination. BMB Rep. 2015, 48, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Umemori, H.; Sato, S.; Yagi, T.; Aizawa, S.; Yamamoto, T. Initial events of myelination involve Fyn tyrosine kinase signalling. Nature 1994, 367, 572–576. [Google Scholar] [CrossRef] [PubMed]
- White, R.; Gonsior, C.; Krämer-Albers, E.M.; Stöhr, N.; Hüttelmaier, S.; Trotter, J. Activation of oligodendroglial Fyn kinase enhances translation of mRNAs transported in hnRNP A2-dependent RNA granules. J. Cell Biol. 2008, 181, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Biffiger, K.; Bartsch, S.; Montag, D.; Aguzzi, A.; Schachner, M.; Bartsch, U. Severe hypomyelination of the murine CNS in the absence of myelin-associated glycoprotein and fyn tyrosine kinase. J. Neurosci. 2000, 20, 7430–7437. [Google Scholar] [CrossRef]
- Sperber, B.R.; Boyle-Walsh, E.A.; Engleka, M.J.; Gadue, P.; Peterson, A.C.; Stein, P.L.; Scherer, S.S.; McMorris, F.A. A unique role for Fyn in CNS myelination. J. Neurosci. 2001, 21, 2039–2047. [Google Scholar] [CrossRef]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef]
- Steinman, L. Multiple sclerosis: A coordinated immunological attack against myelin in the central nervous system. Cell 1996, 85, 299–302. [Google Scholar] [CrossRef]
- Greer, J.M.; Lees, M.B. Myelin proteolipid protein--the first 50 years. Int. J. Biochem. Cell Biol. 2002, 34, 211–215. [Google Scholar] [CrossRef]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef]
- Roach, A.; Boylan, K.; Horvath, S.; Prusiner, S.B.; Hood, L.E. Characterization of cloned cDNA representing rat myelin basic protein: Absence of expression in brain of shiverer mutant mice. Cell 1983, 34, 799–806. [Google Scholar] [CrossRef]
- Roach, A.; Takahashi, N.; Pravtcheva, D.; Ruddle, F.; Hood, L. Chromosomal mapping of mouse myelin basic protein gene and structure and transcription of the partially deleted gene in shiverer mutant mice. Cell 1985, 42, 149–155. [Google Scholar] [CrossRef]
- Readhead, C.; Popko, B.; Takahashi, N.; Shine, H.D.; Saavedra, R.A.; Sidman, R.L.; Hood, L. Expression of a myelin basic protein gene in transgenic shiverer mice: Correction of the dysmyelinating phenotype. Cell 1987, 48, 703–712. [Google Scholar] [CrossRef]
- Lemke, G. Unwrapping the genes of myelin. Neuron 1988, 1, 535–543. [Google Scholar] [CrossRef]
- Umemori, H.; Kadowaki, Y.; Hirosawa, K.; Yoshida, Y.; Hironaka, K.; Okano, H.; Yamamoto, T. Stimulation of Myelin Basic Protein Gene Transcription by Fyn Tyrosine Kinase for Myelination. J. Neurosci. 1999, 19, 1393–1397. [Google Scholar] [CrossRef][Green Version]
- Krämer, E.M.; Klein, C.; Koch, T.; Boytinck, M.; Trotter, J. Compartmentation of Fyn kinase with glycosylphosphatidylinositol-anchored molecules in oligodendrocytes facilitates kinase activation during myelination. J. Biol. Chem. 1999, 274, 29042–29049. [Google Scholar] [CrossRef] [PubMed]
- Osterhout, D.J.; Wolven, A.; Wolf, R.M.; Resh, M.D.; Chao, M.V. Morphological differentiation of oligodendrocytes requires activation of Fyn tyrosine kinase. J. Cell Biol. 1999, 145, 1209–1218. [Google Scholar] [CrossRef]
- Colognato, H.; Ramachandrappa, S.; Olsen, I.M.; ffrench-Constant, C. Integrins direct Src family kinases to regulate distinct phases of oligodendrocyte development. J. Cell Biol. 2004, 167, 365–375. [Google Scholar] [CrossRef]
- Liang, X.; Draghi, N.A.; Resh, M.D. Signaling from integrins to Fyn to Rho family GTPases regulates morphologic differentiation of oligodendrocytes. J. Neurosci. 2004, 24, 7140–7149. [Google Scholar] [CrossRef]
- Nakahara, J.; Tan-Takeuchi, K.; Seiwa, C.; Gotoh, M.; Kaifu, T.; Ujike, A.; Inui, M.; Yagi, T.; Ogawa, M.; Aiso, S.; et al. Signaling via immunoglobulin Fc receptors induces oligodendrocyte precursor cell differentiation. Dev. Cell 2003, 4, 841–852. [Google Scholar] [CrossRef]
- Ninio-Many, L.; Grossman, H.; Shomron, N.; Chuderland, D.; Shalgi, R. microRNA-125a-3p reduces cell proliferation and migration by targeting Fyn. J. Cell Sci. 2013, 126, 2867–2876. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.T. The muddle with MAG. Mol. Cell. Neurosci. 1996, 8, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, S.; Carmichael, S.T. The axon-glia unit in white matter stroke: Mechanisms of damage and recovery. Brain Res. 2015, 1623, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Haroutunian, V.; Katsel, P.; Roussos, P.; Davis, K.L.; Altshuler, L.L.; Bartzokis, G. Myelination, oligodendrocytes, and serious mental illness. Glia 2014, 62, 1856–1877. [Google Scholar] [CrossRef]
- Araque Caballero, M.; Suárez-Calvet, M.; Duering, M.; Franzmeier, N.; Benzinger, T.; Fagan, A.M.; Bateman, R.J.; Jack, C.R.; Levin, J.; Dichgans, M.; et al. White matter diffusion alterations precede symptom onset in autosomal dominant Alzheimer’s disease. Brain 2018, 141, 3065–3080. [Google Scholar] [CrossRef]
- Allen, M.; Wang, X.; Burgess, J.D.; Watzlawik, J.; Serie, D.J.; Younkin, C.S.; Nguyen, T.; Malphrus, K.G.; Lincoln, S.; Carrasquillo, M.M.; et al. Conserved brain myelination networks are altered in Alzheimer’s and other neurodegenerative diseases. Alzheimers Dement. 2018, 14, 352–366. [Google Scholar] [CrossRef]
- Hattori, K.; Fukuzako, H.; Hashiguchi, T.; Hamada, S.; Murata, Y.; Isosaka, T.; Yuasa, S.; Yagi, T. Decreased expression of Fyn protein and disbalanced alternative splicing patterns in platelets from patients with schizophrenia. Psychiatry Res. 2009, 168, 119–128. [Google Scholar] [CrossRef]
- Szczepankiewicz, A.; Rybakowski, J.K.; Skibinska, M.; Dmitrzak-Weglarz, M.; Leszczynska-Rodziewicz, A.; Wilkosc, M.; Hauser, J. FYN kinase gene: Another glutamatergic gene associated with bipolar disorder? Neuropsychobiology 2009, 59, 178–183. [Google Scholar] [CrossRef]
- Szczepankiewicz, A.; Skibinska, M.; Suwalska, A.; Hauser, J.; Rybakowski, J.K. The association study of three FYN polymorphisms with prophylactic lithium response in bipolar patients. Hum. Psychopharmacol. 2009, 24, 287–291. [Google Scholar] [CrossRef]
- Klein, C.; Kramer, E.M.; Cardine, A.M.; Schraven, B.; Brandt, R.; Trotter, J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J. Neurosci. 2002, 22, 698–707. [Google Scholar] [CrossRef]
- Belkadi, A.; LoPresti, P. Truncated Tau with the Fyn-binding domain and without the microtubule-binding domain hinders the myelinating capacity of an oligodendrocyte cell line. J. Neurochem. 2008, 107, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Baranzini, S.E.; Galwey, N.W.; Wang, J.; Khankhanian, P.; Lindberg, R.; Pelletier, D.; Wu, W.; Uitdehaag, B.M.; Kappos, L.; Polman, C.H.; et al. Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum. Mol. Genet. 2009, 18, 2078–2090. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls. Am. J. Hum. Genet. 2013, 92, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.G.; O’Dell, T.J.; Karl, K.A.; Stein, P.L.; Soriano, P.; Kandel, E.R. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 1992, 258, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Aizawa, S.; Tokunaga, T.; Shigetani, Y.; Takeda, N.; Ikawa, Y. A role for Fyn tyrosine kinase in the suckling behaviour of neonatal mice. Nature 1993, 366, 742–745. [Google Scholar] [CrossRef]
- Ohnuma, T.; Kato, H.; Arai, H.; McKenna, P.J.; Emson, P.C. Expression of Fyn, a non-receptor tyrosine kinase in prefrontal cortex from patients with schizophrenia and its correlation with clinical onset. Brain Res. Mol. Brain. Res. 2003, 112, 90–94. [Google Scholar] [CrossRef]
- Schafer, S.T.; Paquola, A.C.M.; Stern, S.; Gosselin, D.; Ku, M.; Pena, M.; Kuret, T.J.M.; Liyanage, M.; Mansour, A.A.; Jaeger, B.N.; et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat. Neurosci. 2019, 22, 243–255. [Google Scholar] [CrossRef]
- Wu, L.; Huang, Y.; Li, J.; Zhao, H.; Du, H.; Jin, Q.; Zhao, X.; Ma, H.; Zhu, G. Association study of the Fyn gene with schizophrenia in the Chinese-Han population. Psychiatr. Genet. 2013, 23, 39–40. [Google Scholar] [CrossRef]
- Matrone, C.; Ciotti, M.T.; Mercanti, D.; Marolda, R.; Calissano, P. NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 13139–13144. [Google Scholar] [CrossRef]
- Matrone, C.; Marolda, R.; Ciafre, S.; Ciotti, M.T.; Mercanti, D.; Calissano, P. Tyrosine kinase nerve growth factor receptor switches from prosurvival to proapoptotic activity via Abeta-mediated phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 11358–11363. [Google Scholar] [CrossRef]
- Houlton, J.; Abumaria, N.; Hinkley, S.F.R.; Clarkson, A.N. Therapeutic Potential of Neurotrophins for Repair After Brain Injury: A Helping Hand From Biomaterials. Front. Neurosci. 2019, 13, 790. [Google Scholar] [CrossRef] [PubMed]
- Peckham, H.; Giuffrida, L.; Wood, R.; Gonsalvez, D.; Ferner, A.; Kilpatrick, T.J.; Murray, S.S.; Xiao, J. Fyn is an intermediate kinase that BDNF utilizes to promote oligodendrocyte myelination. Glia 2016, 64, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Nagappan, G.; Lu, B. Activity-dependent modulation of the BDNF receptor TrkB: Mechanisms and implications. Trends Neurosci. 2005, 28, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Yang, S.Y.; Cho, I.H.; Park, M.; Kim, S.A. Polymorphisms of BDNF gene and autism spectrum disorders: Family based association study with korean trios. Psychiatry Investig. 2014, 11, 319–324. [Google Scholar] [CrossRef]
- Yamada, K.; Nabeshima, T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J. Pharmacol. Sci. 2003, 91, 267–270. [Google Scholar] [CrossRef]
- Mizuno, M.; Yamada, K.; He, J.; Nakajima, A.; Nabeshima, T. Involvement of BDNF receptor TrkB in spatial memory formation. Learn. Mem. 2003, 10, 108–115. [Google Scholar] [CrossRef]
- Yamada, K.; Nabeshima, T. Interaction of BDNF/TrkB signaling with NMDA receptor in learning and memory. Drug News Perspect. 2004, 17, 435–438. [Google Scholar] [CrossRef]
- Chao, M.V. Trophic factors: An evolutionary cul-de-sac or door into higher neuronal function? J. Neurosci. Res. 2000, 59, 353–355. [Google Scholar] [CrossRef]
- Mizuno, M.; Yamada, K.; Olariu, A.; Nawa, H.; Nabeshima, T. Involvement of brain-derived neurotrophic factor in spatial memory formation and maintenance in a radial arm maze test in rats. J. Neurosci. 2000, 20, 7116–7121. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Gay, B.; Wada, K.; Koizumi, S. Association of the Src family tyrosine kinase Fyn with TrkB. J. Neurochem. 1998, 71, 106–111. [Google Scholar] [CrossRef]
- Sasaki, Y.; Cheng, C.; Uchida, Y.; Nakajima, O.; Ohshima, T.; Yagi, T.; Taniguchi, M.; Nakayama, T.; Kishida, R.; Kudo, Y.; et al. Fyn and Cdk5 mediate semaphorin-3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron 2002, 35, 907–920. [Google Scholar] [CrossRef]
- Franco, M.; Tamagnone, L. Tyrosine phosphorylation in semaphorin signalling: Shifting into overdrive. EMBO Rep. 2008, 9, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Suda, S.; Iwata, K.; Shimmura, C.; Kameno, Y.; Anitha, A.; Thanseem, I.; Nakamura, K.; Matsuzaki, H.; Tsuchiya, K.J.; Sugihara, G.; et al. Decreased expression of axon-guidance receptors in the anterior cingulate cortex in autism. Mol. Autism 2011, 2, 14. [Google Scholar] [CrossRef]
- Chen, G.; Sima, J.; Jin, M.; Wang, K.Y.; Xue, X.J.; Zheng, W.; Ding, Y.Q.; Yuan, X.B. Semaphorin-3A guides radial migration of cortical neurons during development. Nat. Neurosci. 2008, 11, 36–44. [Google Scholar] [CrossRef]
- Renaud, J.; Kerjan, G.; Sumita, I.; Zagar, Y.; Georget, V.; Kim, D.; Fouquet, C.; Suda, K.; Sanbo, M.; Suto, F.; et al. Plexin-A2 and its ligand, Sema6A, control nucleus-centrosome coupling in migrating granule cells. Nat. Neurosci. 2008, 11, 440–449. [Google Scholar] [CrossRef]
- Orr, B.O.; Fetter, R.D.; Davis, G.W. Retrograde semaphorin-plexin signalling drives homeostatic synaptic plasticity. Nature 2017, 550, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Tillo, M.; Ruhrberg, C.; Mackenzie, F. Emerging roles for semaphorins and VEGFs in synaptogenesis and synaptic plasticity. Cell Adh. Migr. 2012, 6, 541–546. [Google Scholar] [CrossRef]
- Bagri, A.; Cheng, H.J.; Yaron, A.; Pleasure, S.J.; Tessier-Lavigne, M. Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell 2003, 113, 285–299. [Google Scholar] [CrossRef]
- Low, L.K.; Liu, X.B.; Faulkner, R.L.; Coble, J.; Cheng, H.J. Plexin signaling selectively regulates the stereotyped pruning of corticospinal axons from visual cortex. Proc. Natl. Acad. Sci. USA 2008, 105, 8136–8141. [Google Scholar] [CrossRef]
- Morita, A.; Yamashita, N.; Sasaki, Y.; Uchida, Y.; Nakajima, O.; Nakamura, F.; Yagi, T.; Taniguchi, M.; Usui, H.; Katoh-Semba, R.; et al. Regulation of dendritic branching and spine maturation by semaphorin3A-Fyn signaling. J. Neurosci. 2006, 26, 2971–2980. [Google Scholar] [CrossRef]
- Makihara, H.; Nakai, S.; Ohkubo, W.; Yamashita, N.; Nakamura, F.; Kiyonari, H.; Shioi, G.; Jitsuki-Takahashi, A.; Nakamura, H.; Tanaka, F.; et al. CRMP1 and CRMP2 have synergistic but distinct roles in dendritic development. Genes. Cells 2016, 21, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Ziak, J.; Weissova, R.; Jeřábková, K.; Janikova, M.; Maimon, R.; Petrasek, T.; Pukajova, B.; Kleisnerova, M.; Wang, M.; Brill, M.S.; et al. CRMP2 mediates Sema3F-dependent axon pruning and dendritic spine remodeling. EMBO Rep. 2020, 21, e48512. [Google Scholar] [CrossRef]
- Eixarch, H.; Gutiérrez-Franco, A.; Montalban, X.; Espejo, C. Semaphorins 3A and 7A: Potential immune and neuroregenerative targets in multiple sclerosis. Trends Mol. Med. 2013, 19, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Gil, V.; Del Río, J.A. Functions of Plexins/Neuropilins and Their Ligands during Hippocampal Development and Neurodegeneration. Cells 2019, 8, 206. [Google Scholar] [CrossRef]
- Lee, W.S.; Lee, W.H.; Bae, Y.C.; Suk, K. Axon Guidance Molecules Guiding Neuroinflammation. Exp. Neurobiol. 2019, 28, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Pasterkamp, R.J. Getting neural circuits into shape with semaphorins. Nat. Rev. Neurosci. 2012, 13, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Kenney, J.W.; Frankland, P.W. Making connections. Elife 2014, 3. [Google Scholar] [CrossRef]
- Mah, S.; Nelson, M.R.; Delisi, L.E.; Reneland, R.H.; Markward, N.; James, M.R.; Nyholt, D.R.; Hayward, N.; Handoko, H.; Mowry, B.; et al. Identification of the semaphorin receptor PLXNA2 as a candidate for susceptibility to schizophrenia. Mol. Psychiatry 2006, 11, 471–478. [Google Scholar] [CrossRef]
- Su, S.C.; Tsai, L.H. Cyclin-dependent kinases in brain development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 465–491. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohshima, T.; Sasaki, Y.; Suzuki, H.; Yanai, S.; Yamashita, N.; Nakamura, F.; Takei, K.; Ihara, Y.; Mikoshiba, K.; et al. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: Implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells 2005, 10, 165–179. [Google Scholar] [CrossRef]
- Fukata, Y.; Itoh, T.J.; Kimura, T.; Ménager, C.; Nishimura, T.; Shiromizu, T.; Watanabe, H.; Inagaki, N.; Iwamatsu, A.; Hotani, H.; et al. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 2002, 4, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Chihara, K.; Arimura, N.; Ménager, C.; Kawano, Y.; Matsuo, N.; Nishimura, T.; Amano, M.; Kaibuchi, K. CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci. 2001, 4, 781–782. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Jacobs, T.; Eickholt, B.; Ferrari, G.; Teo, M.; Monfries, C.; Qi, R.Z.; Leung, T.; Lim, L.; Hall, C. Alpha2-chimaerin, cyclin-dependent Kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. J. Neurosci. 2004, 24, 8994–9004. [Google Scholar] [CrossRef] [PubMed]
- Mecollari, V.; Nieuwenhuis, B.; Verhaagen, J. A perspective on the role of class III semaphorin signaling in central nervous system trauma. Front. Cell. Neurosci. 2014, 8, 328. [Google Scholar] [CrossRef]
- Van Battum, E.Y.; Brignani, S.; Pasterkamp, R.J. Axon guidance proteins in neurological disorders. Lancet Neurol. 2015, 14, 532–546. [Google Scholar] [CrossRef]
- Hirsch, E.; Hu, L.J.; Prigent, A.; Constantin, B.; Agid, Y.; Drabkin, H.; Roche, J. Distribution of semaphorin IV in adult human brain. Brain Res. 1999, 823, 67–79. [Google Scholar] [CrossRef]
- Good, P.F.; Alapat, D.; Hsu, A.; Chu, C.; Perl, D.; Wen, X.; Burstein, D.E.; Kohtz, D.S. A role for semaphorin 3A signaling in the degeneration of hippocampal neurons during Alzheimer’s disease. J. Neurochem. 2004, 91, 716–736. [Google Scholar] [CrossRef]
- Villa, C.; Venturelli, E.; Fenoglio, C.; De Riz, M.; Scalabrini, D.; Cortini, F.; Serpente, M.; Cantoni, C.; Bresolin, N.; Scarpini, E.; et al. Candidate gene analysis of semaphorins in patients with Alzheimer’s disease. Neurol. Sci. 2010, 31, 169–173. [Google Scholar] [CrossRef]
- Venkova, K.; Christov, A.; Kamaluddin, Z.; Kobalka, P.; Siddiqui, S.; Hensley, K. Semaphorin 3A signaling through neuropilin-1 is an early trigger for distal axonopathy in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2014, 73, 702–713. [Google Scholar] [CrossRef]
- Kuo, G.; Arnaud, L.; Kronstad-O’Brien, P.; Cooper, J.A. Absence of Fyn and Src causes a reeler-like phenotype. J. Neurosci. 2005, 25, 8578–8586. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Miao, G.G.; Chen, S.C.; Soares, H.D.; Morgan, J.I.; Curran, T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G.; Curran, T. Reeler: New tales on an old mutant mouse. Bioessays 1998, 20, 235–244. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Homayouni, R.; Keshvara, L.; Rice, D.S.; Sheldon, M.; Curran, T. Reelin is a ligand for lipoprotein receptors. Neuron 1999, 24, 471–479. [Google Scholar] [CrossRef]
- Trommsdorff, M.; Gotthardt, M.; Hiesberger, T.; Shelton, J.; Stockinger, W.; Nimpf, J.; Hammer, R.E.; Richardson, J.A.; Herz, J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999, 97, 689–701. [Google Scholar] [CrossRef]
- Miyata, T.; Nakajima, K.; Mikoshiba, K.; Ogawa, M. Regulation of Purkinje cell alignment by reelin as revealed with CR-50 antibody. J. Neurosci. 1997, 17, 3599–3609. [Google Scholar] [CrossRef] [PubMed]
- Goffinet, A.M. The embryonic development of the cerebellum in normal and reeler mutant mice. Anat. Embryol. 1983, 168, 73–86. [Google Scholar] [CrossRef]
- Hong, S.E.; Shugart, Y.Y.; Huang, D.T.; Shahwan, S.A.; Grant, P.E.; Hourihane, J.O.; Martin, N.D.; Walsh, C.A. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 2000, 26, 93–96. [Google Scholar] [CrossRef]
- DiBattista, A.M.; Dumanis, S.B.; Song, J.M.; Bu, G.; Weeber, E.; Rebeck, G.W.; Hoe, H.S. Very low density lipoprotein receptor regulates dendritic spine formation in a RasGRF1/CaMKII dependent manner. Biochim. Biophys. Acta 2015, 1853, 904–917. [Google Scholar] [CrossRef][Green Version]
- Pujadas, L.; Gruart, A.; Bosch, C.; Delgado, L.; Teixeira, C.M.; Rossi, D.; de Lecea, L.; Martínez, A.; Delgado-García, J.M.; Soriano, E. Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J. Neurosci. 2010, 30, 4636–4649. [Google Scholar] [CrossRef]
- Niu, S.; Yabut, O.; D’Arcangelo, G. The Reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J. Neurosci. 2008, 28, 10339–10348. [Google Scholar] [CrossRef]
- Iafrati, J.; Orejarena, M.J.; Lassalle, O.; Bouamrane, L.; Gonzalez-Campo, C.; Chavis, P. Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol. Psychiatry 2014, 19, 417–426. [Google Scholar] [CrossRef]
- Kim, M.; Jeong, Y.; Chang, Y.C. Extracellular matrix protein reelin regulate dendritic spine density through CaMKIIβ. Neurosci. Lett. 2015, 599, 97–101. [Google Scholar] [CrossRef]
- Dazzo, E.; Fanciulli, M.; Serioli, E.; Minervini, G.; Pulitano, P.; Binelli, S.; Di Bonaventura, C.; Luisi, C.; Pasini, E.; Striano, S.; et al. Heterozygous reelin mutations cause autosomal-dominant lateral temporal epilepsy. Am. J. Hum. Genet. 2015, 96, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Groc, L.; Choquet, D.; Stephenson, F.A.; Verrier, D.; Manzoni, O.J.; Chavis, P. NMDA receptor surface trafficking and synaptic subunit composition are developmentally regulated by the extracellular matrix protein Reelin. J. Neurosci. 2007, 27, 10165–10175. [Google Scholar] [CrossRef]
- Ventruti, A.; Kazdoba, T.M.; Niu, S.; D’Arcangelo, G. Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience 2011, 189, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.W.; Lanier, L.M.; Frank, R.; Gertler, F.B.; Cooper, J.A. The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Mol. Cell. Biol. 1999, 19, 5179–5188. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.W.; Herrick, T.M.; Hildebrand, J.D.; Zhang, Y.; Cooper, J.A. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr. Biol. 2000, 10, 877–885. [Google Scholar] [CrossRef]
- Howell, B.W.; Gertler, F.B.; Cooper, J.A. Mouse disabled (mDab1): A Src binding protein implicated in neuronal development. EMBO J. 1997, 16, 121–132. [Google Scholar] [CrossRef]
- Sheldon, M.; Rice, D.S.; D’Arcangelo, G.; Yoneshima, H.; Nakajima, K.; Mikoshiba, K.; Howell, B.W.; Cooper, J.A.; Goldowitz, D.; Curran, T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 1997, 389, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Yoneshima, H.; Nagata, E.; Matsumoto, M.; Yamada, M.; Nakajima, K.; Miyata, T.; Ogawa, M.; Mikoshiba, K. A novel neurological mutant mouse, yotari, which exhibits reeler-like phenotype but expresses CR-50 antigen/reelin. Neurosci. Res. 1997, 29, 217–223. [Google Scholar] [CrossRef]
- Ware, M.L.; Fox, J.W.; González, J.L.; Davis, N.M.; Lambert de Rouvroit, C.; Russo, C.J.; Chua, S.C., Jr.; Goffinet, A.M.; Walsh, C.A. Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler mouse. Neuron 1997, 19, 239–249. [Google Scholar] [CrossRef]
- Kojima, N.; Wang, J.; Mansuy, I.M.; Grant, S.G.; Mayford, M.; Kandel, E.R. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc. Natl. Acad. Sci. USA 1997, 94, 4761–4765. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Nakajima, K.; Mikoshiba, K. The disabled 1 gene is disrupted by a replacement with L1 fragment in yotari mice. Brain. Res. Mol. Brain Res. 2000, 75, 121–127. [Google Scholar] [CrossRef]
- Beffert, U.; Weeber, E.J.; Durudas, A.; Qiu, S.; Masiulis, I.; Sweatt, J.D.; Li, W.P.; Adelmann, G.; Frotscher, M.; Hammer, R.E.; et al. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron 2005, 47, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Beffert, U.; Ertunc, M.; Tang, T.S.; Kavalali, E.T.; Bezprozvanny, I.; Herz, J. Reelin modulates NMDA receptor activity in cortical neurons. J. Neurosci. 2005, 25, 8209–8216. [Google Scholar] [CrossRef]
- Hoe, H.S.; Lee, K.J.; Carney, R.S.; Lee, J.; Markova, A.; Lee, J.Y.; Howell, B.W.; Hyman, B.T.; Pak, D.T.; Bu, G.; et al. Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J. Neurosci. 2009, 29, 7459–7473. [Google Scholar] [CrossRef]
- Homayouni, R.; Rice, D.S.; Sheldon, M.; Curran, T. Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J. Neurosci. 1999, 19, 7507–7515. [Google Scholar] [CrossRef]
- Durakoglugil, M.S.; Chen, Y.; White, C.L.; Kavalali, E.T.; Herz, J. Reelin signaling antagonizes beta-amyloid at the synapse. Proc. Natl. Acad. Sci. USA 2009, 106, 15938–15943. [Google Scholar] [CrossRef]
- Krstic, D.; Rodriguez, M.; Knuesel, I. Regulated proteolytic processing of Reelin through interplay of tissue plasminogen activator (tPA), ADAMTS-4, ADAMTS-5, and their modulators. PLoS ONE 2012, 7, e47793. [Google Scholar] [CrossRef]
- Pujadas, L.; Rossi, D.; Andrés, R.; Teixeira, C.M.; Serra-Vidal, B.; Parcerisas, A.; Maldonado, R.; Giralt, E.; Carulla, N.; Soriano, E. Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat. Commun. 2014, 5, 3443. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Gingrich, J.R.; Salter, M.W. Src in synaptic transmission and plasticity. Oncogene 2004, 23, 8007–8016. [Google Scholar] [CrossRef]
- Ohnishi, H.; Murata, Y.; Okazawa, H.; Matozaki, T. Src family kinases: Modulators of neurotransmitter receptor function and behavior. Trends Neurosci. 2011, 34, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, H.B. Targeting Fyn Kinase in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, A.C.; Salazar, S.V.; Haas, L.T.; Yang, J.; Kostylev, M.A.; Jeng, A.T.; Robinson, S.A.; Gunther, E.C.; van Dyck, C.H.; Nygaard, H.B.; et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. 2015, 77, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, H.B.; van Dyck, C.H.; Strittmatter, S.M. Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar]
- Mao, L.M.; Guo, M.L.; Jin, D.Z.; Fibuch, E.E.; Choe, E.S.; Wang, J.Q. Post-translational modification biology of glutamate receptors and drug addiction. Front. Neuroanat. 2011, 5, 19. [Google Scholar] [CrossRef]
- Suzuki, T.; Okumura-Noji, K. NMDA receptor subunits epsilon 1 (NR2A) and epsilon 2 (NR2B) are substrates for Fyn in the postsynaptic density fraction isolated from the rat brain. Biochem. Biophys. Res. Commun. 1995, 216, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Trepanier, C.H.; Jackson, M.F.; MacDonald, J.F. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J. 2012, 279, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Trepanier, C.; Sidhu, B.; Xie, Y.F.; Li, H.; Lei, G.; Salter, M.W.; Orser, B.A.; Nakazawa, T.; Yamamoto, T.; et al. Metaplasticity gated through differential regulation of GluN2A versus GluN2B receptors by Src family kinases. EMBO J. 2012, 31, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Komai, S.; Tezuka, T.; Hisatsune, C.; Umemori, H.; Semba, K.; Mishina, M.; Manabe, T.; Yamamoto, T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 2001, 276, 693–699. [Google Scholar] [CrossRef]
- Sato, Y.; Tao, Y.X.; Su, Q.; Johns, R.A. Post-synaptic density-93 mediates tyrosine-phosphorylation of the N-methyl-D-aspartate receptors. Neuroscience 2008, 153, 700–708. [Google Scholar] [CrossRef][Green Version]
- Tezuka, T.; Umemori, H.; Akiyama, T.; Nakanishi, S.; Yamamoto, T. PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. Proc. Natl. Acad. Sci. USA 1999, 96, 435–440. [Google Scholar] [CrossRef]
- Nakazawa, T.; Tezuka, T.; Yamamoto, T. Regulation of NMDA receptor function by Fyn-mediated tyrosine phosphorylation. Nihon Shinkei Seishin Yakurigaku Zasshi 2002, 22, 165–167. [Google Scholar]
- Hou, X.Y.; Zhang, G.Y.; Yan, J.Z.; Liu, Y. Increased tyrosine phosphorylation of alpha(1C) subunits of L-type voltage-gated calcium channels and interactions among Src/Fyn, PSD-95 and alpha(1C) in rat hippocampus after transient brain ischemia. Brain Res. 2003, 979, 43–50. [Google Scholar] [CrossRef]
- Pei, L.; Teves, R.L.; Wallace, M.C.; Gurd, J.W. Transient cerebral ischemia increases tyrosine phosphorylation of the synaptic RAS-GTPase activating protein, SynGAP. J. Cereb. Blood Flow Metab. 2001, 21, 955–963. [Google Scholar] [CrossRef]
- Zhang, M.; Li, Q.; Chen, L.; Li, J.; Zhang, X.; Chen, X.; Zhang, Q.; Shao, Y.; Xu, Y. PSD-93 deletion inhibits Fyn-mediated phosphorylation of NR2B and protects against focal cerebral ischemia. Neurobiol. Dis. 2014, 68, 104–111. [Google Scholar] [CrossRef]
- Nada, S.; Shima, T.; Yanai, H.; Husi, H.; Grant, S.G.; Okada, M.; Akiyama, T. Identification of PSD-93 as a substrate for the Src family tyrosine kinase Fyn. J. Biol. Chem. 2003, 278, 47610–47621. [Google Scholar] [CrossRef]
- Prybylowski, K.; Chang, K.; Sans, N.; Kan, L.; Vicini, S.; Wenthold, R.J. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 2005, 47, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.J.; Evans, P.R. A structural explanation for the recognition of tyrosine-based endocytotic signals. Science 1998, 282, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Lavezzari, G.; McCallum, J.; Lee, R.; Roche, K.W. Differential binding of the AP-2 adaptor complex and PSD-95 to the C-terminus of the NMDA receptor subunit NR2B regulates surface expression. Neuropharmacology 2003, 45, 729–737. [Google Scholar] [CrossRef]
- Goebel-Goody, S.M.; Davies, K.D.; Alvestad Linger, R.M.; Freund, R.K.; Browning, M.D. Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience 2009, 158, 1446–1459. [Google Scholar] [CrossRef] [PubMed]
- Knox, R.; Zhao, C.; Miguel-Perez, D.; Wang, S.; Yuan, J.; Ferriero, D.; Jiang, X. Enhanced NMDA receptor tyrosine phosphorylation and increased brain injury following neonatal hypoxia-ischemia in mice with neuronal Fyn overexpression. Neurobiol. Dis. 2013, 51, 113–119. [Google Scholar] [CrossRef]
- Hayashi, T.; Huganir, R.L. Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J. Neurosci. 2004, 24, 6152–6160. [Google Scholar] [CrossRef] [PubMed]
- Seamans, J.K.; Yang, C.R. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog. Neurobiol. 2004, 74, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wolf, M.E. Dopamine receptors regulate NMDA receptor surface expression in prefrontal cortex neurons. J. Neurochem. 2008, 106, 2489–2501. [Google Scholar] [CrossRef]
- Jin, D.Z.; Guo, M.L.; Xue, B.; Fibuch, E.E.; Choe, E.S.; Mao, L.M.; Wang, J.Q. Phosphorylation and feedback regulation of metabotropic glutamate receptor 1 by calcium/calmodulin-dependent protein kinase II. J. Neurosci. 2013, 33, 3402–3412. [Google Scholar] [CrossRef]
- Jin, D.Z.; Mao, L.M.; Wang, J.Q. An Essential Role of Fyn in the Modulation of Metabotropic Glutamate Receptor 1 in Neurons. Eneuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.F.; Kojima, N.; Tomizawa, K.; Moriwaki, A.; Matsushita, M.; Obata, K.; Matsui, H. Enhanced synaptic transmission and reduced threshold for LTP induction in fyn-transgenic mice. Eur. J. Neurosci. 1999, 11, 75–82. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, T.J.; Hawkins, R.D.; Kandel, E.R.; Arancio, O. Tests of the roles of two diffusible substances in long-term potentiation: Evidence for nitric oxide as a possible early retrograde messenger. Proc. Natl. Acad. Sci. USA 1991, 88, 11285–11289. [Google Scholar] [CrossRef]
- Rosenblum, K.; Dudai, Y.; Richter-Levin, G. Long-term potentiation increases tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit 2B in rat dentate gyrus in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 10457–10460. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lu, W.; Ali, D.W.; Pelkey, K.A.; Pitcher, G.M.; Lu, Y.M.; Aoto, H.; Roder, J.C.; Sasaki, T.; Salter, M.W.; et al. CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 2001, 29, 485–496. [Google Scholar] [CrossRef]
- Qian, D.; Lev, S.; van Oers, N.S.; Dikic, I.; Schlessinger, J.; Weiss, A. Tyrosine phosphorylation of Pyk2 is selectively regulated by Fyn during TCR signaling. J. Exp. Med. 1997, 185, 1253–1259. [Google Scholar] [CrossRef]
- Yang, K.; Belrose, J.; Trepanier, C.H.; Lei, G.; Jackson, M.F.; MacDonald, J.F. Fyn, a potential target for Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 243–252. [Google Scholar] [CrossRef]
- Boehm, S.L.; Peden, L.; Chang, R.; Harris, R.A.; Blednov, Y.A. Deletion of the fyn-kinase gene alters behavioral sensitivity to ethanol. Alcohol. Clin. Exp. Res. 2003, 27, 1033–1040. [Google Scholar] [CrossRef]
- Boehm, J. A ‘danse macabre’: Tau and Fyn in STEP with amyloid beta to facilitate induction of synaptic depression and excitotoxicity. Eur. J. Neurosci. 2013, 37, 1925–1930. [Google Scholar] [CrossRef]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Lesné, S.E. The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J. Neurosci. 2012, 32, 16857–16871. [Google Scholar] [CrossRef]
- Poulsen, E.T.; Iannuzzi, F.; Rasmussen, H.F.; Maier, T.J.; Enghild, J.J.; Jørgensen, A.L.; Matrone, C. An Aberrant Phosphorylation of Amyloid Precursor Protein Tyrosine Regulates Its Trafficking and the Binding to the Clathrin Endocytic Complex in Neural Stem Cells of Alzheimer’s Disease Patients. Front. Mol. Neurosci. 2017, 10, 59. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X. Recent developments with tau-based drug discovery. Expert Opin. Drug Discov. 2018, 13, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Thangavel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; Van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of tau by fyn: Implications for Alzheimer’s disease. J. Neurosci. 2004, 24, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Petrov, D.; Ettcheto, M.; Pedrós, I.; Abad, S.; Beas-Zarate, C.; Lazarowski, A.; Marin, M.; Olloquequi, J.; Auladell, C.; et al. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev. Neurother. 2015, 15, 587–596. [Google Scholar] [CrossRef]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.P.; et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimers Res. Ther. 2011, 3, 16. [Google Scholar] [CrossRef]
- Haass, C.; Mandelkow, E. Fyn-tau-amyloid: A toxic triad. Cell 2010, 142, 356–358. [Google Scholar] [CrossRef]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111 Pt 21, 3167–3177. [Google Scholar]
- Hernandez, P.; Lee, G.; Sjoberg, M.; Maccioni, R.B. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25-35): Involvement of lipid rafts. J. Alzheimers Dis. 2009, 16, 149–156. [Google Scholar] [CrossRef]
- Shirazi, S.K.; Wood, J.G. The protein tyrosine kinase, fyn, in Alzheimer’s disease pathology. Neuroreport 1993, 4, 435–437. [Google Scholar] [CrossRef]
- Ho, O.H.; Delgado, J.Y.; O’Dell, T.J. Phosphorylation of proteins involved in activity-dependent forms of synaptic plasticity is altered in hippocampal slices maintained in vitro. J. Neurochem. 2004, 91, 1344–1357. [Google Scholar] [CrossRef] [PubMed]
- Lambert de Rouvroit, C.; Goffinet, A.M. The reeler mouse as a model of brain development. Adv. Anat. Embryol. Cell Biol. 1998, 150, 1–106. [Google Scholar] [PubMed]
- Chin, J.; Palop, J.J.; Yu, G.Q.; Kojima, N.; Masliah, E.; Mucke, L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J. Neurosci. 2004, 24, 4692–4697. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhao, J.; Lu, G. miR-106b inhibits tau phosphorylation at Tyr18 by targeting Fyn in a model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2016, 478, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Millard, S.; Lutz, F.; Li, G.; Galasko, D.R.; Farlow, M.R.; Quinn, J.F.; Kaye, J.A.; Leverenz, J.B.; Tsuang, D.W.; et al. Tau phosphorylation pathway genes and cerebrospinal fluid tau levels in Alzheimer’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.E.; Schwartzberg, P.L.; Grider, T.L.; Fink, D.W.; Nussbaum, R.L. alpha-synuclein is phosphorylated by members of the Src family of protein-tyrosine kinases. J. Biol. Chem. 2001, 276, 3879–3884. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Blasco, S.; Bordone, M.P.; Damianich, A.; Gomez, G.; Bernardi, M.A.; Isaja, L.; Taravini, I.R.; Hanger, D.P.; Avale, M.E.; Gershanik, O.S.; et al. The Kinase Fyn As a Novel Intermediate in L-DOPA-Induced Dyskinesia in Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 5125–5136. [Google Scholar] [CrossRef]
- Dunah, A.W.; Sirianni, A.C.; Fienberg, A.A.; Bastia, E.; Schwarzschild, M.A.; Standaert, D.G. Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol. Pharmacol. 2004, 65, 121–129. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol and Name Gene | Support in Autism | N° of Studies Reporting the Evidence | Supporting Evidence |
|---|---|---|---|
| PLEXA4 (Plexin A4) | Functional | 3 | Copy number variations (CNVs) involving the PLXN-A4 gene were identified in two unrelated ASD case |
| SEMA5A (Semaphorin 5A) | Functional | 15 | Expression of the SEMA5A gene has been shown to be downregulated in some autistic individuals |
| FRK (Fyn-related kinase) | Genetic association | 3 | Genetic association has been found between the FRK gene and autism in two large cohorts (AGRE and ACC) of European ancestry and replicate in two other cohort (CAP and CART) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matrone, C.; Petrillo, F.; Nasso, R.; Ferretti, G. Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions. Int. J. Mol. Sci. 2020, 21, 4444. https://doi.org/10.3390/ijms21124444
Matrone C, Petrillo F, Nasso R, Ferretti G. Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions. International Journal of Molecular Sciences. 2020; 21(12):4444. https://doi.org/10.3390/ijms21124444
Chicago/Turabian StyleMatrone, Carmela, Federica Petrillo, Rosarita Nasso, and Gabriella Ferretti. 2020. "Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions" International Journal of Molecular Sciences 21, no. 12: 4444. https://doi.org/10.3390/ijms21124444
APA StyleMatrone, C., Petrillo, F., Nasso, R., & Ferretti, G. (2020). Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions. International Journal of Molecular Sciences, 21(12), 4444. https://doi.org/10.3390/ijms21124444