Mechanisms of Taxane Resistance

 and
and
Simple Summary
Abstract
1. Introduction
1.1. Taxane Family of Chemotherapy
1.1.1. Paclitaxel
1.1.2. Docetaxel
1.1.3. Cabazitaxel
1.2. The Problem of Taxane Resistance
1.2.1. Taxane-Metabolizing Enzymes
1.2.2. ATP-Dependent Pumps
1.2.3. Tubulin Subunit Expression
1.2.4. Hypoxia Response Pathway
2. Taxane Resistance in Breast Cancer
2.1. Drug Transport and Efflux
2.2. Drug Metabolism
2.3. Alteration of Microtubule Regulatory Proteins and Tubulin Isotypes
2.3.1. Microtubule (MT) and MT Dynamics
2.3.2. MT Regulators in Mitosis and Cell Cycle Progression
2.4. Non-Coding RNAs
2.4.1. MicroRNAs (miRs)
2.4.2. Long Non-Coding RNAs (lncRNAs)
2.5. Tumor Suppressor Genes
2.5.1. Breast Cancer 1 (BRCA1)
2.5.2. Adenomatous Polyposis Coli (APC)
2.5.3. p16
2.5.4. Human Expanded (hEx)
2.5.5. Yes-Associated Protein (YAP)
2.5.6. Leucine Zipper Tumor Suppressor 1 (LZTS1)
2.6. Hypoxia Response Pathway
3. Taxane Resistance in Ovarian Cancer
3.1. Drug Transport and Efflux
3.2. Drug Metabolism by CYP Enzymes
3.3. Alteration of Microtubule Regulatory Proteins and Tubulin Isotypes
3.3.1. MAPs and MAPKs
3.3.2. Tubulin Isotypes
3.4. Cell Cycle Progression
3.4.1. Cyclin E1 Amplification
3.4.2. Cyclin A1
3.4.3. Spindle Assembly Checkpoint
3.4.4. Mitotic Exit
3.5. Pro-Survival and Anti-Apoptotic Proteins
3.5.1. BCL-2 Family
3.5.2. IAP Family
3.6. Signal Transduction Pathways
3.6.1. PI3K/AKT/mTOR Pathway
3.6.2. Src Family Kinases
3.7. Non-Coding RNA
3.7.1. Micro-RNAs
3.7.2. Long Non-Coding RNAs
3.8. Hypoxia Response Pathway
4. Taxane Resistance in Prostate Cancer
4.1. Intracellular Drug Concentration and Transport
4.1.1. MDR1
4.1.2. SLCO Genes and Drug Influx
4.2. Microtubule Dynamics and AR Signaling Pathway
4.2.1. β-Tubulin Isotypes and Mutations
4.2.2. AR Signaling
4.3. EMT Phenotype
4.4. Pro-Survival and Anti-Apoptotic Pathways
4.4.1. BCL-2 Protein Family
4.4.2. PI3K/Akt Pathway
4.5. Non-Coding RNAs
4.5.1. miRNAs
4.5.2. lncRNAs
4.6. Hypoxia Response Pathway
5. Taxane Resistance in Other Cancers
5.1. Non-Small-Cell Lung Cancer
5.2. Cervical Cancer
5.3. Pancreatic Cancer
5.4. Head and Neck Cancer
5.5. Nasopharyngeal Cancer
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg. Med. Chem. 2014, 22, 5050–5059. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Khanna, C.; Rosenberg, M.; Vail, D.M. A Review of paclitaxel and novel formulations including those suitable for use in dogs. J. Vet. Intern. Med. 2015, 29, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Jibodh, R.A.; Lagas, J.S.; Nuijen, B.; Beijnen, J.H.; Schellens, J.H. Taxanes: Old drugs, new oral formulations. Eur. J. Pharmacol. 2013, 717, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kiguchi, K.; Mikami, M.; Aoki, D.; Iwamori, M. Involvement of the MDR1 gene and glycolipids in anticancer drug-resistance of human ovarian carcinoma-derived cells. Hum. Cell 2019, 32, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Farjadian, F.; Ghasemi, A.; Gohari, O.; Roointan, A.; Karimi, M.; Hamblin, M.R. Nanopharmaceuticals and nanomedicines currently on the market: Challenges and opportunities. Nanomedicine 2019, 14, 93–126. [Google Scholar] [CrossRef] [PubMed]
- Viudez, A.; Ramirez, N.; Hernandez-Garcia, I.; Carvalho, F.L.; Vera, R.; Hidalgo, M. Nab-paclitaxel: A flattering facelift. Crit. Rev. Oncol. Hematol. 2014, 92, 166–180. [Google Scholar] [CrossRef]
- De Luca, R.; Profita, G.; Cicero, G. Nab-paclitaxel in pretreated metastatic breast cancer: Evaluation of activity, safety, and quality of life. Oncol. Targets Ther. 2019, 12, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Bachet, J.B.; Chibaudel, B.; Bonnetain, F.; Validire, P.; Hammel, P.; Andre, T.; Louvet, C.; GERCOR Group. A randomized phase II study of weekly nab-paclitaxel plus gemcitabine or simplified LV5FU2 as first-line therapy in patients with metastatic pancreatic cancer: The AFUGEM GERCOR trial. BMC Cancer 2015, 15, 653. [Google Scholar] [CrossRef] [PubMed]
- Joyce, C. Taxol: Search for a cancer drug: Natural products chemistry vindicated and conservation concerns balanced in the quest for plant-based medicines. BioScience 1993, 43, 133–136. [Google Scholar] [CrossRef][Green Version]
- Heinig, U.; Scholz, S.; Jennewein, S. Getting to the bottom of Taxol biosynthesis by fungi. Fungal. Divers. 2013, 60, 161–170. [Google Scholar] [CrossRef]
- Gligorov, J.; Lotz, J.P. Preclinical pharmacology of the taxanes: Implications of the differences. Oncologist 2004, 9 (Suppl. 2), 3–8. [Google Scholar] [CrossRef]
- Fromes, Y.; Gounon, P.; Veitia, R.; Bissery, M.C.; Fellous, A. Influence of microtubule-associated proteins on the differential effects of paclitaxel and docetaxel. J. Protein. Chem. 1996, 15, 377–388. [Google Scholar] [CrossRef]
- Verweij, J.; Clavel, M.; Chevalier, B. Paclitaxel (Taxol) and docetaxel (Taxotere): Not simply two of a kind. Ann. Oncol. 1994, 5, 495–505. [Google Scholar] [CrossRef]
- Azarenko, O.; Smiyun, G.; Mah, J.; Wilson, L.; Jordan, M.A. Antiproliferative mechanism of action of the novel taxane cabazitaxel as compared with the parent compound docetaxel in MCF7 breast cancer cells. Mol. Cancer Ther. 2014, 13, 2092–2103. [Google Scholar] [CrossRef]
- Paller, C.J.; Antonarakis, E.S. Cabazitaxel: A novel second-line treatment for metastatic castration-resistant prostate cancer. Drug Des. Devel. Ther. 2011, 5, 117–124. [Google Scholar] [CrossRef]
- van Eijk, M.; Boosman, R.J.; Schinkel, A.H.; Huitema, A.D.R.; Beijnen, J.H. Cytochrome P450 3A4, 3A5, and 2C8 expression in breast, prostate, lung, endometrial, and ovarian tumors: Relevance for resistance to taxanes. Cancer Chemother. Pharmacol. 2019, 84, 487–499. [Google Scholar] [CrossRef]
- Kamel, A.; Harriman, S. Inhibition of cytochrome P450 enzymes and biochemical aspects of mechanism-based inactivation (MBI). Drug Discov. Today Technol. 2013, 10, e177–e189. [Google Scholar] [CrossRef] [PubMed]
- Augustin, E.; Borowa-Mazgaj, B.; Kikulska, A.; Kordalewska, M.; Pawlowska, M. CYP3A4 overexpression enhances the cytotoxicity of the antitumor triazoloacridinone derivative C-1305 in CHO cells. Acta Pharmacol. Sin. 2013, 34, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Leonessa, F.; Clarke, R. ATP binding cassette transporters and drug resistance in breast cancer. Endocr. Relat. Cancer 2003, 10, 43–73. [Google Scholar] [CrossRef]
- Dumontet, C.; Sikic, B.I. Mechanisms of action of and resistance to antitubulin agents: Microtubule dynamics, drug transport, and cell death. J. Clin. Oncol. 1999, 17, 1061–1070. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Mayer, U.; Wagenaar, E.; Mol, C.A.; van Deemter, L.; Smit, J.J.; van der Valk, M.A.; Voordouw, A.C.; Spits, H.; van Tellingen, O.; et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc. Natl. Acad. Sci. USA 1997, 94, 4028–4033. [Google Scholar] [CrossRef]
- Litman, T.; Druley, T.E.; Stein, W.D.; Bates, S.E. From MDR to MXR: New understanding of multidrug resistance systems, their properties and clinical significance. Cell. Mol. Life Sci. 2001, 58, 931–959. [Google Scholar] [CrossRef]
- Waghray, D.; Zhang, Q. Inhibit or evade multidrug resistance P-Glycoprotein in cancer treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Kumar, S.; Mahdi, H.; Bryant, C.; Shah, J.P.; Garg, G.; Munkarah, A. Clinical trials and progress with paclitaxel in ovarian cancer. Int. J. Womens Health 2010, 2, 411–427. [Google Scholar] [CrossRef]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.L.; Teo, W.S.; McCarroll, J.A.; Kavallaris, M. An emerging role for tubulin isotypes in modulating cancer biology and chemotherapy resistance. Int. J. Mol. Sci. 2017, 18, 1434. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.; Miller, H.P.; Banerjee, A.; Luduena, R.F.; Wilson, L. Microtubule dynamics in vitro are regulated by the tubulin isotype composition. Proc. Natl. Acad. Sci. USA 1994, 91, 11358–11362. [Google Scholar] [CrossRef] [PubMed]
- Derry, W.B.; Wilson, L.; Khan, I.A.; Luduena, R.F.; Jordan, M.A. Taxol differentially modulates the dynamics of microtubules assembled from unfractionated and purified beta-tubulin isotypes. Biochemistry 1997, 36, 3554–3562. [Google Scholar] [CrossRef]
- Yang, C.H.; Yap, E.H.; Xiao, H.; Fiser, A.; Horwitz, S.B. 2-(m-Azidobenzoyl)taxol binds differentially to distinct beta-tubulin isotypes. Proc. Natl. Acad. Sci. USA 2016, 113, 11294–11299. [Google Scholar] [CrossRef]
- McCarroll, J.A.; Sharbeen, G.; Liu, J.; Youkhana, J.; Goldstein, D.; McCarthy, N.; Limbri, L.F.; Dischl, D.; Ceyhan, G.O.; Erkan, M.; et al. betaIII-tubulin: A novel mediator of chemoresistance and metastases in pancreatic cancer. Oncotarget 2015, 6, 2235–2249. [Google Scholar] [CrossRef]
- Bumbaca, B.; Li, W. Taxane resistance in castration-resistant prostate cancer: Mechanisms and therapeutic strategies. Acta Pharm. Sin. B 2018, 8, 518–529. [Google Scholar] [CrossRef]
- Mozzetti, S.; Ferlini, C.; Concolino, P.; Filippetti, F.; Raspaglio, G.; Prislei, S.; Gallo, D.; Martinelli, E.; Ranelletti, F.O.; Ferrandina, G.; et al. Class III beta-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin. Cancer Res. 2005, 11, 298–305. [Google Scholar]
- Zhang, H.L.; Ruan, L.; Zheng, L.M.; Whyte, D.; Tzeng, C.M.; Zhou, X.W. Association between class III beta-tubulin expression and response to paclitaxel/vinorebine-based chemotherapy for non-small cell lung cancer: A meta-analysis. Lung Cancer 2012, 77, 9–15. [Google Scholar] [CrossRef]
- Hasegawa, S.; Miyoshi, Y.; Egawa, C.; Ishitobi, M.; Taguchi, T.; Tamaki, Y.; Monden, M.; Noguchi, S. Prediction of response to docetaxel by quantitative analysis of class I and III beta-tubulin isotype mRNA expression in human breast cancers. Clin. Cancer Res. 2003, 9, 2992–2997. [Google Scholar]
- Tommasi, S.; Mangia, A.; Lacalamita, R.; Bellizzi, A.; Fedele, V.; Chiriatti, A.; Thomssen, C.; Kendzierski, N.; Latorre, A.; Lorusso, V.; et al. Cytoskeleton and paclitaxel sensitivity in breast cancer: The role of beta-tubulins. Int. J. Cancer 2007, 120, 2078–2085. [Google Scholar] [CrossRef] [PubMed]
- Duran, G.E.; Wang, Y.C.; Francisco, E.B.; Rose, J.C.; Martinez, F.J.; Coller, J.; Brassard, D.; Vrignaud, P.; Sikic, B.I. Mechanisms of resistance to cabazitaxel. Mol. Cancer Ther. 2015, 14, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Kampan, N.C.; Madondo, M.T.; McNally, O.M.; Quinn, M.; Plebanski, M. Paclitaxel and its evolving role in the management of ovarian cancer. BioMed. Res. Int. 2015, 2015, 413076. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J. Taxanes for the treatment of metastatic breast cancer. Breast Cancer 2012, 6, 159–171. [Google Scholar] [CrossRef]
- Mahon, K.L.; Henshall, S.M.; Sutherland, R.L.; Horvath, L.G. Pathways of chemotherapy resistance in castration-resistant prostate cancer. Endocr. Relat. Cancer 2011, 18, R103–R123. [Google Scholar] [CrossRef]
- Pucci, P.; Rescigno, P.; Sumanasuriya, S.; de Bono, J.; Crea, F. Hypoxia and noncoding RNAs in Taxane resistance. Trends Pharmacol. Sci. 2018, 39, 695–709. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef]
- Xia, Y.; Jiang, L.; Zhong, T. The role of HIF-1alpha in chemo-/radioresistant tumors. OncoTargets Ther. 2018, 11, 3003–3011. [Google Scholar] [CrossRef]
- Das, V.; Stepankova, J.; Hajduch, M.; Miller, J.H. Role of tumor hypoxia in acquisition of resistance to microtubule-stabilizing drugs. Biochim. Biophys. Acta 2015, 1855, 172–182. [Google Scholar] [CrossRef]
- Karakashev, S.V.; Reginato, M.J. Progress toward overcoming hypoxia-induced resistance to solid tumor therapy. Cancer Manag. Res. 2015, 7, 253–264. [Google Scholar] [CrossRef]
- Sermeus, A.; Genin, M.; Maincent, A.; Fransolet, M.; Notte, A.; Leclere, L.; Riquier, H.; Arnould, T.; Michiels, C. Hypoxia-induced modulation of apoptosis and BCL-2 family proteins in different cancer cell types. PLoS ONE 2012, 7, e47519. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Wang, M.; Yu, M.; Zhang, J.; Bristow, R.G.; Hill, R.P.; Tannock, I.F. Role of autophagy as a survival mechanism for hypoxic cells in tumors. Neoplasia 2016, 18, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Tang, B.; Sun, X. Development of inhibitors targeting hypoxia-inducible factor 1 and 2 for cancer therapy. Yonsei Med. J. 2017, 58, 489–496. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Lequeux, A.; Xiao, M.; Duhem, C.; Chouaib, S.; Berchem, G.; Janji, B. Improving cancer immunotherapy by targeting the hypoxic tumor microenvironment: New opportunities and challenges. Cells 2019, 8, 1083. [Google Scholar] [CrossRef] [PubMed]
- Wigerup, C.; Pahlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.S.; Ansari, J.; Spooner, D.; Hussain, S.A. Chemotherapy for breast cancer (Review). Oncol. Rep. 2010, 24, 1121–1131. [Google Scholar] [CrossRef]
- O’Driscoll, L.; Clynes, M. Biomarkers and multiple drug resistance in breast cancer. Curr. Cancer Drug Targets 2006, 6, 365–384. [Google Scholar] [CrossRef]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of chemotherapy resistance in triple-negative breast cancer-how we can rise to the challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Gomez-Miragaya, J.; Palafox, M.; Pare, L.; Yoldi, G.; Ferrer, I.; Vila, S.; Galvan, P.; Pellegrini, P.; Perez-Montoyo, H.; Igea, A.; et al. Resistance to taxanes in triple-negative breast cancer associates with the dynamics of a CD49f+ Tumor-initiating population. Stem Cell Rep. 2017, 8, 1392–1407. [Google Scholar] [CrossRef]
- Kuo, M.T. Roles of multidrug resistance genes in breast cancer chemoresistance. Adv. Exp. Med. Biol. 2007, 608, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhao, P.; Li, Y.; Yang, D.; Hu, P.; Li, L.; Cheng, Y.; Yao, H. Reversal of P-glycoprotein-mediated multidrug resistance by novel curcumin analogues in paclitaxel-resistant human breast cancer cells. Biochem. Cell Biol. 2020, 98, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Pusztai, L. Markers predicting clinical benefit in breast cancer from microtubule-targeting agents. Ann. Oncol. 2007, 18 (Suppl. 12), xii15–xii20. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.S.; Ku, J.M.; Lee, H.J.; Woo, J.K.; Cheon, C.; Kim, M.; Jang, B.H.; Shin, Y.C.; Ko, S.G. SH003 reverses drug resistance by blocking signal transducer and activator of transcription 3 (STAT3) signaling in breast cancer cells. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Shi, J.F.; Zhang, Q.; Xue, B.; Sun, Y.J.; Li, C.J. Egr-1 enhances drug resistance of breast cancer by modulating MDR1 expression in a GGPPS-independent manner. Biomed. Pharmacother. 2013, 67, 197–202. [Google Scholar] [CrossRef]
- Mei, M.; Xie, D.; Zhang, Y.; Jin, J.; You, F.; Li, Y.; Dai, J.; Chen, X. A new 2alpha,5alpha,10beta,14beta-tetraacetoxy-4(20),11-taxadiene (SIA) derivative overcomes paclitaxel resistance by inhibiting MAPK signaling and increasing paclitaxel accumulation in breast cancer cells. PLoS ONE 2014, 9, e104317. [Google Scholar] [CrossRef]
- Chen, J.; Lu, L.; Feng, Y.; Wang, H.; Dai, L.; Li, Y.; Zhang, P. PKD2 mediates multi-drug resistance in breast cancer cells through modulation of P-glycoprotein expression. Cancer Lett. 2011, 300, 48–56. [Google Scholar] [CrossRef]
- Domanitskaya, N.; Wangari-Talbot, J.; Jacobs, J.; Peiffer, E.; Mahdaviyeh, Y.; Paulose, C.; Malofeeva, E.; Foster, K.; Cai, K.Q.; Zhou, Y.; et al. Abcc10 status affects mammary tumour growth, metastasis, and docetaxel treatment response. Br. J. Cancer 2014, 111, 696–707. [Google Scholar] [CrossRef]
- Engels, F.K.; Ten Tije, A.J.; Baker, S.D.; Lee, C.K.; Loos, W.J.; Vulto, A.G.; Verweij, J.; Sparreboom, A. Effect of cytochrome P450 3A4 inhibition on the pharmacokinetics of docetaxel. Clin. Pharmacol. Ther. 2004, 75, 448–454. [Google Scholar] [CrossRef]
- Cresteil, T.; Monsarrat, B.; Dubois, J.; Sonnier, M.; Alvinerie, P.; Gueritte, F. Regioselective metabolism of taxoids by human CYP3A4 and 2C8: Structure-activity relationship. Drug Metab. Dispos. 2002, 30, 438–445. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Ando, A.; Takamura, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Prediction of response to docetaxel by CYP3A4 mRNA expression in breast cancer tissues. Int. J. Cancer 2002, 97, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Taguchi, T.; Kim, S.J.; Tamaki, Y.; Noguchi, S. Prediction of response to docetaxel by immunohistochemical analysis of CYP3A4 expression in human breast cancers. Breast Cancer 2005, 12, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhao, R.; He, Y.; Fu, X.; Fu, L.; Zhu, Z.; Fu, L.; Dong, J.T. MicroRNA 100 sensitizes luminal A breast cancer cells to paclitaxel treatment in part by targeting mTOR. Oncotarget 2016, 7, 5702–5714. [Google Scholar] [CrossRef] [PubMed]
- Lobert, S.; Jefferson, B.; Morris, K. Regulation of beta-tubulin isotypes by micro-RNA 100 in MCF7 breast cancer cells. Cytoskeleton 2011, 68, 355–362. [Google Scholar] [CrossRef]
- McGrogan, B.T.; Gilmartin, B.; Carney, D.N.; McCann, A. Taxanes, microtubules and chemoresistant breast cancer. Biochim. Biophys. Acta 2008, 1785, 96–132. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005, 6, 204. [Google Scholar] [CrossRef]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef]
- Hait, W.N.; Yang, J.M. The individualization of cancer therapy: The unexpected role of p53. Trans. Am. Clin. Climatol. Assoc. 2006, 117, 85–101. [Google Scholar]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef]
- Rouzier, R.; Rajan, R.; Wagner, P.; Hess, K.R.; Gold, D.L.; Stec, J.; Ayers, M.; Ross, J.S.; Zhang, P.; Buchholz, T.A.; et al. Microtubule-associated protein tau: A marker of paclitaxel sensitivity in breast cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 8315–8320. [Google Scholar] [CrossRef]
- Bowen, J.R.; Hwang, D.; Bai, X.; Roy, D.; Spiliotis, E.T. Septin GTPases spatially guide microtubule organization and plus end dynamics in polarizing epithelia. J. Cell Biol. 2011, 194, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Pous, C.; Klipfel, L.; Baillet, A. Cancer-related functions and subcellular localizations of septins. Front. Cell Dev. Biol. 2016, 4, 126. [Google Scholar] [CrossRef] [PubMed]
- Targa, B.; Klipfel, L.; Cantaloube, I.; Salameh, J.; Benoit, B.; Pous, C.; Baillet, A. Septin filament coalignment with microtubules depends on SEPT9_i1 and tubulin polyglutamylation, and is an early feature of acquired cell resistance to paclitaxel. Cell Death Dis. 2019, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Lewis, S.A.; Feierbach, B.; Stearns, T.; Rommelaere, H.; Ampe, C.; Cowan, N.J. Tubulin subunits exist in an activated conformational state generated and maintained by protein cofactors. J. Cell Biol. 1997, 138, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Le, X.F.; Bast, R.C., J. Src family kinases and paclitaxel sensitivity. Cancer Biol. Ther. 2011, 12, 260–269. [Google Scholar] [CrossRef]
- Hage-Sleiman, R.; Herveau, S.; Matera, E.L.; Laurier, J.F.; Dumontet, C. Tubulin binding cofactor C (TBCC) suppresses tumor growth and enhances chemosensitivity in human breast cancer cells. BMC Cancer 2010, 10, 135. [Google Scholar] [CrossRef]
- Roberts, M.S.; Sahni, J.M.; Schrock, M.S.; Piemonte, K.M.; Weber-Bonk, K.L.; Seachrist, D.D.; Avril, S.; Anstine, L.J.; Singh, S.; Sizemore, S.T.; et al. LIN9 and NEK2 Are core regulators of mitotic fidelity that can be therapeutically targeted to overcome taxane resistance. Cancer Res. 2020, 80, 1693–1706. [Google Scholar] [CrossRef]
- Uzbekov, R.; Kireyev, I.; Prigent, C. Centrosome separation: Respective role of microtubules and actin filaments. Biol. Cell 2002, 94, 275–288. [Google Scholar] [CrossRef]
- Lai, H.; Wang, R.; Li, S.; Shi, Q.; Cai, Z.; Li, Y.; Liu, Y. LIN9 confers paclitaxel resistance in triple negative breast cancer cells by upregulating CCSAP. Sci. China Life Sci. 2020, 63, 419–428. [Google Scholar] [CrossRef]
- Manna, T.; Thrower, D.; Miller, H.P.; Curmi, P.; Wilson, L. Stathmin strongly increases the minus end catastrophe frequency and induces rapid treadmilling of bovine brain microtubules at steady state in vitro. J. Biol. Chem. 2006, 281, 2071–2078. [Google Scholar] [CrossRef]
- Du, Q.; Taylor, L.; Compton, D.A.; Macara, I.G. LGN blocks the ability of NuMA to bind and stabilize microtubules. A mechanism for mitotic spindle assembly regulation. Curr. Biol. 2002, 12, 1928–1933. [Google Scholar] [CrossRef]
- Fukukawa, C.; Ueda, K.; Nishidate, T.; Katagiri, T.; Nakamura, Y. Critical roles of LGN/GPSM2 phosphorylation by PBK/TOPK in cell division of breast cancer cells. Genes Chromosomes Cancer 2010, 49, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Z.; Deng, M.; Liu, B.; Xin, X.; Zhao, Z.; Zhang, Y.; Lv, Q. Downregulation of GPSM2 is associated with primary resistance to paclitaxel in breast cancer. Oncol. Rep. 2020, 43, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Curmi, P.A.; Gavet, O.; Charbaut, E.; Ozon, S.; Lachkar-Colmerauer, S.; Manceau, V.; Siavoshian, S.; Maucuer, A.; Sobel, A. Stathmin and its phosphoprotein family: General properties, biochemical and functional interaction with tubulin. Cell Struct. Funct. 1999, 24, 345–357. [Google Scholar] [CrossRef]
- Cassimeris, L. The oncoprotein 18/stathmin family of microtubule destabilizers. Curr. Opin. Cell Biol. 2002, 14, 18–24. [Google Scholar] [CrossRef]
- Alli, E.; Bash-Babula, J.; Yang, J.M.; Hait, W.N. Effect of stathmin on the sensitivity to antimicrotubule drugs in human breast cancer. Cancer Res. 2002, 62, 6864–6869. [Google Scholar]
- Michal, A.M.; So, C.H.; Beeharry, N.; Shankar, H.; Mashayekhi, R.; Yen, T.J.; Benovic, J.L. G Protein-coupled receptor kinase 5 is localized to centrosomes and regulates cell cycle progression. J. Biol. Chem. 2012, 287, 6928–6940. [Google Scholar] [CrossRef]
- Lagman, J.; Sayegh, P.; Lee, C.S.; Sulon, S.M.; Jacinto, A.Z.; Sok, V.; Peng, N.; Alp, D.; Benovic, J.L.; So, C.H. G protein-coupled receptor kinase 5 modifies cancer cell resistance to paclitaxel. Mol. Cell. Biochem. 2019, 461, 103–118. [Google Scholar] [CrossRef]
- Sha, L.Y.; Zhang, Y.; Wang, W.; Sui, X.; Liu, S.K.; Wang, T.; Zhang, H. MiR-18a upregulation decreases Dicer expression and confers paclitaxel resistance in triple negative breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2201–2208. [Google Scholar]
- Calvano Filho, C.M.; Calvano-Mendes, D.C.; Carvalho, K.C.; Maciel, G.A.; Ricci, M.D.; Torres, A.P.; Filassi, J.R.; Baracat, E.C. Triple-negative and luminal A breast tumors: Differential expression of miR-18a-5p, miR-17-5p, and miR-20a-5p. Tumour Biol. 2014, 35, 7733–7741. [Google Scholar] [CrossRef]
- Hu, H.; Li, S.; Cui, X.; Lv, X.; Jiao, Y.; Yu, F.; Yao, H.; Song, E.; Chen, Y.; Wang, M.; et al. The overexpression of hypomethylated miR-663 induces chemotherapy resistance in human breast cancer cells by targeting heparin sulfate proteoglycan 2 (HSPG2). J. Biol. Chem. 2013, 288, 10973–10985. [Google Scholar] [CrossRef]
- Zhang, X.; Zhong, S.; Xu, Y.; Yu, D.; Ma, T.; Chen, L.; Zhao, Y.; Chen, X.; Yang, S.; Wu, Y.; et al. MicroRNA-3646 contributes to docetaxel resistance in human breast cancer cells by GSK-3beta/beta-Catenin signaling pathway. PLoS ONE 2016, 11, e0153194. [Google Scholar] [CrossRef]
- Campos-Parra, A.D.; Mitznahuatl, G.C.; Pedroza-Torres, A.; Romo, R.V.; Reyes, F.I.P.; Lopez-Urrutia, E.; Perez-Plasencia, C. Micro-RNAs as potential predictors of response to breast cancer systemic therapy: Future clinical implications. Int. J. Mol. Sci. 2017, 18, 1182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, Z.; Zhao, Y.; Ding, Y.; Liu, H.; Xi, Y.; Xiong, W.; Li, G.; Lu, J.; Fodstad, O.; et al. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J. Biol. Chem. 2010, 285, 21496–21507. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wang, Y.; Lu, X.; Zhao, Z.; Zhu, L.; Chen, S.; Wu, Q.; Chen, C.; Wang, Z. MiR-125b regulates epithelial-mesenchymal transition via targeting Sema4C in paclitaxel-resistant breast cancer cells. Oncotarget 2015, 6, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Toyota, M.; Itoh, F.; Kikuchi, T.; Obata, T.; Sasaki, Y.; Suzuki, H.; Yawata, A.; Kusano, M.; Fujita, M.; et al. DNA methylation and histone deacetylation associated with silencing DAP kinase gene expression in colorectal and gastric cancers. Br. J. Cancer 2002, 86, 1817–1823. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.J.; Yang, L.; Hong, Q.; Kuang, X.Y.; Di, G.H.; Shao, Z.M. MicroRNA-200a confers chemoresistance by antagonizing TP53INP1 and YAP1 in human breast cancer. BMC Cancer 2018, 18, 74. [Google Scholar] [CrossRef]
- Gu, X.; Xue, J.Q.; Han, S.J.; Qian, S.Y.; Zhang, W.H. Circulating microRNA-451 as a predictor of resistance to neoadjuvant chemotherapy in breast cancer. Cancer Biomark. 2016, 16, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Dong, L.; Zhang, B.; Dai, J.; Zhang, Y.; Wang, Y.; Qin, S. Long noncoding RNA CASC2 promotes paclitaxel resistance in breast cancer through regulation of miR-18a-5p/CDK19. Histochem. Cell Biol. 2019, 152, 281–291. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Z.; Li, Y.; Zhao, Y.; Zhai, W.; Yang, L.; Kong, D.; Wu, C.; Chen, Z.; Teng, C.B. miR-18a counteracts AKT and ERK activation to inhibit the proliferation of pancreatic progenitor cells. Sci. Rep. 2017, 7, 45002. [Google Scholar] [CrossRef]
- Du, T.; Shi, Y.; Xu, S.; Wan, X.; Sun, H.; Liu, B. Long non-coding RNAs in drug resistance of breast cancer. Onco. Targets Ther. 2020, 13, 7075–7087. [Google Scholar] [CrossRef] [PubMed]
- Ao, X.; Nie, P.; Wu, B.; Xu, W.; Zhang, T.; Wang, S.; Chang, H.; Zou, Z. Decreased expression of microRNA-17 and microRNA-20b promotes breast cancer resistance to taxol therapy by upregulation of NCOA3. Cell Death Dis. 2016, 7, e2463. [Google Scholar] [CrossRef] [PubMed]
- Campos-Parra, A.D.; Lopez-Urrutia, E.; Orozco Moreno, L.T.; Lopez-Camarillo, C.; Meza-Menchaca, T.; Figueroa Gonzalez, G.; Bustamante Montes, L.P.; Perez-Plasencia, C. Long non-coding RNAs as new master regulators of resistance to systemic treatments in breast cancer. Int. J. Mol. Sci. 2018, 19, 2711. [Google Scholar] [CrossRef] [PubMed]
- Si, X.; Zang, R.; Zhang, E.; Liu, Y.; Shi, X.; Zhang, E.; Shao, L.; Li, A.; Yang, N.; Han, X.; et al. LncRNA H19 confers chemoresistance in ERalpha-positive breast cancer through epigenetic silencing of the pro-apoptotic gene BIK. Oncotarget 2016, 7, 81452–81462. [Google Scholar] [CrossRef]
- Han, J.; Han, B.; Wu, X.; Hao, J.; Dong, X.; Shen, Q.; Pang, H. Knockdown of lncRNA H19 restores chemo-sensitivity in paclitaxel-resistant triple-negative breast cancer through triggering apoptosis and regulating Akt signaling pathway. Toxicol. Appl. Pharmacol. 2018, 359, 55–61. [Google Scholar] [CrossRef]
- Bida, O.; Gidoni, M.; Ideses, D.; Efroni, S.; Ginsberg, D. A novel mitosis-associated lncRNA, MA-linc1, is required for cell cycle progression and sensitizes cancer cells to Paclitaxel. Oncotarget 2015, 6, 27880–27890. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Liu, Y.R.; Xu, X.E.; Jin, X.; Hu, X.; Yu, K.D.; Shao, Z.M. Transcriptome Analysis of triple-negative breast cancer reveals an integrated mRNA-lncRNA signature with predictive and prognostic value. Cancer Res. 2016, 76, 2105–2114. [Google Scholar] [CrossRef]
- Huang, P.; Li, F.; Li, L.; You, Y.; Luo, S.; Dong, Z.; Gao, Q.; Wu, S.; Brunner, N.; Stenvang, J. lncRNA profile study reveals the mRNAs and lncRNAs associated with docetaxel resistance in breast cancer cells. Sci. Rep. 2018, 8, 17970. [Google Scholar] [CrossRef]
- Pang, M.F.; Georgoudaki, A.M.; Lambut, L.; Johansson, J.; Tabor, V.; Hagikura, K.; Jin, Y.; Jansson, M.; Alexander, J.S.; Nelson, C.M.; et al. TGF-beta1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene 2016, 35, 748–760. [Google Scholar] [CrossRef]
- Yousefi, H.; Maheronnaghsh, M.; Molaei, F.; Mashouri, L.; Reza Aref, A.; Momeny, M.; Alahari, S.K. Long noncoding RNAs and exosomal lncRNAs: Classification, and mechanisms in breast cancer metastasis and drug resistance. Oncogene 2020, 39, 953–974. [Google Scholar] [CrossRef]
- Hou, P.; Zhao, Y.; Li, Z.; Yao, R.; Ma, M.; Gao, Y.; Zhao, L.; Zhang, Y.; Huang, B.; Lu, J. LincRNA-ROR induces epithelial-to-mesenchymal transition and contributes to breast cancer tumorigenesis and metastasis. Cell Death Dis. 2014, 5, e1287. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Liu, Y.; Wei, H.Y.; Lv, K.Z.; Fu, P. Linc-ROR induces epithelial-mesenchymal transition and contributes to drug resistance and invasion of breast cancer cells. Tumour Biol. 2016, 37, 10861–10870. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Wu, L.; Li, X.; Dai, H.; Zhang, Z. Large intergenic non-coding RNA-ROR as a potential biomarker for the diagnosis and dynamic monitoring of breast cancer. Cancer Biomark. 2017, 20, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Pan, Y.; Cheng, Y.; Yang, F.; Yao, Z.; Wang, O. Knockdown of LncRNA MAPT-AS1 inhibites proliferation and migration and sensitizes cancer cells to paclitaxel by regulating MAPT expression in ER-negative breast cancers. Cell Biosci. 2018, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, T.; Yang, Z.; Jiang, C.; Seng, J. Long non-coding RNA FTH1P3 activates paclitaxel resistance in breast cancer through miR-206/ABCB1. J. Cell. Mol. Med. 2018, 22, 4068–4075. [Google Scholar] [CrossRef]
- Gu, M.; Zheng, W.; Zhang, M.; Dong, X.; Zhao, Y.; Wang, S.; Jiang, H.; Zheng, X. LncRNA NONHSAT141924 promotes paclitaxel chemotherapy resistance through p-CREB/Bcl-2 apoptosis signaling pathway in breast cancer. J. Cancer 2020, 11, 3645–3654. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Zarrabi, A.; Hushmandi, K.; Zarrin, V.; Moghadam, E.R.; Zabolian, A.; Tavakol, S.; Samarghandian, S.; Najafi, M. PD-1/PD-L1 axis regulation in cancer therapy: The role of long non-coding RNAs and microRNAs. Life Sci. 2020, 256, 117899. [Google Scholar] [CrossRef]
- Mullan, P.B.; Quinn, J.E.; Gilmore, P.M.; McWilliams, S.; Andrews, H.; Gervin, C.; McCabe, N.; McKenna, S.; White, P.; Song, Y.H.; et al. BRCA1 and GADD45 mediated G2/M cell cycle arrest in response to antimicrotubule agents. Oncogene 2001, 20, 6123–6131. [Google Scholar] [CrossRef][Green Version]
- Gilmore, P.M.; McCabe, N.; Quinn, J.E.; Kennedy, R.D.; Gorski, J.J.; Andrews, H.N.; McWilliams, S.; Carty, M.; Mullan, P.B.; Duprex, W.P.; et al. BRCA1 interacts with and is required for paclitaxel-induced activation of mitogen-activated protein kinase kinase kinase 3. Cancer Res. 2004, 64, 4148–4154. [Google Scholar] [CrossRef]
- Prasad, C.P.; Mirza, S.; Sharma, G.; Prashad, R.; DattaGupta, S.; Rath, G.; Ralhan, R. Epigenetic alterations of CDH1 and APC genes: Relationship with activation of Wnt/beta-catenin pathway in invasive ductal carcinoma of breast. Life Sci. 2008, 83, 318–325. [Google Scholar] [CrossRef]
- VanKlompenberg, M.K.; Bedalov, C.O.; Soto, K.F.; Prosperi, J.R. APC selectively mediates response to chemotherapeutic agents in breast cancer. BMC Cancer 2015, 15, 457. [Google Scholar] [CrossRef]
- Astarita, E.M.; Hoover, C.A.; Maloney, S.M.; Nair, T.M.; Prosperi, J.R. Adenomatous polyposis coli loss controls cell cycle regulators and response to paclitaxel. Submitt. Sci. Rep. 2020. [Google Scholar] [CrossRef]
- Holleman, A.; Chung, I.; Olsen, R.R.; Kwak, B.; Mizokami, A.; Saijo, N.; Parissenti, A.; Duan, Z.; Voest, E.E.; Zetter, B.R. miR-135a contributes to paclitaxel resistance in tumor cells both in vitro and in vivo. Oncogene 2011, 30, 4386–4398. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Hu, S.L.; Shen, G.D.; Shen, G. Tumor suppressor genes and their underlying interactions in paclitaxel resistance in cancer therapy. Cancer Cell Int. 2016, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Hayashi, N.; Hayashi, H.; Sasaki, M.; Kai, K.; Sugihara, E.; Abe, E.; Yoshida, A.; Mikami, S.; Nakamura, S.; et al. Loss of p16 expression is associated with the stem cell characteristics of surface markers and therapeutic resistance in estrogen receptor-negative breast cancer. Int. J. Cancer 2012, 130, 2568–2579. [Google Scholar] [CrossRef]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef]
- Visser-Grieve, S.; Hao, Y.; Yang, X. Human homolog of Drosophila expanded, hEx, functions as a putative tumor suppressor in human cancer cell lines independently of the Hippo pathway. Oncogene 2012, 31, 1189–1195. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, X. The Hippo pathway in chemotherapeutic drug resistance. Int. J. Cancer 2015, 137, 2767–2773. [Google Scholar] [CrossRef]
- Yuan, M.; Tomlinson, V.; Lara, R.; Holliday, D.; Chelala, C.; Harada, T.; Gangeswaran, R.; Manson-Bishop, C.; Smith, P.; Danovi, S.A.; et al. Yes-associated protein (YAP) functions as a tumor suppressor in breast. Cell Death Differ. 2008, 15, 1752–1759. [Google Scholar] [CrossRef]
- Lovat, F.; Ishii, H.; Schiappacassi, M.; Fassan, M.; Barbareschi, M.; Galligioni, E.; Gasparini, P.; Baldassarre, G.; Croce, C.M.; Vecchione, A. LZTS1 downregulation confers paclitaxel resistance and is associated with worse prognosis in breast cancer. Oncotarget 2014, 5, 970–977. [Google Scholar] [CrossRef]
- Notte, A.; Ninane, N.; Arnould, T.; Michiels, C. Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast cancer cells: Role of autophagy and JNK activation. Cell Death Dis. 2013, 4, e638. [Google Scholar] [CrossRef]
- Flamant, L.; Notte, A.; Ninane, N.; Raes, M.; Michiels, C. Anti-apoptotic role of HIF-1 and AP-1 in paclitaxel exposed breast cancer cells under hypoxia. Mol. Cancer 2010, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Maggiolini, M.; Musti, A.M. Crosstalk between notch, HIF-1alpha and GPER in breast cancer EMT. Int. J. Mol. Sci. 2018, 19, 2011. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Liu, F.; Han, L.; Zhao, L.; Chen, J.; Olopade, O.I.; He, M.; Wei, M. HIF-2alpha promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J. Exp. Clin. Cancer Res. 2018, 37, 256. [Google Scholar] [CrossRef]
- Fekete, J.T.; Osz, A.; Pete, I.; Nagy, G.R.; Vereczkey, I.; Gyorffy, B. Predictive biomarkers of platinum and taxane resistance using the transcriptomic data of 1816 ovarian cancer patients. Gynecol. Oncol. 2020, 156, 654–661. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, C.; Chen, Y.; Zhang, X.; Wang, X.; Xie, X. Down-regulation of UTP23 promotes paclitaxel resistance and predicts poorer prognosis in ovarian cancer. Pathol. Res. Pract. 2019, 215, 152625. [Google Scholar] [CrossRef]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Pastan, I.H. The role of multidrug resistance efflux pumps in cancer: Revisiting a JNCI publication exploring expression of the MDR1 (P-glycoprotein) gene. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Ha, J.S.; Byun, J.; Ahn, D.R. Overcoming doxorubicin resistance of cancer cells by Cas9-mediated gene disruption. Sci. Rep. 2016, 6, 22847. [Google Scholar] [CrossRef]
- Zhang, X.; Yashiro, M.; Qiu, H.; Nishii, T.; Matsuzaki, T.; Hirakawa, K. Establishment and characterization of multidrug-resistant gastric cancer cell lines. Anticancer. Res. 2010, 30, 915–921. [Google Scholar] [PubMed]
- Januchowski, R.; Wojtowicz, K.; Sujka-Kordowska, P.; Andrzejewska, M.; Zabel, M. MDR gene expression analysis of six drug-resistant ovarian cancer cell lines. BioMed. Res. Int. 2013, 2013, 241763. [Google Scholar] [CrossRef] [PubMed]
- Stover, E.H.; Baco, M.B.; Cohen, O.; Li, Y.Y.; Christie, E.L.; Bagul, M.; Goodale, A.; Lee, Y.; Pantel, S.; Rees, M.G.; et al. Pooled genomic screens identify anti-apoptotic genes as targetable mediators of chemotherapy resistance in ovarian cancer. Mol. Cancer Res. 2019, 17, 2281–2293. [Google Scholar] [CrossRef] [PubMed]
- Seborova, K.; Vaclavikova, R.; Soucek, P.; Elsnerova, K.; Bartakova, A.; Cernaj, P.; Bouda, J.; Rob, L.; Hruda, M.; Dvorak, P. Association of ABC gene profiles with time to progression and resistance in ovarian cancer revealed by bioinformatics analyses. Cancer Med. 2019, 8, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Cornelison, R.; Llaneza, D.C.; Landen, C.N. Emerging Therapeutics to overcome chemoresistance in epithelial ovarian cancer: A mini-review. Int. J. Mol. Sci. 2017, 18, 2171. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef]
- Johnatty, S.E.; Beesley, J.; Gao, B.; Chen, X.; Lu, Y.; Law, M.H.; Henderson, M.J.; Russell, A.J.; Hedditch, E.L.; Emmanuel, C.; et al. ABCB1 (MDR1) polymorphisms and ovarian cancer progression and survival: A comprehensive analysis from the Ovarian Cancer Association Consortium and the cancer genome atlas. Gynecol. Oncol. 2013, 131, 8–14. [Google Scholar] [CrossRef]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Gao, B.; Russell, A.; Beesley, J.; Chen, X.Q.; Healey, S.; Henderson, M.; Wong, M.; Emmanuel, C.; Galletta, L.; Johnatty, S.E.; et al. Paclitaxel sensitivity in relation to ABCB1 expression, efflux and single nucleotide polymorphisms in ovarian cancer. Sci. Rep. 2014, 4, 4669. [Google Scholar] [CrossRef]
- Peethambaram, P.; Fridley, B.L.; Vierkant, R.A.; Larson, M.C.; Kalli, K.R.; Elliott, E.A.; Oberg, A.L.; White, K.L.; Rider, D.N.; Keeney, G.L.; et al. Polymorphisms in ABCB1 and ERCC2 associated with ovarian cancer outcome. Int. J. Mol. Epidemiol. Genet. 2011, 2, 185–195. [Google Scholar]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun. 2019, 10, 1295. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Cai, J.; Yang, Q.; Zhu, Y.; Zhao, S.; Wang, Z. Prognostic value and implication for chemotherapy treatment of ABCB1 in epithelial ovarian cancer: A meta-analysis. PLoS ONE 2016, 11, e0166058. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.H.; Chen, X.; Cai, L.Y.; Nan, X.W.; Chen, J.H.; Chen, X.X.; Yang, Y.; Xing, Z.H.; Wei, M.N.; Li, Y.; et al. Erastin reverses ABCB1-mediated docetaxel resistance in ovarian cancer. Front. Oncol. 2019, 9, 1398. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Andruska, N.; Lambrecht, M.J.; He, S.; Parissenti, A.; Hergenrother, P.J.; Nelson, E.R.; Shapiro, D.J. Targeting multidrug-resistant ovarian cancer through estrogen receptor alpha dependent ATP depletion caused by hyperactivation of the unfolded protein response. Oncotarget 2018, 9, 14741–14753. [Google Scholar] [CrossRef]
- Alzahrani, A.M.; Rajendran, P. The multifarious link between cytochrome P450s and cancer. Oxid. Med. Cell. Longev. 2020, 2020, 3028387. [Google Scholar] [CrossRef]
- Jiang, H.; Zhong, F.; Sun, L.; Feng, W.; Huang, Z.X.; Tan, X. Structural and functional insights into polymorphic enzymes of cytochrome P450 2C8. Amino Acids 2011, 40, 1195–1204. [Google Scholar] [CrossRef]
- Lambrechts, S.; Lambrechts, D.; Despierre, E.; Van Nieuwenhuysen, E.; Smeets, D.; Debruyne, P.R.; Renard, V.; Vroman, P.; Luyten, D.; Neven, P.; et al. Genetic variability in drug transport, metabolism or DNA repair affecting toxicity of chemotherapy in ovarian cancer. BMC Pharmacol. Toxicol. 2015, 16, 2. [Google Scholar] [CrossRef]
- Green, H.; Khan, M.S.; Jakobsen-Falk, I.; Avall-Lundqvist, E.; Peterson, C. Impact of CYP3A5*3 and CYP2C8-HapC on paclitaxel/carboplatin-induced myelosuppression in patients with ovarian cancer. J. Pharm. Sci. 2011, 100, 4205–4209. [Google Scholar] [CrossRef]
- Leskela, S.; Jara, C.; Leandro-Garcia, L.J.; Martinez, A.; Garcia-Donas, J.; Hernando, S.; Hurtado, A.; Vicario, J.C.; Montero-Conde, C.; Landa, I.; et al. Polymorphisms in cytochromes P450 2C8 and 3A5 are associated with paclitaxel neurotoxicity. Pharm. J. 2011, 11, 121–129. [Google Scholar] [CrossRef]
- Piotrowska, H.; Kucinska, M.; Murias, M. Expression of CYP1A1, CYP1B1 and MnSOD in a panel of human cancer cell lines. Mol. Cell. Biochem. 2013, 383, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Mu, Y.; Qi, C.; Wang, J.; Xi, G.; Guo, J.; Mi, R.; Zhao, F. CYP1B1 enhances the resistance of epithelial ovarian cancer cells to paclitaxel in vivo and in vitro. Int. J. Mol. Med. 2015, 35, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, A.R.; Chow, H.S.; Martinez, J.A. Effects of resveratrol on drug- and carcinogen-metabolizing enzymes, implications for cancer prevention. Pharmacol. Res. Perspect. 2017, 5, e00294. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A double-edged sword in health benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef]
- Piotrowska, H.; Myszkowski, K.; Ziolkowska, A.; Kulcenty, K.; Wierzchowski, M.; Kaczmarek, M.; Murias, M.; Kwiatkowska-Borowczyk, E.; Jodynis-Liebert, J. Resveratrol analogue 3,4,4’,5-tetramethoxystilbene inhibits growth, arrests cell cycle and induces apoptosis in ovarian SKOV-3 and A-2780 cancer cells. Toxicol. Appl. Pharmacol. 2012, 263, 53–60. [Google Scholar] [CrossRef]
- Piotrowska-Kempisty, H.; Klupczynska, A.; Trzybulska, D.; Kulcenty, K.; Sulej-Suchomska, A.M.; Kucinska, M.; Mikstacka, R.; Wierzchowski, M.; Murias, M.; Baer-Dubowska, W.; et al. Role of CYP1A1 in the biological activity of methylated resveratrol analogue, 3,4,5,4’-tetramethoxystilbene (DMU-212) in ovarian cancer A-2780 and non-cancerous HOSE cells. Toxicol. Lett. 2017, 267, 59–66. [Google Scholar] [CrossRef]
- Szekeres, T.; Fritzer-Szekeres, M.; Saiko, P.; Jager, W. Resveratrol and resveratrol analogues--structure-activity relationship. Pharm. Res. 2010, 27, 1042–1048. [Google Scholar] [CrossRef]
- Detampel, P.; Beck, M.; Krahenbuhl, S.; Huwyler, J. Drug interaction potential of resveratrol. Drug Metab. Rev. 2012, 44, 253–265. [Google Scholar] [CrossRef]
- Piotrowska-Kempisty, H.; Rucinski, M.; Borys, S.; Kucinska, M.; Kaczmarek, M.; Zawierucha, P.; Wierzchowski, M.; Lazewski, D.; Murias, M.; Jodynis-Liebert, J. 3′-hydroxy-3,4,5,4′-tetramethoxystilbene, the metabolite of resveratrol analogue DMU-212, inhibits ovarian cancer cell growth in vitro and in a mice xenograft model. Sci. Rep. 2016, 6, 32627. [Google Scholar] [CrossRef]
- Smoter, M.; Bodnar, L.; Duchnowska, R.; Stec, R.; Grala, B.; Szczylik, C. The role of Tau protein in resistance to paclitaxel. Cancer Chemother. Pharmacol. 2011, 68, 553–557. [Google Scholar] [CrossRef]
- Smoter, M.; Bodnar, L.; Grala, B.; Stec, R.; Zieniuk, K.; Kozlowski, W.; Szczylik, C. Tau protein as a potential predictive marker in epithelial ovarian cancer patients treated with paclitaxel/platinum first-line chemotherapy. J. Exp. Clin. Cancer Res. 2013, 32, 25. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, A.; Kobayashi, A.; Oikiri, H.; Yokoyama, Y. Functional role of the Tau protein in epithelial ovarian cancer cells. Reprod. Med. Biol. 2017, 16, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Gurler, H.; Yu, Y.; Choi, J.; Kajdacsy-Balla, A.A.; Barbolina, M.V. Three-dimensional collagen type I matrix up-regulates nuclear isoforms of the microtubule associated protein tau implicated in resistance to paclitaxel therapy in ovarian carcinoma. Int. J. Mol. Sci. 2015, 16, 3419–3433. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gaillard, S.; Phillip, J.M.; Huang, T.C.; Pinto, S.M.; Tessarollo, N.G.; Zhang, Z.; Pandey, A.; Wirtz, D.; Ayhan, A.; et al. Inhibition of spleen tyrosine kinase potentiates paclitaxel-induced cytotoxicity in ovarian cancer cells by stabilizing microtubules. Cancer Cell 2015, 28, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Mooneyham, A.; Iizuka, Y.; Yang, Q.; Coombes, C.; McClellan, M.; Shridhar, V.; Emmings, E.; Shetty, M.; Chen, L.; Ai, T.; et al. UNC-45A Is a novel microtubule-associated protein and regulator of paclitaxel sensitivity in ovarian cancer cells. Mol. Cancer Res. 2019, 17, 370–383. [Google Scholar] [CrossRef]
- Habicht, J.; Mooneyham, A.; Shetty, M.; Zhang, X.; Shridhar, V.; Winterhoff, B.; Zhang, Y.; Cepela, J.; Starr, T.; Lou, E.; et al. UNC-45A is preferentially expressed in epithelial cells and binds to and co-localizes with interphase MTs. Cancer Biol. Ther. 2019, 20, 1304–1313. [Google Scholar] [CrossRef]
- Yang, H.; Mao, W.; Rodriguez-Aguayo, C.; Mangala, L.S.; Bartholomeusz, G.; Iles, L.R.; Jennings, N.B.; Ahmed, A.A.; Sood, A.K.; Lopez-Berestein, G.; et al. Paclitaxel sensitivity of ovarian cancer can be enhanced by knocking down pairs of kinases that regulate MAP4 Phosphorylation and microtubule stability. Clin. Cancer Res. 2018, 24, 5072–5084. [Google Scholar] [CrossRef]
- Theriault, B.L.; Pajovic, S.; Bernardini, M.Q.; Shaw, P.A.; Gallie, B.L. Kinesin family member 14: An independent prognostic marker and potential therapeutic target for ovarian cancer. Int. J. Cancer. 2012, 130, 1844–1854. [Google Scholar] [CrossRef]
- Du, J.; Li, B.; Fang, Y.; Liu, Y.; Wang, Y.; Li, J.; Zhou, W.; Wang, X. Overexpression of Class III beta-tubulin, Sox2, and nuclear Survivin is predictive of taxane resistance in patients with stage III ovarian epithelial cancer. BMC Cancer 2015, 15, 536. [Google Scholar] [CrossRef]
- Duran, G.E.; Wang, Y.C.; Moisan, F.; Francisco, E.B.; Sikic, B.I. Decreased levels of baseline and drug-induced tubulin polymerisation are hallmarks of resistance to taxanes in ovarian cancer cells and are associated with epithelial-to-mesenchymal transition. Br. J. Cancer 2017, 116, 1318–1328. [Google Scholar] [CrossRef]
- Roque, D.M.; Buza, N.; Glasgow, M.; Bellone, S.; Bortolomai, I.; Gasparrini, S.; Cocco, E.; Ratner, E.; Silasi, D.A.; Azodi, M.; et al. Class III beta-tubulin overexpression within the tumor microenvironment is a prognostic biomarker for poor overall survival in ovarian cancer patients treated with neoadjuvant carboplatin/paclitaxel. Clin. Exp. Metastasis 2014, 31, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Kanakkanthara, A.; Northcote, P.T.; Miller, J.H. betaII-tubulin and betaIII-tubulin mediate sensitivity to peloruside A and laulimalide, but not paclitaxel or vinblastine, in human ovarian carcinoma cells. Mol. Cancer Ther. 2012, 11, 393–404. [Google Scholar] [CrossRef]
- Kumbhar, B.V.; Bhandare, V.V. Exploring the interaction of Peloruside-A with drug resistant alphabetaII and alphabetaIII tubulin isotypes in human ovarian carcinoma using a molecular modeling approach. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Grant, G.D.; Cook, J.G. The temporal regulation of S phase proteins during G1. Adv. Exp. Med. Biol. 2017, 1042, 335–369. [Google Scholar] [CrossRef]
- Gorski, J.W.; Ueland, F.R.; Kolesar, J.M. CCNE1 amplification as a predictive biomarker of chemotherapy resistance in epithelial ovarian cancer. Diagnostics 2020, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, N.; Nakayama, K.; Shamima, Y.; Ishikawa, M.; Katagiri, A.; Iida, K.; Miyazaki, K. Gene amplification CCNE1 is related to poor survival and potential therapeutic target in ovarian cancer. Cancer 2010, 116, 2621–2634. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Wang, T.L.; Doberstein, K.; Bahadirli-Talbott, A.; Ayhan, A.; Sehdev, A.S.; Drapkin, R.; Kurman, R.J.; Shih Ie, M. CCNE1 amplification and centrosome number abnormality in serous tubal intraepithelial carcinoma: Further evidence supporting its role as a precursor of ovarian high-grade serous carcinoma. Mod. Pathol. 2016, 29, 1254–1261. [Google Scholar] [CrossRef]
- Kanska, J.; Zakhour, M.; Taylor-Harding, B.; Karlan, B.Y.; Wiedemeyer, W.R. Cyclin E as a potential therapeutic target in high grade serous ovarian cancer. Gynecol. Oncol. 2016, 143, 152–158. [Google Scholar] [CrossRef]
- Au-Yeung, G.; Lang, F.; Azar, W.J.; Mitchell, C.; Jarman, K.E.; Lackovic, K.; Aziz, D.; Cullinane, C.; Pearson, R.B.; Mileshkin, L.; et al. Selective targeting of cyclin E1-amplified high-grade serous ovarian cancer by cyclin-dependent kinase 2 and AKT inhibition. Clin. Cancer Res. 2017, 23, 1862–1874. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; Au-Yeung, G.; Wall, M.; Mitchell, C.; Kansara, M.; Loehrer, E.; Batzios, C.; George, J.; Ftouni, S.; Weir, B.A.; et al. Resistance to CDK2 inhibitors is associated with selection of polyploid cells in CCNE1-amplified ovarian cancer. Clin. Cancer Res. 2013, 19, 5960–5971. [Google Scholar] [CrossRef]
- Noack, S.; Raab, M.; Matthess, Y.; Sanhaji, M.; Kramer, A.; Gyorffy, B.; Kaderali, L.; El-Balat, A.; Becker, S.; Strebhardt, K. Synthetic lethality in CCNE1-amplified high grade serous ovarian cancer through combined inhibition of Polo-like kinase 1 and microtubule dynamics. Oncotarget 2018, 9, 25842–25859. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Li, Y.; Zhao, C.; Jiang, X.; Chen, H.; Lang, M.F.; Sun, J. Cyclin A1 is expressed in mouse ovary. Int. J. Med. Sci. 2014, 11, 754–757. [Google Scholar] [CrossRef] [PubMed]
- Arsenic, R.; Braicu, E.I.; Letsch, A.; Dietel, M.; Sehouli, J.; Keilholz, U.; Ochsenreither, S. Cancer-testis antigen cyclin A1 is broadly expressed in ovarian cancer and is associated with prolonged time to tumor progression after platinum-based therapy. BMC Cancer 2015, 15, 784. [Google Scholar] [CrossRef]
- Huang, K.C.; Yang, J.; Ng, M.C.; Ng, S.K.; Welch, W.R.; Muto, M.G.; Berkowitz, R.S.; Ng, S.W. Cyclin A1 expression and paclitaxel resistance in human ovarian cancer cells. Eur. J. Cancer 2016, 67, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Syed Khaja, A.S.; Dizeyi, N.; Kopparapu, P.K.; Anagnostaki, L.; Harkonen, P.; Persson, J.L. Cyclin A1 modulates the expression of vascular endothelial growth factor and promotes hormone-dependent growth and angiogenesis of breast cancer. PLoS ONE 2013, 8, e72210. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.Q.; Wang, X.H.; Hou, L.J.; Jia, W.H.; Yang, Q.; Li, Y.X.; Liu, M.; Li, X.; Tang, H. MicroRNA-372 is down-regulated and targets cyclin-dependent kinase 2 (CDK2) and cyclin A1 in human cervical cancer, which may contribute to tumorigenesis. J. Biol. Chem. 2011, 286, 25556–25563. [Google Scholar] [CrossRef] [PubMed]
- Lara-Gonzalez, P.; Westhorpe, F.G.; Taylor, S.S. The spindle assembly checkpoint. Curr. Biol. 2012, 22, R966–R980. [Google Scholar] [CrossRef] [PubMed]
- Hayward, D.; Alfonso-Perez, T.; Gruneberg, U. Orchestration of the spindle assembly checkpoint by CDK1-cyclin B1. FEBS Lett. 2019, 593, 2889–2907. [Google Scholar] [CrossRef] [PubMed]
- Chong, T.; Sarac, A.; Yao, C.Q.; Liao, L.; Lyttle, N.; Boutros, P.C.; Bartlett, J.M.S.; Spears, M. Deregulation of the spindle assembly checkpoint is associated with paclitaxel resistance in ovarian cancer. J. Ovarian Res. 2018, 11, 27. [Google Scholar] [CrossRef]
- Tipton, A.R.; Wang, K.; Link, L.; Bellizzi, J.J.; Huang, H.; Yen, T.; Liu, S.T. BUBR1 and closed MAD2 (C-MAD2) interact directly to assemble a functional mitotic checkpoint complex. J. Biol. Chem. 2011, 286, 21173–21179. [Google Scholar] [CrossRef]
- Hao, X.; Zhou, Z.; Ye, S.; Zhou, T.; Lu, Y.; Ma, D.; Wang, S. Effect of Mad2 on paclitaxel-induced cell death in ovarian cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2010, 30, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Sumi, T.; Teramae, M.; Morishita, M.; Fukuda, T.; Terada, H.; Yoshida, H.; Matsumoto, Y.; Yasui, T.; Ishiko, O. Expression of the mitotic-arrest deficiency 2 is associated with chemotherapy resistance in ovarian serous adenocarcinoma. Oncol. Rep. 2012, 28, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Sumi, T.; Morishita, M.; Fukuda, T.; Nobeyama, H.; Yoshida, H.; Matsumoto, Y.; Yasui, T.; Ishiko, O. Mitotic arrest deficiency 2 induces carcinogenesis in mucinous ovarian tumors. Oncol. Lett. 2012, 3, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Byrne, T.; Coleman, H.G.; Cooper, J.A.; McCluggage, W.G.; McCann, A.; Furlong, F. The association between MAD2 and prognosis in cancer: A systematic review and meta-analyses. Oncotarget 2017, 8, 102223–102234. [Google Scholar] [CrossRef]
- Zeng, X.; Sigoillot, F.; Gaur, S.; Choi, S.; Pfaff, K.L.; Oh, D.C.; Hathaway, N.; Dimova, N.; Cuny, G.D.; King, R.W. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 2010, 18, 382–395. [Google Scholar] [CrossRef]
- Raab, M.; Sanhaji, M.; Zhou, S.; Rodel, F.; El-Balat, A.; Becker, S.; Strebhardt, K. Blocking mitotic exit of ovarian cancer cells by pharmaceutical inhibition of the anaphase-promoting complex reduces chromosomal instability. Neoplasia 2019, 21, 363–375. [Google Scholar] [CrossRef]
- Mc Gee, M.M. Targeting the mitotic catastrophe signaling pathway in cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef]
- Giovinazzi, S.; Bellapu, D.; Morozov, V.M.; Ishov, A.M. Targeting mitotic exit with hyperthermia or APC/C inhibition to increase paclitaxel efficacy. Cell Cycle 2013, 12, 2598–2607. [Google Scholar] [CrossRef]
- Affatato, R.; Carrassa, L.; Chila, R.; Lupi, M.; Restelli, V.; Damia, G. Identification of PLK1 as a new therapeutic target in mucinous ovarian carcinoma. Cancers 2020, 12, 672. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A potential target for cancer therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef]
- Binju, M.; Amaya-Padilla, M.A.; Wan, G.; Gunosewoyo, H.; Suryo Rahmanto, Y.; Yu, Y. Therapeutic inducers of apoptosis in ovarian cancer. Cancers 2019, 11, 1786. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL2 family: Key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Habata, S.; Iwasaki, M.; Sugio, A.; Suzuki, M.; Tamate, M.; Satohisa, S.; Tanaka, R.; Saito, T. BAG3-mediated Mcl-1 stabilization contributes to drug resistance via interaction with USP9X in ovarian cancer. Int. J. Oncol. 2016, 49, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Mojsa, B.; Lassot, I.; Desagher, S. Mcl-1 ubiquitination: Unique regulation of an essential survival protein. Cells 2014, 3, 418–437. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Giorgi, C.; Balestra, D.; Marchi, S.; Perrone, D.; Pinotti, M.; Pinton, P. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol. Biol. Cell 2016, 27, 20–34. [Google Scholar] [CrossRef]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, M.; Jing, Y.; Yin, X.; Ma, P.; Zhang, Z.; Wang, X.; Di, W.; Zhuang, G. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat. Commun. 2018, 9, 215. [Google Scholar] [CrossRef]
- Wu, X.; Luo, Q.; Zhao, P.; Chang, W.; Wang, Y.; Shu, T.; Ding, F.; Li, B.; Liu, Z. MGMT-activated DUB3 stabilizes MCL1 and drives chemoresistance in ovarian cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 2961–2966. [Google Scholar] [CrossRef]
- Al-Alem, L.F.; Baker, A.T.; Pandya, U.M.; Eisenhauer, E.L.; Rueda, B.R. Understanding and Targeting apoptotic pathways in ovarian cancer. Cancers 2019, 11, 1631. [Google Scholar] [CrossRef]
- Yokoyama, T.; Kohn, E.C.; Brill, E.; Lee, J.M. Apoptosis is augmented in high-grade serous ovarian cancer by the combined inhibition of Bcl-2/Bcl-xL and PARP. Int. J. Oncol. 2017. [Google Scholar] [CrossRef]
- Wong, M.; Tan, N.; Zha, J.; Peale, F.V.; Yue, P.; Fairbrother, W.J.; Belmont, L.D. Navitoclax (ABT-263) reduces Bcl-x(L)-mediated chemoresistance in ovarian cancer models. Mol. Cancer Ther. 2012, 11, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hamo, R.; Zilberberg, A.; Cohen, H.; Bahar-Shany, K.; Wachtel, C.; Korach, J.; Aviel-Ronen, S.; Barshack, I.; Barash, D.; Levanon, K.; et al. Resistance to paclitaxel is associated with a variant of the gene BCL2 in multiple tumor types. NPJ Precis. Oncol. 2019, 3, 12. [Google Scholar] [CrossRef]
- Shi, J.; Zhou, Y.; Huang, H.C.; Mitchison, T.J. Navitoclax (ABT-263) accelerates apoptosis during drug-induced mitotic arrest by antagonizing Bcl-xL. Cancer Res. 2011, 71, 4518–4526. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M.; Takano, M.; Iwaya, K.; Shinomiya, N.; Kato, M.; Aoyama, T.; Sasaki, N.; Goto, T.; Suzuki, A.; Hitrata, J.; et al. X-chromosome-linked inhibitor of apoptosis as a key factor for chemoresistance in clear cell carcinoma of the ovary. Br. J. Cancer 2014, 110, 2881–2886. [Google Scholar] [CrossRef] [PubMed]
- Thibault, B.; Genre, L.; Le Naour, A.; Broca, C.; Mery, E.; Vuagniaux, G.; Delord, J.P.; Wiedemann, N.; Couderc, B. DEBIO 1143, an IAP inhibitor, reverses carboplatin resistance in ovarian cancer cells and triggers apoptotic or necroptotic cell death. Sci. Rep. 2018, 8, 17862. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, E.; Pasquini, L.; Bernabei, M.; Saulle, E.; Biffoni, M.; Accarpio, F.; Sibio, S.; Di Giorgio, A.; Di Donato, V.; Casorelli, A.; et al. A small molecule SMAC mimic LBW242 potentiates TRAIL- and anticancer drug-mediated cell death of ovarian cancer cells. PLoS ONE 2012, 7, e35073. [Google Scholar] [CrossRef]
- Gatti, L.; De Cesare, M.; Ciusani, E.; Corna, E.; Arrighetti, N.; Cominetti, D.; Belvisi, L.; Potenza, D.; Moroni, E.; Vasile, F.; et al. Antitumor activity of a novel homodimeric SMAC mimetic in ovarian carcinoma. Mol. Pharm. 2014, 11, 283–293. [Google Scholar] [CrossRef]
- Yue, C.; Li, R.H.; Chen, C.; Liu, H. Study on the relationship between XIAP gene and resistance of taxol in ovarian cancer. Sichuan Da Xue Xue Bao Yi Xue Ban 2018, 49, 337–341. [Google Scholar]
- de Almagro, M.C.; Vucic, D. The inhibitor of apoptosis (IAP) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Exp. Oncol. 2012, 34, 200–211. [Google Scholar]
- Bai, L.; Smith, D.C.; Wang, S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol. Ther. 2014, 144, 82–95. [Google Scholar] [CrossRef]
- Altieri, D.C. Survivin and IAP proteins in cell-death mechanisms. Biochem. J. 2010, 430, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Luo, R.Y.; Yang, J.; Cheng, Y.X. Knockdown of survivin contributes to antitumor activity in cisplatin-resistant ovarian cancer cells. Mol. Med. Rep. 2013, 7, 425–430. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, S.; Chan, H.F.; Lu, H.; Lin, Z.; He, C.; Chen, M. Dihydromyricetin Induces apoptosis and reverses drug resistance in ovarian cancer cells by p53-mediated downregulation of survivin. Sci. Rep. 2017, 7, 46060. [Google Scholar] [CrossRef] [PubMed]
- Vivas-Mejia, P.E.; Rodriguez-Aguayo, C.; Han, H.D.; Shahzad, M.M.; Valiyeva, F.; Shibayama, M.; Chavez-Reyes, A.; Sood, A.K.; Lopez-Berestein, G. Silencing survivin splice variant 2B leads to antitumor activity in taxane--resistant ovarian cancer. Clin. Cancer Res. 2011, 17, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wang, Q.; Wu, Z.; Tian, X.; Yan, H.; Wang, B.; Dong, P.; Watari, H.; Pfeffer, L.M.; Guo, Y.; et al. Ovarian primary and metastatic tumors suppressed by survivin knockout or a novel survivin inhibitor. Mol. Cancer Ther. 2019, 18, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Adamson, V. Fitness at work schemes. Occup. Health 1990, 42, 242. [Google Scholar]
- Ghoneum, A.; Said, N. PI3K-AKT-mTOR and NFkappaB pathways in ovarian cancer: Implications for targeted therapeutics. Cancers 2019, 11, 949. [Google Scholar] [CrossRef]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy resistance in advanced ovarian cancer patients. Biomark. Cancer 2019, 11, 1179299X19860815. [Google Scholar] [CrossRef]
- Cheaib, B.; Auguste, A.; Leary, A. The PI3K/Akt/mTOR pathway in ovarian cancer: Therapeutic opportunities and challenges. Chin. J. Cancer 2015, 34, 4–16. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Honig, A.; Hahne, J.C.; Meyer, S.; Kranke, P.; Hausler, S.; Diessner, J.; Dietl, J.; Engel, J.B. PI3K inhibitor D-116883 is effective in in vitro models of ovarian cancer. Anticancer Res. 2012, 32, 2035–2041. [Google Scholar] [PubMed]
- Jeong, J.Y.; Kim, K.S.; Moon, J.S.; Song, J.A.; Choi, S.H.; Kim, K.I.; Kim, T.H.; An, H.J. Targeted inhibition of phosphatidyl inositol-3-kinase p110beta, but not p110alpha, enhances apoptosis and sensitivity to paclitaxel in chemoresistant ovarian cancers. Apoptosis 2013, 18, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Huang, Y.F.; Chen, C.C.; Huang, C.Y.; Chou, C.Y. Comparing PI3K/Akt inhibitors used in ovarian cancer treatment. Front. Pharmacol. 2020, 11, 206. [Google Scholar] [CrossRef] [PubMed]
- De Wolf, E.; De Wolf, C.; Richardson, A. ABT-737 and pictilisib synergistically enhance pitavastatin-induced apoptosis in ovarian cancer cells. Oncol. Lett. 2018, 15, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Zervantonakis, I.K.; Iavarone, C.; Chen, H.Y.; Selfors, L.M.; Palakurthi, S.; Liu, J.F.; Drapkin, R.; Matulonis, U.; Leverson, J.D.; Sampath, D.; et al. Systems analysis of apoptotic priming in ovarian cancer identifies vulnerabilities and predictors of drug response. Nat. Commun. 2017, 8, 365. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, A.; Finn, R.S. SRC: A century of science brought to the clinic. Neoplasia 2010, 12, 599–607. [Google Scholar] [CrossRef]
- Kim, H.S.; Han, H.D.; Armaiz-Pena, G.N.; Stone, R.L.; Nam, E.J.; Lee, J.W.; Shahzad, M.M.; Nick, A.M.; Lee, S.J.; Roh, J.W.; et al. Functional roles of Src and Fgr in ovarian carcinoma. Clin. Cancer Res. 2011, 17, 1713–1721. [Google Scholar] [CrossRef]
- Huang, Y.W.; Chen, C.; Xu, M.M.; Li, J.D.; Xiao, J.; Zhu, X.F. Expression of c-Src and phospho-Src in epithelial ovarian carcinoma. Mol. Cell. Biochem. 2013, 376, 73–79. [Google Scholar] [CrossRef]
- Kadife, E.; Chan, E.; Luwor, R.; Kannourakis, G.; Findlay, J.; Ahmed, N. Paclitaxel-induced Src activation is inhibited by dasatinib treatment, independently of cancer stem cell properties, in a mouse model of ovarian cancer. Cancers 2019, 11, 243. [Google Scholar] [CrossRef]
- Le, X.F.; Mao, W.; Lu, Z.; Carter, B.Z.; Bast, R.C., J. Dasatinib induces autophagic cell death in human ovarian cancer. Cancer 2010, 116, 4980–4990. [Google Scholar] [CrossRef]
- Gnoni, A.; Marech, I.; Silvestris, N.; Vacca, A.; Lorusso, V. Dasatinib: An anti-tumour agent via Src inhibition. Curr. Drug Targets 2011, 12, 563–578. [Google Scholar] [CrossRef]
- Schilder, R.J.; Brady, W.E.; Lankes, H.A.; Fiorica, J.V.; Shahin, M.S.; Zhou, X.C.; Mannel, R.S.; Pathak, H.B.; Hu, W.; Alpaugh, R.K.; et al. Phase II evaluation of dasatinib in the treatment of recurrent or persistent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 127, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Teoh, D.; Ayeni, T.A.; Rubatt, J.M.; Adams, D.J.; Grace, L.; Starr, M.D.; Barry, W.T.; Berchuck, A.; Murphy, S.K.; Secord, A.A. Dasatinib (BMS-35482) has synergistic activity with paclitaxel and carboplatin in ovarian cancer cells. Gynecol. Oncol. 2011, 121, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Xu, M.; Hou, T.; Huang, Y.; Yang, C.; Li, J. Dasatinib enhances antitumor activity of paclitaxel in ovarian cancer through Src signaling. Mol. Med. Rep. 2015, 12, 3249–3256. [Google Scholar] [CrossRef] [PubMed]
- Secord, A.A.; Teoh, D.K.; Barry, W.T.; Yu, M.; Broadwater, G.; Havrilesky, L.J.; Lee, P.S.; Berchuck, A.; Lancaster, J.; Wenham, R.M. A phase I trial of dasatinib, an SRC-family kinase inhibitor, in combination with paclitaxel and carboplatin in patients with advanced or recurrent ovarian cancer. Clin. Cancer Res. 2012, 18, 5489–5498. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, X.; Han, X.; Yang, T.; Fu, J.; Zhang, Y.; Gou, W. An ovarian cancer model with positive ER: Reversion of ER antagonist resistance by Src blockade. Oncol. Rep. 2014, 32, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T.; Yang, Y.J.; Zhang, Z.D.; An, Q.; Li, N.; Liu, W.; Yang, B. MiR-1307 promotes ovarian cancer cell chemoresistance by targeting the ING5 expression. J. Ovarian Res. 2017, 10, 1. [Google Scholar] [CrossRef]
- Weiner-Gorzel, K.; Dempsey, E.; Milewska, M.; McGoldrick, A.; Toh, V.; Walsh, A.; Lindsay, S.; Gubbins, L.; Cannon, A.; Sharpe, D.; et al. Overexpression of the microRNA miR-433 promotes resistance to paclitaxel through the induction of cellular senescence in ovarian cancer cells. Cancer Med. 2015, 4, 745–758. [Google Scholar] [CrossRef]
- Gordon, R.R.; Nelson, P.S. Cellular senescence and cancer chemotherapy resistance. Drug Resist. Updates 2012, 15, 123–131. [Google Scholar] [CrossRef]
- Eoh, K.J.; Lee, S.H.; Kim, H.J.; Lee, J.Y.; Kim, S.; Kim, S.W.; Kim, Y.T.; Nam, E.J. MicroRNA-630 inhibitor sensitizes chemoresistant ovarian cancer to chemotherapy by enhancing apoptosis. Biochem. Biophys. Res. Commun. 2018, 497, 513–520. [Google Scholar] [CrossRef]
- Cui, Y.; She, K.; Tian, D.; Zhang, P.; Xin, X. miR-146a inhibits proliferation and enhances chemosensitivity in epithelial ovarian cancer via reduction of SOD2. Oncol. Res. 2016, 23, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Sawada, K.; Miyamoto, M.; Kinose, Y.; Yoshimura, A.; Ishida, K.; Kobayashi, M.; Shimizu, A.; Nakatsuka, E.; Hashimoto, K.; et al. Downregulation of miR-194-5p induces paclitaxel resistance in ovarian cancer cells by altering MDM2 expression. Oncotarget 2019, 10, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Jiao, J.W.; Sun, K.X.; Zong, Z.H.; Zhao, Y. MicroRNA-133b targets glutathione S-transferase pi expression to increase ovarian cancer cell sensitivity to chemotherapy drugs. Drug Des. Devel. Ther. 2015, 9, 5225–5235. [Google Scholar] [CrossRef] [PubMed]
- Sawers, L.; Ferguson, M.J.; Ihrig, B.R.; Young, H.C.; Chakravarty, P.; Wolf, C.R.; Smith, G. Glutathione S-transferase P1 (GSTP1) directly influences platinum drug chemosensitivity in ovarian tumour cell lines. Br. J. Cancer 2014, 111, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, Y.; Shen, X.; Ruan, Y.; Lu, Y.; Jin, X.; Song, P.; Guo, Y.; Zhang, X.; Qu, H.; et al. CLDN6 promotes chemoresistance through GSTP1 in human breast cancer. J. Exp. Clin. Cancer Res. 2017, 36, 157. [Google Scholar] [CrossRef]
- Huh, J.H.; Kim, T.H.; Kim, K.; Song, J.A.; Jung, Y.J.; Jeong, J.Y.; Lee, M.J.; Kim, Y.K.; Lee, D.H.; An, H.J. Dysregulation of miR-106a and miR-591 confers paclitaxel resistance to ovarian cancer. Br. J. Cancer 2013, 109, 452–461. [Google Scholar] [CrossRef]
- Cittelly, D.M.; Dimitrova, I.; Howe, E.N.; Cochrane, D.R.; Jean, A.; Spoelstra, N.S.; Post, M.D.; Lu, X.; Broaddus, R.R.; Spillman, M.A.; et al. Restoration of miR-200c to ovarian cancer reduces tumor burden and increases sensitivity to paclitaxel. Mol. Cancer Ther. 2012, 11, 2556–2565. [Google Scholar] [CrossRef]
- Brozovic, A.; Duran, G.E.; Wang, Y.C.; Francisco, E.B.; Sikic, B.I. The miR-200 family differentially regulates sensitivity to paclitaxel and carboplatin in human ovarian carcinoma OVCAR-3 and MES-OV cells. Mol. Oncol. 2015, 9, 1678–1693. [Google Scholar] [CrossRef]
- Lopez-Urrutia, E.; Bustamante Montes, L.P.; Ladron de Guevara Cervantes, D.; Perez-Plasencia, C.; Campos-Parra, A.D. Crosstalk between long non-coding RNAs, Micro-RNAs and mRNAs: Deciphering molecular mechanisms of master regulators in cancer. Front. Oncol. 2019, 9, 669. [Google Scholar] [CrossRef]
- Abildgaard, C.; Do Canto, L.M.; Steffensen, K.D.; Rogatto, S.R. Long non-coding RNAs involved in resistance to chemotherapy in ovarian cancer. Front. Oncol. 2019, 9, 1549. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zou, B.; Tian, T.; Luo, X.; Mao, B.; Zhang, X.; Lei, H. Overexpression of the lncRNA FER1L4 inhibits paclitaxel tolerance of ovarian cancer cells via the regulation of the MAPK signaling pathway. J. Cell Biochem. 2018, 120, 7581–7589. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ye, C.; Liu, J.; Hu, Y. UCA1 confers paclitaxel resistance to ovarian cancer through miR-129/ABCB1 axis. Biochem. Biophys. Res. Commun. 2018, 501, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Wang, M. LINC01118 modulates paclitaxel resistance of epithelial ovarian cancer by regulating miR-134/ABCC1. Med. Sci. Monit. 2018, 24, 8831–8839. [Google Scholar] [CrossRef]
- An, J.; Lv, W.; Zhang, Y. LncRNA NEAT1 contributes to paclitaxel resistance of ovarian cancer cells by regulating ZEB1 expression via miR-194. Oncol. Targets Ther. 2017, 10, 5377–5390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef]
- Huang, L.; Ao, Q.; Zhang, Q.; Yang, X.; Xing, H.; Li, F.; Chen, G.; Zhou, J.; Wang, S.; Xu, G.; et al. Hypoxia induced paclitaxel resistance in human ovarian cancers via hypoxia-inducible factor 1alpha. J. Cancer Res. Clin. Oncol. 2010, 136, 447–456. [Google Scholar] [CrossRef]
- Lee, H.Y.; Lee, T.; Lee, N.; Yang, E.G.; Lee, C.; Lee, J.; Moon, E.Y.; Ha, J.; Park, H. Src activates HIF-1alpha not through direct phosphorylation of HIF-1alpha specific prolyl-4 hydroxylase 2 but through activation of the NADPH oxidase/Rac pathway. Carcinogenesis 2011, 32, 703–712. [Google Scholar] [CrossRef][Green Version]
- Guo, Q.; Lu, L.; Liao, Y.; Wang, X.; Zhang, Y.; Liu, Y.; Huang, S.; Sun, H.; Li, Z.; Zhao, L. Influence of c-Src on hypoxic resistance to paclitaxel in human ovarian cancer cells and reversal of FV-429. Cell Death Dis. 2018, 8, e3178. [Google Scholar] [CrossRef]
- Xie, Z.; Cao, L.; Zhang, J. miR-21 modulates paclitaxel sensitivity and hypoxia-inducible factor-1alpha expression in human ovarian cancer cells. Oncol. Lett. 2013, 6, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Parmakhtiar, B.; Burger, R.A.; Kim, J.H.; Fruehauf, J.P. HIF Inactivation of p53 in ovarian cancer can be reversed by topotecan, restoring cisplatin and paclitaxel sensitivity. Mol. Cancer Res. 2019, 17, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Raffeld, M.; Juwara, L.; Horneffer, Y.; Strassberger, A.; Allen, D.; Steinberg, S.M.; Rapisarda, A.; Spencer, S.D.; Figg, W.D.; et al. Multihistology, target-driven pilot trial of oral topotecan as an inhibitor of hypoxia-inducible factor-1alpha in advanced solid tumors. Clin. Cancer Res. 2011, 17, 5123–5131. [Google Scholar] [CrossRef]
- Sehouli, J.; Chekerov, R.; Reinthaller, A.; Richter, R.; Gonzalez-Martin, A.; Harter, P.; Woopen, H.; Petru, E.; Hanker, L.C.; Keil, E.; et al. Topotecan plus carboplatin versus standard therapy with paclitaxel plus carboplatin (PC) or gemcitabine plus carboplatin (GC) or pegylated liposomal doxorubicin plus carboplatin (PLDC): A randomized phase III trial of the NOGGO-AGO-Study Group-AGO Austria and GEICO-ENGOT-GCIG intergroup study (HECTOR). Ann. Oncol. Off. J. Eur. Soc. Med Oncol. ESMO 2016, 27, 2236–2241. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Sartor, O.; Halstead, M.; Katz, L. Improving outcomes with recent advances in chemotherapy for castrate-resistant prostate cancer. Clin. Genitourin. Cancer 2010, 8, 23–28. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, phase III study of androgen deprivation therapy with enzalutamide or placebo in men with metastatic hormone-sensitive prostate cancer. J. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef]
- Scott, L.J. Enzalutamide: A review in castration-resistant prostate cancer. Drugs 2018, 78, 1913–1924. [Google Scholar] [CrossRef]
- Mostaghel, E.A. Abiraterone in the treatment of metastatic castration-resistant prostate cancer. Cancer Manag. Res. 2014, 6, 39–51. [Google Scholar] [CrossRef][Green Version]
- Rathkopf, D.; Scher, H.I. Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J. 2013, 19, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Clegg, N.J.; Scher, H.I. Anti-androgens and androgen-depleting therapies in prostate cancer: New agents for an established target. Lancet. Oncol. 2009, 10, 981–991. [Google Scholar] [CrossRef]
- Lohiya, V.; Aragon-Ching, J.B.; Sonpavde, G. Role of chemotherapy and mechanisms of resistance to chemotherapy in metastatic castration-resistant prostate cancer. Clin. Med. Insights Oncol. 2016, 10, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.T.; Chen, S.; He, H.H. Landscape of noncoding RNA in prostate cancer. Trends Genet. 2019, 35, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Kopczynska, E. Role of microRNAs in the resistance of prostate cancer to docetaxel and paclitaxel. Contemp. Oncol. 2015, 19, 423–427. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, C.; Nadiminty, N.; Lou, W.; Tummala, R.; Evans, C.P.; Gao, A.C. Inhibition of ABCB1 expression overcomes acquired docetaxel resistance in prostate cancer. Mol. Cancer Ther. 2013, 12, 1829–1836. [Google Scholar] [CrossRef]
- Lombard, A.P.; Liu, C.; Armstrong, C.M.; Cucchiara, V.; Gu, X.; Lou, W.; Evans, C.P.; Gao, A.C. ABCB1 mediates cabazitaxel-docetaxel cross-resistance in advanced prostate cancer. Mol. Cancer Ther. 2017, 16, 2257–2266. [Google Scholar] [CrossRef]
- O’Neill, A.J.; Prencipe, M.; Dowling, C.; Fan, Y.; Mulrane, L.; Gallagher, W.M.; O’Connor, D.; O’Connor, R.; Devery, A.; Corcoran, C.; et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol. Cancer 2011, 10, 126. [Google Scholar] [CrossRef]
- Saupe, M.; Rauschenberger, L.; Preuss, M.; Oswald, S.; Fussek, S.; Zimmermann, U.; Walther, R.; Knabbe, C.; Burchardt, M.; Stope, M.B. Differential expression of the multidrug resistance 1 (MDR1) protein in prostate cancer cells is independent from anticancer drug treatment and Y box binding protein 1 (YB-1) activity. World J. Urol. 2015, 33, 1481–1486. [Google Scholar] [CrossRef]
- Oprea-Lager, D.E.; Bijnsdorp, I.V.; Reindert J, V.A.N.M.; Alfons J, V.D.E.; Hoekstra, O.S.; Geldof, A.A. ABCC4 decreases docetaxel and not cabazitaxel efficacy in prostate cancer cells in vitro. Anticancer Res. 2013, 33, 387–391. [Google Scholar]
- Hour, T.C.; Chung, S.D.; Kang, W.Y.; Lin, Y.C.; Chuang, S.J.; Huang, A.M.; Wu, W.J.; Huang, S.P.; Huang, C.Y.; Pu, Y.S. EGFR mediates docetaxel resistance in human castration-resistant prostate cancer through the Akt-dependent expression of ABCB1 (MDR1). Arch. Toxicol. 2015, 89, 591–605. [Google Scholar] [CrossRef]
- Li, L.; Wei, X.H.; Pan, Y.P.; Li, H.C.; Yang, H.; He, Q.H.; Pang, Y.; Shan, Y.; Xiong, F.X.; Shao, G.Z.; et al. LAPTM4B: A novel cancer-associated gene motivates multidrug resistance through efflux and activating PI3K/AKT signaling. Oncogene 2010, 29, 5785–5795. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert. Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef]
- Lin, J.Z.; Hameed, I.; Xu, Z.; Yu, Y.; Ren, Z.Y.; Zhu, J.G. Efficacy of gefitinibcelecoxib combination therapy in docetaxelresistant prostate cancer. Oncol. Rep. 2018, 40, 2242–2250. [Google Scholar] [CrossRef]
- Kato, T.; Fujita, Y.; Nakane, K.; Kojima, T.; Nozawa, Y.; Deguchi, T.; Ito, M. ETS1 promotes chemoresistance and invasion of paclitaxel-resistant, hormone-refractory PC3 prostate cancer cells by up-regulating MDR1 and MMP9 expression. Biochem. Biophys. Res. Commun. 2012, 417, 966–971. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Z.; Chen, C.Z.; Liu, C.; Evans, C.P.; Gao, A.C.; Zhou, F.; Chen, H.W. Therapeutic targeting of MDR1 expression by RORgamma antagonists resensitizes cross-resistant CRPC to taxane via coordinated induction of cell death programs. Mol. Cancer Ther. 2020, 19, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Mizutani, K.; Kameyama, K.; Kawakami, K.; Fujita, Y.; Nakane, K.; Kanimoto, Y.; Ehara, H.; Ito, H.; Seishima, M.; et al. Serum exosomal P-glycoprotein is a potential marker to diagnose docetaxel resistance and select a taxoid for patients with prostate cancer. Urol. Oncol. 2015, 33, 385.e15–385.e20. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, C.; Rani, S.; O’Brien, K.; O’Neill, A.; Prencipe, M.; Sheikh, R.; Webb, G.; McDermott, R.; Watson, W.; Crown, J.; et al. Docetaxel-resistance in prostate cancer: Evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PLoS ONE 2012, 7, e50999. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, A.K.; Vogel, P.D.; Wise, J.G. Prolonged inhibition of P-glycoprotein after exposure to chemotherapeutics increases cell mortality in multidrug resistant cultured cancer cells. PLoS ONE 2019, 14, e0217940. [Google Scholar] [CrossRef]
- Sanchez, C.; Mercado, A.; Contreras, H.R.; Mendoza, P.; Cabezas, J.; Acevedo, C.; Huidobro, C.; Castellon, E.A. Chemotherapy sensitivity recovery of prostate cancer cells by functional inhibition and knock down of multidrug resistance proteins. Prostate 2011, 71, 1810–1817. [Google Scholar] [CrossRef]
- Toner, A.P.; McLaughlin, F.; Giles, F.J.; Sullivan, F.J.; O’Connell, E.; Carleton, L.A.; Breen, L.; Dunne, G.; Gorman, A.M.; Lewis, J.D.; et al. The novel toluidine sulphonamide EL102 shows pre-clinical in vitro and in vivo activity against prostate cancer and circumvents MDR1 resistance. Br. J. Cancer 2013, 109, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, C.; Armstrong, C.; Lou, W.; Sandher, A.; Gao, A.C. Antiandrogens Inhibit ABCB1 efflux and ATPase activity and reverse docetaxel resistance in advanced prostate cancer. Clin. Cancer Res. 2015, 21, 4133–4142. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Machioka, K.; Izumi, K.; Kadono, Y.; Iwamoto, H.; Naito, R.; Makino, T.; Kadomoto, S.; Natsagdorj, A.; Keller, E.T.; Zhang, J.; et al. Establishment and characterization of two cabazitaxel-resistant prostate cancer cell lines. Oncotarget 2018, 9, 16185–16196. [Google Scholar] [CrossRef]
- Natsagdorj, A.; Izumi, K.; Hiratsuka, K.; Machioka, K.; Iwamoto, H.; Naito, R.; Makino, T.; Kadomoto, S.; Shigehara, K.; Kadono, Y.; et al. CCL2 induces resistance to the antiproliferative effect of cabazitaxel in prostate cancer cells. Cancer Sci. 2019, 110, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Njiaju, U.O.; Gamazon, E.R.; Gorsic, L.K.; Delaney, S.M.; Wheeler, H.E.; Im, H.K.; Dolan, M.E. Whole-genome studies identify solute carrier transporters in cellular susceptibility to paclitaxel. Pharm. Genom. 2012, 22, 498–507. [Google Scholar] [CrossRef]
- Sun, R.; Ying, Y.; Tang, Z.; Liu, T.; Shi, F.; Li, H.; Guo, T.; Huang, S.; Lai, R. The emerging role of the SLCO1B3 protein in cancer resistance. Protein Pept. Lett. 2020, 27, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Montgomery, R.B.; Mostaghel, E.A. Minireview: SLCO and ABC transporters: A role for steroid transport in prostate cancer progression. Endocrinology 2014, 155, 4124–4132. [Google Scholar] [CrossRef]
- Iusuf, D.; Hendrikx, J.J.; van Esch, A.; van de Steeg, E.; Wagenaar, E.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. Human OATP1B1, OATP1B3 and OATP1A2 can mediate the in vivo uptake and clearance of docetaxel. Int. J. Cancer 2015, 136, 225–233. [Google Scholar] [CrossRef]
- Schulte, R.R.; Ho, R.H. Organic anion transporting polypeptides: Emerging roles in cancer pharmacology. Mol. Pharmacol. 2019, 95, 490–506. [Google Scholar] [CrossRef]
- Lee, H.H.; Leake, B.F.; Teft, W.; Tirona, R.G.; Kim, R.B.; Ho, R.H. Contribution of hepatic organic anion-transporting polypeptides to docetaxel uptake and clearance. Mol. Cancer Ther. 2015, 14, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- de Morree, E.S.; Bottcher, R.; van Soest, R.J.; Aghai, A.; de Ridder, C.M.; Gibson, A.A.; Mathijssen, R.H.; Burger, H.; Wiemer, E.A.; Sparreboom, A.; et al. Loss of SLCO1B3 drives taxane resistance in prostate cancer. Br. J. Cancer 2016, 115, 674–681. [Google Scholar] [CrossRef] [PubMed]
- de Morree, E.; van Soest, R.; Aghai, A.; de Ridder, C.; de Bruijn, P.; Ghobadi Moghaddam-Helmantel, I.; Burger, H.; Mathijssen, R.; Wiemer, E.; de Wit, R.; et al. Understanding taxanes in prostate cancer; importance of intratumoral drug accumulation. Prostate 2016, 76, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Ushio, K.; Nishiwaki, M.; Kouno, J.; Araki, H.; Hikichi, Y.; Hattori, M.; Imai, Y.; Yamaoka, M. A mutation in beta-tubulin and a sustained dependence on androgen receptor signalling in a newly established docetaxel-resistant prostate cancer cell line. Cell Biol. Int. 2010, 34, 177–184. [Google Scholar] [CrossRef]
- Fanale, D.; Bronte, G.; Passiglia, F.; Calo, V.; Castiglia, M.; Di Piazza, F.; Barraco, N.; Cangemi, A.; Catarella, M.T.; Insalaco, L.; et al. Stabilizing versus destabilizing the microtubules: A double-edge sword for an effective cancer treatment option? Anal. Cell Pathol. 2015, 2015, 690916. [Google Scholar] [CrossRef]
- Ploussard, G.; Terry, S.; Maille, P.; Allory, Y.; Sirab, N.; Kheuang, L.; Soyeux, P.; Nicolaiew, N.; Coppolani, E.; Paule, B.; et al. Class III beta-tubulin expression predicts prostate tumor aggressiveness and patient response to docetaxel-based chemotherapy. Cancer Res. 2010, 70, 9253–9264. [Google Scholar] [CrossRef]
- Maahs, L.; Sanchez, B.E.; Gupta, N.; Van Harn, M.; Barrack, E.R.; Reddy, P.V.; Hwang, C. Class III beta-tubulin expression as a predictor of docetaxel-resistance in metastatic castration-resistant prostate cancer. PLoS ONE 2019, 14, e0222510. [Google Scholar] [CrossRef]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4, 153. [Google Scholar] [CrossRef]
- Kim, M.; Rejniak, K.A. Mechanical aspects of microtubule bundling in taxane-treated circulating tumor cells. Biophys. J. 2014, 107, 1236–1246. [Google Scholar] [CrossRef]
- Gjyrezi, A.; Xie, F.; Voznesensky, O.; Khanna, P.; Calagua, C.; Bai, Y.; Kung, J.; Wu, J.; Corey, E.; Montgomery, B.; et al. Taxane resistance in prostate cancer is mediated by decreased drug-target engagement. J. Clin. Investig. 2020, 130, 3287–3298. [Google Scholar] [CrossRef] [PubMed]
- Sekino, Y.; Han, X.; Kawaguchi, T.; Babasaki, T.; Goto, K.; Inoue, S.; Hayashi, T.; Teishima, J.; Shiota, M.; Yasui, W.; et al. TUBB3 reverses resistance to docetaxel and cabazitaxel in prostate cancer. Int. J. Mol. Sci. 2019, 20, 3936. [Google Scholar] [CrossRef] [PubMed]
- Smiyun, G.; Azarenko, O.; Miller, H.; Rifkind, A.; LaPointe, N.E.; Wilson, L.; Jordan, M.A. betaIII-tubulin enhances efficacy of cabazitaxel as compared with docetaxel. Cancer Chemother. Pharmacol. 2017, 80, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhang, C.; Lu, X.; Song, C.; Wang, C.; Zhang, M.; Xie, Y.; Schaefer, H.F., 3rd. Binding modes of cabazitaxel with the different human beta-tubulin isotypes: DFT and MD studies. J. Mol. Model. 2020, 26, 162. [Google Scholar] [CrossRef]
- Eder, I.E.; Culig, Z.; Putz, T.; Nessler-Menardi, C.; Bartsch, G.; Klocker, H. Molecular biology of the androgen receptor: From molecular understanding to the clinic. Eur. Urol. 2001, 40, 241–251. [Google Scholar] [CrossRef]
- Jiang, J.; Huang, H. Targeting the androgen receptor by taxol in castration-resistant prostate cancer. Mol. Cell Pharmacol. 2010, 2, 1–5. [Google Scholar]
- Augello, M.A.; Den, R.B.; Knudsen, K.E. AR function in promoting metastatic prostate cancer. Cancer Metastasis Rev. 2014, 33, 399–411. [Google Scholar] [CrossRef]
- Darshan, M.S.; Loftus, M.S.; Thadani-Mulero, M.; Levy, B.P.; Escuin, D.; Zhou, X.K.; Gjyrezi, A.; Chanel-Vos, C.; Shen, R.; Tagawa, S.T.; et al. Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res. 2011, 71, 6019–6029. [Google Scholar] [CrossRef]
- Zhu, M.L.; Horbinski, C.M.; Garzotto, M.; Qian, D.Z.; Beer, T.M.; Kyprianou, N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. 2010, 70, 7992–8002. [Google Scholar] [CrossRef]
- Thadani-Mulero, M.; Nanus, D.M.; Giannakakou, P. Androgen receptor on the move: Boarding the microtubule expressway to the nucleus. Cancer Res. 2012, 72, 4611–4615. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Tagawa, S.T.; Galletti, G.; Worroll, D.; Ballman, K.; Vanhuyse, M.; Sonpavde, G.; North, S.; Albany, C.; Tsao, C.K.; et al. Randomized, noncomparative, phase II trial of early switch from docetaxel to cabazitaxel or vice versa, with integrated biomarker analysis, in men with chemotherapy-naive, metastatic, castration-resistant prostate cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3181–3188. [Google Scholar] [CrossRef] [PubMed]
- Komura, K.; Jeong, S.H.; Hinohara, K.; Qu, F.; Wang, X.; Hiraki, M.; Azuma, H.; Lee, G.S.; Kantoff, P.W.; Sweeney, C.J. Resistance to docetaxel in prostate cancer is associated with androgen receptor activation and loss of KDM5D expression. Proc. Natl. Acad. Sci. USA 2016, 113, 6259–6264. [Google Scholar] [CrossRef] [PubMed]
- Thadani-Mulero, M.; Portella, L.; Sun, S.; Sung, M.; Matov, A.; Vessella, R.L.; Corey, E.; Nanus, D.M.; Plymate, S.R.; Giannakakou, P. Androgen receptor splice variants determine taxane sensitivity in prostate cancer. Cancer Res. 2014, 74, 2270–2282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, X.; Li, J.; Ledet, E.; Alvarez, X.; Qi, Y.; Fu, X.; Sartor, O.; Dong, Y.; Zhang, H. Androgen receptor splice variants circumvent AR blockade by microtubule-targeting agents. Oncotarget 2015, 6, 23358–23371. [Google Scholar] [CrossRef]
- Nakazawa, M.; Lu, C.; Chen, Y.; Paller, C.J.; Carducci, M.A.; Eisenberger, M.A.; Luo, J.; Antonarakis, E.S. Serial blood-based analysis of AR-V7 in men with advanced prostate cancer. Ann. Oncol. 2015, 26, 1859–1865. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar] [CrossRef]
- Shimizu, Y.; Tamada, S.; Kato, M.; Hirayama, Y.; Takeyama, Y.; Iguchi, T.; Sadar, M.D.; Nakatani, T. Androgen receptor splice variant 7 drives the growth of castration resistant prostate cancer without being involved in the efficacy of taxane chemotherapy. J. Clin. Med. 2018, 7, 444. [Google Scholar] [CrossRef]
- Davey, R.A.; Grossmann, M. Androgen receptor structure, function and biology: From bench to bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Galletti, G.; Matov, A.; Beltran, H.; Fontugne, J.; Miguel Mosquera, J.; Cheung, C.; MacDonald, T.Y.; Sung, M.; O’Toole, S.; Kench, J.G.; et al. ERG induces taxane resistance in castration-resistant prostate cancer. Nat. Commun. 2014, 5, 5548. [Google Scholar] [CrossRef]
- Reig, O.; Marin-Aguilera, M.; Carrera, G.; Jimenez, N.; Pare, L.; Garcia-Recio, S.; Gaba, L.; Pereira, M.V.; Fernandez, P.; Prat, A.; et al. TMPRSS2-ERG in blood and docetaxel resistance in metastatic castration-resistant prostate cancer. Eur. Urol. 2016, 70, 709–713. [Google Scholar] [CrossRef]
- Yu, J.; Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Marin-Aguilera, M.; Reig, O.; Mila-Guasch, M.; Font, A.; Domenech, M.; Rodriguez-Vida, A.; Carles, J.; Suarez, C.; Del Alba, A.G.; Jimenez, N.; et al. The influence of treatment sequence in the prognostic value of TMPRSS2-ERG as biomarker of taxane resistance in castration-resistant prostate cancer. Int. J. Cancer 2019, 145, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Qiu, Y. Current opinion and mechanistic interpretation of combination therapy for castration-resistant prostate cancer. Asian J. Androl. 2019, 21, 270–278. [Google Scholar] [CrossRef]
- de Bono, J.S.; Chowdhury, S.; Feyerabend, S.; Elliott, T.; Grande, E.; Melhem-Bertrandt, A.; Baron, B.; Hirmand, M.; Werbrouck, P.; Fizazi, K. Antitumour activity and safety of enzalutamide in patients with metastatic castration-resistant prostate cancer previously treated with abiraterone acetate plus prednisone for >/=24 weeks in Europe. Eur. Urol. 2018, 74, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Mateo, J.; Loriot, Y.; Pezaro, C.; Albiges, L.; Mehra, N.; Varga, A.; Bianchini, D.; Ryan, C.J.; Petrylak, D.P.; et al. Phase I/II trial of cabazitaxel plus abiraterone in patients with metastatic castration-resistant prostate cancer (mCRPC) progressing after docetaxel and abiraterone. Ann. Oncol. 2017, 28, 90–95. [Google Scholar] [CrossRef]
- Komura, K.; Sweeney, C.J.; Inamoto, T.; Ibuki, N.; Azuma, H.; Kantoff, P.W. Current treatment strategies for advanced prostate cancer. Int. J. Urol. 2018, 25, 220–231. [Google Scholar] [CrossRef]
- Linder, S.; van der Poel, H.G.; Bergman, A.M.; Zwart, W.; Prekovic, S. Enzalutamide therapy for advanced prostate cancer: Efficacy, resistance and beyond. Endocr. Relat. Cancer 2018, 26, R31–R52. [Google Scholar] [CrossRef]
- Massard, C.; Fizazi, K. Targeting continued androgen receptor signaling in prostate cancer. Clin. Cancer Res. 2011, 17, 3876–3883. [Google Scholar] [CrossRef]
- Shiota, M.; Dejima, T.; Yamamoto, Y.; Takeuchi, A.; Imada, K.; Kashiwagi, E.; Inokuchi, J.; Tatsugami, K.; Kajioka, S.; Uchiumi, T.; et al. Collateral resistance to taxanes in enzalutamide-resistant prostate cancer through aberrant androgen receptor and its variants. Cancer Sci. 2018, 109, 3224–3234. [Google Scholar] [CrossRef]
- van Soest, R.J.; de Morree, E.S.; Kweldam, C.F.; de Ridder, C.M.A.; Wiemer, E.A.C.; Mathijssen, R.H.J.; de Wit, R.; van Weerden, W.M. Targeting the androgen receptor confers in vivo cross-resistance between enzalutamide and docetaxel, but not cabazitaxel, in castration-resistant prostate cancer. Eur. Urol. 2015, 67, 981–985. [Google Scholar] [CrossRef]
- Lombard, A.P.; Liu, L.; Cucchiara, V.; Liu, C.; Armstrong, C.M.; Zhao, R.; Yang, J.C.; Lou, W.; Evans, C.P.; Gao, A.C. Intra versus inter cross-resistance determines treatment sequence between taxane and AR-targeting therapies in advanced prostate cancer. Mol. Cancer Ther. 2018, 17, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Al Nakouzi, N.; Le Moulec, S.; Albiges, L.; Wang, C.; Beuzeboc, P.; Gross-Goupil, M.; de La Motte Rouge, T.; Guillot, A.; Gajda, D.; Massard, C.; et al. Cabazitaxel remains active in patients progressing after docetaxel followed by novel androgen receptor pathway targeted therapies. Eur. Urol. 2015, 68, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Pezaro, C.J.; Omlin, A.G.; Altavilla, A.; Lorente, D.; Ferraldeschi, R.; Bianchini, D.; Dearnaley, D.; Parker, C.; de Bono, J.S.; Attard, G. Activity of cabazitaxel in castration-resistant prostate cancer progressing after docetaxel and next-generation endocrine agents. Eur. Urol. 2014, 66, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Dicken, H.; Hensley, P.J.; Kyprianou, N. Prostate tumor neuroendocrine differentiation via EMT: The road less traveled. Asian J. Urol. 2019, 6, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef]
- Seruga, B.; Ocana, A.; Tannock, I.F. Drug resistance in metastatic castration-resistant prostate cancer. Nat. Rev. Clin. Oncol. 2011, 8, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Marin-Aguilera, M.; Codony-Servat, J.; Reig, O.; Lozano, J.J.; Fernandez, P.L.; Pereira, M.V.; Jimenez, N.; Donovan, M.; Puig, P.; Mengual, L.; et al. Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol. Cancer Ther. 2014, 13, 1270–1284. [Google Scholar] [CrossRef]
- Roche, J. The Epithelial-to-mesenchymal transition in cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Schafer, G.; Erb, H.H.; Oh, S.J.; Klocker, H.; Heidegger, I.; Neuwirt, H.; Culig, Z. Epithelial-to-mesenchymal transition leads to docetaxel resistance in prostate cancer and is mediated by reduced expression of miR-200c and miR-205. Am. J. Pathol. 2012, 181, 2188–2201. [Google Scholar] [CrossRef]
- Hanrahan, K.; O’Neill, A.; Prencipe, M.; Bugler, J.; Murphy, L.; Fabre, A.; Puhr, M.; Culig, Z.; Murphy, K.; Watson, R.W. The role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Mol. Oncol. 2017, 11, 251–265. [Google Scholar] [CrossRef]
- Nakazawa, M.; Kyprianou, N. Epithelial-mesenchymal-transition regulators in prostate cancer: Androgens and beyond. J. Steroid Biochem. Mol. Biol. 2017, 166, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Johansson, S.L.; Batra, S.K. Marked improvement of cytotoxic effects induced by docetaxel on highly metastatic and androgen-independent prostate cancer cells by downregulating macrophage inhibitory cytokine-1. Br. J. Cancer 2013, 108, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Paller, C.; Pu, H.; Begemann, D.E.; Wade, C.A.; Hensley, P.J.; Kyprianou, N. TGF-beta receptor I inhibitor enhances response to enzalutamide in a pre-clinical model of advanced prostate cancer. Prostate 2019, 79, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.L.; Kyprianou, N. Role of androgens and the androgen receptor in epithelial-mesenchymal transition and invasion of prostate cancer cells. FASEB J. 2010, 24, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Pu, H.; Penticuff, J.C.; Cao, Z.; Horbinski, C.; Kyprianou, N. Multinucleation and mesenchymal-to-epithelial transition alleviate resistance to combined cabazitaxel and antiandrogen therapy in advanced prostate cancer. Cancer Res. 2016, 76, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.H.; Placzek, W.J. Regulating the BCL2 family to improve sensitivity to microtubule targeting agents. Cells 2019, 8, 346. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P. BH3 mimetics for the treatment of prostate cancer. Front. Pharmacol. 2017, 8, 557. [Google Scholar] [CrossRef]
- Tamaki, H.; Harashima, N.; Hiraki, M.; Arichi, N.; Nishimura, N.; Shiina, H.; Naora, K.; Harada, M. Bcl-2 family inhibition sensitizes human prostate cancer cells to docetaxel and promotes unexpected apoptosis under caspase-9 inhibition. Oncotarget 2014, 5, 11399–11412. [Google Scholar] [CrossRef]
- Karnak, D.; Xu, L. Chemosensitization of prostate cancer by modulating Bcl-2 family proteins. Curr. Drug Targets 2010, 11, 699–707. [Google Scholar] [CrossRef][Green Version]
- Asmarinah, A.; Paradowska-Dogan, A.; Kodariah, R.; Tanuhardja, B.; Waliszewski, P.; Mochtar, C.A.; Weidner, W.; Hinsch, E. Expression of the Bcl-2 family genes and complexes involved in the mitochondrial transport in prostate cancer cells. Int. J. Oncol. 2014, 45, 1489–1496. [Google Scholar] [CrossRef]
- Calastretti, A.; Gatti, G.; Quaresmini, C.; Bevilacqua, A. Down-modulation of Bcl-2 sensitizes PTEN-mutated prostate cancer cells to starvation and taxanes. Prostate 2014, 74, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Jonas, O.; Whitman, M.A.; Corey, E.; Balk, S.P.; Chen, S. Tyrosine kinase inhibitors increase MCL1 degradation and in combination with BCLXL/BCL2 inhibitors drive prostate cancer apoptosis. Clin. Cancer Res. 2018, 24, 5458–5470. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Huang, S.B.; Yang, M.C.; Lin, Y.T.; Chu, I.H.; Shen, Y.N.; Chiu, Y.H.; Hung, S.H.; Kang, L.; Hong, Y.R.; et al. Combining paclitaxel with ABT-263 has a synergistic effect on paclitaxel resistant prostate cancer cells. PLoS ONE 2015, 10, e0120913. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Tanaka, Y.; Tamaki, H.; Harada, M. Docetaxel induces Bcl-2- and pro-apoptotic caspase-independent death of human prostate cancer DU145 cells. Int. J. Oncol. 2016, 48, 2330–2338. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.F.; Zhang, Y.C.; Peng, X.Q.; Long, Z.; Ming, Y.Z.; He, L.Y. Silencing Notch-1 induces apoptosis and increases the chemosensitivity of prostate cancer cells to docetaxel through Bcl-2 and Bax. Oncol. Lett. 2012, 3, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Parrondo, R.; de Las Pozas, A.; Reiner, T.; Perez-Stable, C. ABT-737, a small molecule Bcl-2/Bcl-xL antagonist, increases antimitotic-mediated apoptosis in human prostate cancer cells. PeerJ 2013, 1, e144. [Google Scholar] [CrossRef]
- Perez-Stable, C. 2-Methoxyestradiol and paclitaxel have similar effects on the cell cycle and induction of apoptosis in prostate cancer cells. Cancer Lett. 2006, 231, 49–64. [Google Scholar] [CrossRef]
- Terrano, D.T.; Upreti, M.; Chambers, T.C. Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol. Cell Biol. 2010, 30, 640–656. [Google Scholar] [CrossRef]
- Sonpavde, G.; Matveev, V.; Burke, J.M.; Caton, J.R.; Fleming, M.T.; Hutson, T.E.; Galsky, M.D.; Berry, W.R.; Karlov, P.; Holmlund, J.T.; et al. Randomized phase II trial of docetaxel plus prednisone in combination with placebo or AT-101, an oral small molecule Bcl-2 family antagonist, as first-line therapy for metastatic castration-resistant prostate cancer. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. ESMO 2012, 23, 1803–1808. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Papa, A.; Pandolfi, P.P. The PTEN(-)PI3K axis in cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yang, F.; Chen, D.; Zhao, Q.; Chen, D.; Ping, H.; Xing, N. Quercetin reverses docetaxel resistance in prostate cancer via androgen receptor and PI3K/Akt signaling pathways. Int. J. Biol. Sci. 2020, 16, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Bitting, R.L.; Armstrong, A.J. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr. Relat. Cancer 2013, 20, R83–R99. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.M.; Koreckij, T.D.; Corey, E. Targeted therapy for advanced prostate cancer: Inhibition of the PI3K/Akt/mTOR pathway. Curr. Cancer Drug Targets 2009, 9, 237–249. [Google Scholar] [CrossRef]
- Drake, J.M.; Huang, J. PIP5K1alpha inhibition as a therapeutic strategy for prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 12578–12579. [Google Scholar] [CrossRef]
- Avan, A.; Narayan, R.; Giovannetti, E.; Peters, G.J. Role of Akt signaling in resistance to DNA-targeted therapy. World J. Clin. Oncol. 2016, 7, 352–369. [Google Scholar] [CrossRef]
- Gan, Y.; Chen, Q.; Lei, Y. Regulation of paclitaxel sensitivity in prostate cancer cells by PTEN/maspin signaling. Oncol. Lett. 2017, 14, 4977–4982. [Google Scholar] [CrossRef][Green Version]
- Ward, A.B.; Mir, H.; Kapur, N.; Gales, D.N.; Carriere, P.P.; Singh, S. Quercetin inhibits prostate cancer by attenuating cell survival and inhibiting anti-apoptotic pathways. World J. Surg. Oncol. 2018, 16, 108. [Google Scholar] [CrossRef]
- Kosaka, T.; Miyajima, A.; Shirotake, S.; Suzuki, E.; Kikuchi, E.; Oya, M. Long-term androgen ablation and docetaxel up-regulate phosphorylated Akt in castration resistant prostate cancer. J. Urol. 2011, 185, 2376–2381. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.Z.; Rademacher, B.L.; Pittsenbarger, J.; Huang, C.Y.; Myrthue, A.; Higano, C.S.; Garzotto, M.; Nelson, P.S.; Beer, T.M. CCL2 is induced by chemotherapy and protects prostate cancer cells from docetaxel-induced cytotoxicity. Prostate 2010, 70, 433–442. [Google Scholar] [CrossRef]
- Yasumizu, Y.; Miyajima, A.; Kosaka, T.; Miyazaki, Y.; Kikuchi, E.; Oya, M. Dual PI3K/mTOR inhibitor NVP-BEZ235 sensitizes docetaxel in castration resistant prostate cancer. J. Urol. 2014, 191, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Manola, J.B.; Elfiky, A.A.; Ross, R.; Oh, W.K.; Yap, J.T.; Van den Abbeele, A.D.; Ryan, C.W.; Beer, T.M.; Loda, M.; et al. A phase I study of everolimus and docetaxel in patients with castration-resistant prostate cancer. Clin. Genitourin. Cancer 2015, 13, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Lo, U.G.; Yang, D.; Hsieh, J.T. The role of microRNAs in prostate cancer progression. Transl. Androl. Urol. 2013, 2, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Labbe, M.; Hoey, C.; Ray, J.; Potiron, V.; Supiot, S.; Liu, S.K.; Fradin, D. microRNAs identified in prostate cancer: Correlative studies on response to ionizing radiation. Mol. Cancer 2020, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.M.; Nikolic, I.; Yang, J.; Castillo, L.; Deng, N.; Chan, C.L.; Yeung, N.K.; Dodson, E.; Elsworth, B.; Spielman, C.; et al. MicroRNAs as potential therapeutics to enhance chemosensitivity in advanced prostate cancer. Sci. Rep. 2018, 8, 7820. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.M.; Castillo, L.; Mahon, K.L.; Chiam, K.; Lee, B.Y.; Nguyen, Q.; Boyer, M.J.; Stockler, M.R.; Pavlakis, N.; Marx, G.; et al. Circulating microRNAs are associated with docetaxel chemotherapy outcome in castration-resistant prostate cancer. Br. J. Cancer 2014, 110, 2462–2471. [Google Scholar] [CrossRef]
- Li, F.; Mahato, R.I. MicroRNAs and drug resistance in prostate cancers. Mol. Pharm. 2014, 11, 2539–2552. [Google Scholar] [CrossRef]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenetics 2019, 11, 25. [Google Scholar] [CrossRef]
- Xu, B.; Niu, X.; Zhang, X.; Tao, J.; Wu, D.; Wang, Z.; Li, P.; Zhang, W.; Wu, H.; Feng, N.; et al. miR-143 decreases prostate cancer cells proliferation and migration and enhances their sensitivity to docetaxel through suppression of KRAS. Mol. Cell. Biochem. 2011, 350, 207–213. [Google Scholar] [CrossRef]
- Liu, X.; Luo, X.; Wu, Y.; Xia, D.; Chen, W.; Fang, Z.; Deng, J.; Hao, Y.; Yang, X.; Zhang, T.; et al. MicroRNA-34a attenuates paclitaxel resistance in prostate cancer cells via direct suppression of JAG1/Notch1 axis. Cell Physiol. Biochem. 2018, 50, 261–276. [Google Scholar] [CrossRef]
- Giacinti, S.; Poti, G.; Roberto, M.; Macrini, S.; Bassanelli, M.; Francesca, D.I.P.; Aschelter, A.M.; Ceribelli, A.; Ruggeri, E.M.; Marchetti, P. Molecular Basis of Drug Resistance and Insights for New Treatment Approaches in mCRPC. Anticancer Res. 2018, 38, 6029–6039. [Google Scholar] [CrossRef]
- Shi, G.H.; Ye, D.W.; Yao, X.D.; Zhang, S.L.; Dai, B.; Zhang, H.L.; Shen, Y.J.; Zhu, Y.; Zhu, Y.P.; Xiao, W.J.; et al. Involvement of microRNA-21 in mediating chemo-resistance to docetaxel in androgen-independent prostate cancer PC3 cells. Acta Pharmacol. Sin. 2010, 31, 867–873. [Google Scholar] [CrossRef]
- Wang, L.; Cho, K.B.; Li, Y.; Tao, G.; Xie, Z.; Guo, B. Long noncoding RNA (lncRNA)-Mediated competing endogenous RNA networks provide novel potential biomarkers and therapeutic targets for colorectal cancer. Int. J. Mol. Sci. 2019, 20, 5758. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Gao, L.; Ma, X.; Huang, J.J.; Chen, J.; Zeng, L.; Ashby, C.R., J.; Zou, C.; Chen, Z.S. Long non-coding RNAs regulate drug resistance in cancer. Mol. Cancer 2020, 19, 54. [Google Scholar] [CrossRef] [PubMed]
- Corra, F.; Agnoletto, C.; Minotti, L.; Baldassari, F.; Volinia, S. The network of non-coding RNAs in cancer drug resistance. Front. Oncol. 2018, 8, 327. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Guo, S.; Zhang, Y.; Zhao, Y.; Li, X.; Jia, Y.; Xu, Y.; Ma, B. LncRNA NEAT1 promotes docetaxel resistance in prostate cancer by regulating ACSL4 via sponging miR-34a-5p and miR-204-5p. Cell. Signal. 2020, 65, 109422. [Google Scholar] [CrossRef]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lin, J.F.; Tsai, T.F.; Chou, K.Y.; Chen, H.E.; Hwang, T.I. Tumor suppressor miRNA-204-5p promotes apoptosis by targeting BCL2 in prostate cancer cells. Asian J. Surg. 2017, 40, 396–406. [Google Scholar] [CrossRef]
- He, J.; Sun, M.; Geng, H.; Tian, S. Long non-coding RNA Linc00518 promotes paclitaxel resistance of the human prostate cancer by sequestering miR-216b-5p. Biol. Cell 2019, 111, 39–50. [Google Scholar] [CrossRef]
- You, Y.; Tan, J.; Gong, Y.; Dai, H.; Chen, H.; Xu, X.; Yang, A.; Zhang, Y.; Bie, P. MicroRNA-216b-5p FUNCTIONS as a tumor-suppressive RNA by targeting TPT1 in pancreatic cancer cells. J. Cancer 2017, 8, 2854–2865. [Google Scholar] [CrossRef][Green Version]
- Cao, C.; Sun, G.; Liu, C. Long non-coding RNA SNHG6 regulates the sensitivity of prostate cancer cells to paclitaxel by sponging miR-186. Cancer Cell Int. 2020, 20, 381. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, M.S.; Chen, P.J.; Ren, Q.; Bai, P. miRNA-186 inhibits prostate cancer cell proliferation and tumor growth by targeting YY1 and CDK6. Exp. Ther. Med. 2017, 13, 3309–3314. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Liu, C.; Wang, C.; Ling, Z.; Wang, Y.; Wang, Y.; Zhang, M.; Chen, S.; Xu, B.; Guan, H.; et al. LncRNA CCAT1 promotes prostate cancer cell proliferation by interacting with DDX5 and MIR-28-5P. Mol. Cancer Ther. 2019, 18, 2469–2479. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Han, X.; Wei, P.; Yang, J.; Sun, J. Knockdown of lncRNA CCAT1 enhances sensitivity of paclitaxel in prostate cancer via regulating miR-24-3p and FSCN1. Cancer Biol. Ther. 2020, 21, 452–462. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, Y.; Du, L.; Li, J.; Qu, A.; Zhang, X.; Wang, L.; Wang, C. Down-regulation of miR-24-3p in colorectal cancer is associated with malignant behavior. Med. Oncol. 2015, 32, 362. [Google Scholar] [CrossRef]
- Shu, Y.; Ren, L.; Xie, B.; Liang, Z.; Chen, J. MiR-204 enhances mitochondrial apoptosis in doxorubicin-treated prostate cancer cells by targeting SIRT1/p53 pathway. Oncotarget 2017, 8, 97313–97322. [Google Scholar] [CrossRef]
- Wang, X.; Yang, B.; Ma, B. The UCA1/miR-204/Sirt1 axis modulates docetaxel sensitivity of prostate cancer cells. Cancer Chemother. Pharmacol. 2016, 78, 1025–1031. [Google Scholar] [CrossRef]
- Misawa, A.; Takayama, K.; Urano, T.; Inoue, S. Androgen-induced long noncoding RNA (lncRNA) SOCS2-AS1 promotes cell growth and inhibits apoptosis in prostate cancer cells. J. Biol. Chem. 2016, 291, 17861–17880. [Google Scholar] [CrossRef]
- Gu, P.; Chen, X.; Xie, R.; Han, J.; Xie, W.; Wang, B.; Dong, W.; Chen, C.; Yang, M.; Jiang, J.; et al. lncRNA HOXD-AS1 regulates proliferation and chemo-resistance of castration-resistant prostate cancer via recruiting WDR5. Mol. Ther. 2017, 25, 1959–1973. [Google Scholar] [CrossRef]
- Lu, K.; Tao, H.; Si, X.; Chen, Q. The histone H3 Lysine 4 presenter WDR5 as an oncogenic protein and novel epigenetic target in cancer. Front. Oncol. 2018, 8, 502. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y.; Zhang, X.; Huang, Q.; Diao, Y.; Yin, H.; Liu, H. Long non-coding RNA HOXD-AS1 in cancer. Clin. Chim. Acta 2018, 487, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Banerjee, T.; Vinckevicius, A.; Luo, Q.; Parker, J.B.; Baker, M.R.; Radhakrishnan, I.; Wei, J.J.; Barish, G.D.; Chakravarti, D. A role for WDR5 in integrating threonine 11 phosphorylation to lysine 4 methylation on histone H3 during androgen signaling and in prostate cancer. Mol. Cell 2014, 54, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Pucci, P.; Venalainen, E.; Alborelli, I.; Quagliata, L.; Hawkes, C.; Mather, R.; Romero, I.; Rigas, S.H.; Wang, Y.; Crea, F. LncRNA HORAS5 promotes taxane resistance in castration-resistant prostate cancer via a BCL2A1-dependent mechanism. Epigenomics 2020, 12, 1123–1138. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, W.K.; Xiao, L.; Kovac, S.; Chang, M.; Michiels, C.; Bolton, D.; Shulkes, A.; Baldwin, G.S.; Patel, O. The role of hypoxia-inducible factor 1alpha in determining the properties of castrate-resistant prostate cancers. PLoS ONE 2013, 8, e54251. [Google Scholar] [CrossRef]
- Carbonaro, M.; Escuin, D.; O’Brate, A.; Thadani-Mulero, M.; Giannakakou, P. Microtubules regulate hypoxia-inducible factor-1alpha protein trafficking and activity: Implications for taxane therapy. J. Biol. Chem. 2012, 287, 11859–11869. [Google Scholar] [CrossRef]
- Chan, Y.C.; Banerjee, J.; Choi, S.Y.; Sen, C.K. miR-210: The master hypoxamir. Microcirculation 2012, 19, 215–223. [Google Scholar] [CrossRef]
- Cheng, H.H.; Mitchell, P.S.; Kroh, E.M.; Dowell, A.E.; Chery, L.; Siddiqui, J.; Nelson, P.S.; Vessella, R.L.; Knudsen, B.S.; Chinnaiyan, A.M.; et al. Circulating microRNA profiling identifies a subset of metastatic prostate cancer patients with evidence of cancer-associated hypoxia. PLoS ONE 2013, 8, e69239. [Google Scholar] [CrossRef]
- Qian, J.; Shen, S.; Chen, W.; Chen, N. Propofol reversed hypoxia-induced docetaxel resistance in prostate cancer cells by preventing epithelial-mesenchymal transition by inhibiting hypoxia-inducible factor 1alpha. BioMed Res. Int. 2018, 2018, 4174232. [Google Scholar] [CrossRef]
- Forde, J.C.; Perry, A.S.; Brennan, K.; Martin, L.M.; Lawler, M.P.; Lynch, T.H.; Hollywood, D.; Marignol, L. Docetaxel maintains its cytotoxic activity under hypoxic conditions in prostate cancer cells. Urol. Oncol. 2012, 30, 912–919. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, J.D.; Wang, J.; Ahluwalia, D.; Baker, A.F.; Cranmer, L.D.; Ferraro, D.; Wang, Y.; Duan, J.X.; Ammons, W.S.; et al. TH-302, a hypoxia-activated prodrug with broad in vivo preclinical combination therapy efficacy: Optimization of dosing regimens and schedules. Cancer Chemother. Pharmacol. 2012, 69, 1487–1498. [Google Scholar] [CrossRef]
- Saggar, J.K.; Tannock, I.F. Activity of the hypoxia-activated pro-drug TH-302 in hypoxic and perivascular regions of solid tumors and its potential to enhance therapeutic effects of chemotherapy. Int. J. Cancer 2014, 134, 2726–2734. [Google Scholar] [CrossRef] [PubMed]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef] [PubMed]
- Sosa Iglesias, V.; Giuranno, L.; Dubois, L.J.; Theys, J.; Vooijs, M. Drug resistance in non-small cell lung cancer: A potential for NOTCH targeting? Front. Oncol. 2018, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Wang, R.; Chang, S.; Li, K.; Sun, R.; Wang, M.; Li, Z. Long non-coding RNA in drug resistance of non-small cell lung cancer: A mini review. Front. Pharmacol. 2019, 10, 1457. [Google Scholar] [CrossRef]
- Wang, X.C.; Tian, L.L.; Jiang, X.Y.; Wang, Y.Y.; Li, D.G.; She, Y.; Chang, J.H.; Meng, A.M. The expression and function of miRNA-451 in non-small cell lung cancer. Cancer Lett. 2011, 311, 203–209. [Google Scholar] [CrossRef]
- Huang, J.; Chen, Y.; Li, J.; Zhang, K.; Chen, J.; Chen, D.; Feng, B.; Song, H.; Feng, J.; Wang, R.; et al. Notch-1 confers chemoresistance in lung adenocarcinoma to taxanes through AP-1/microRNA-451 mediated regulation of MDR-1. Mol. Ther. Nucleic Acids 2016, 5, e375. [Google Scholar] [CrossRef]
- Dumontet, C.; Isaac, S.; Souquet, P.J.; Bejui-Thivolet, F.; Pacheco, Y.; Peloux, N.; Frankfurter, A.; Luduena, R.; Perol, M. Expression of class III beta tubulin in non-small cell lung cancer is correlated with resistance to taxane chemotherapy. Bull. Cancer 2005, 92, E25–E30. [Google Scholar]
- Li, Z.; Qing, Y.; Guan, W.; Li, M.; Peng, Y.; Zhang, S.; Xiong, Y.; Wang, D. Predictive value of APE1, BRCA1, ERCC1 and TUBB3 expression in patients with advanced non-small cell lung cancer (NSCLC) receiving first-line platinum-paclitaxel chemotherapy. Cancer Chemother. Pharmacol. 2014, 74, 777–786. [Google Scholar] [CrossRef]
- Della Corte, L.; Barra, F.; Foreste, V.; Giampaolino, P.; Evangelisti, G.; Ferrero, S.; Bifulco, G. Advances in paclitaxel combinations for treating cervical cancer. Expert Opin. Pharmacother. 2020, 21, 663–677. [Google Scholar] [CrossRef]
- Fan, Z.; Cui, H.; Yu, H.; Ji, Q.; Kang, L.; Han, B.; Wang, J.; Dong, Q.; Li, Y.; Yan, Z.; et al. MiR-125a promotes paclitaxel sensitivity in cervical cancer through altering STAT3 expression. Oncogenesis 2016, 5, e197. [Google Scholar] [CrossRef]
- Liu, J.J.; Ho, J.Y.; Lee, H.W.; Baik, M.W.; Kim, O.; Choi, Y.J.; Hur, S.Y. Inhibition of phosphatidylinositol 3-kinase (PI3K) signaling synergistically potentiates antitumor efficacy of paclitaxel and overcomes paclitaxel-mediated resistance in cervical cancer. Int. J. Mol. Sci. 2019, 20, 3383. [Google Scholar] [CrossRef] [PubMed]
- Scharer, C.D.; Laycock, N.; Osunkoya, A.O.; Logani, S.; McDonald, J.F.; Benigno, B.B.; Moreno, C.S. Aurora kinase inhibitors synergize with paclitaxel to induce apoptosis in ovarian cancer cells. J. Transl. Med. 2008, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Sato, N.; Yanai, K.; Akiyoshi, T.; Nagai, S.; Wada, J.; Koga, K.; Mibu, R.; Nakamura, M.; Katano, M. Enhancement of paclitaxel-induced apoptosis by inhibition of mitogen-activated protein kinase pathway in colon cancer cells. Anticancer Res. 2009, 29, 261–270. [Google Scholar] [PubMed]
- Peng, X.; Gong, F.; Chen, Y.; Jiang, Y.; Liu, J.; Yu, M.; Zhang, S.; Wang, M.; Xiao, G.; Liao, H. Autophagy promotes paclitaxel resistance of cervical cancer cells: Involvement of Warburg effect activated hypoxia-induced factor 1-alpha-mediated signaling. Cell Death Dis. 2014, 5, e1367. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Le Large, T.Y.S.; El Hassouni, B.; Funel, N.; Kok, B.; Piersma, S.R.; Pham, T.V.; Olive, K.P.; Kazemier, G.; van Laarhoven, H.W.M.; Jimenez, C.R.; et al. Proteomic analysis of gemcitabine-resistant pancreatic cancer cells reveals that microtubule-associated protein 2 upregulation associates with taxane treatment. Ther. Adv. Med. Oncol. 2019, 11, 1758835919841233. [Google Scholar] [CrossRef]
- Lemstrova, R.; Melichar, B.; Mohelnikova-Duchonova, B. Therapeutic potential of taxanes in the treatment of metastatic pancreatic cancer. Cancer Chemother. Pharmacol. 2016, 78, 1101–1111. [Google Scholar] [CrossRef]
- Dong, Q.G.; Sclabas, G.M.; Fujioka, S.; Schmidt, C.; Peng, B.; Wu, T.; Tsao, M.S.; Evans, D.B.; Abbruzzese, J.L.; McDonnell, T.J.; et al. The function of multiple IkappaB: NF-kappaB complexes in the resistance of cancer cells to Taxol-induced apoptosis. Oncogene 2002, 21, 6510–6519. [Google Scholar] [CrossRef]
- Li, Z.; Xuan, Z.; Chen, J.; Song, W.; Zhang, S.; Jin, C.; Zhou, M.; Zheng, S.; Song, P. Inhibiting the NF-kappaB pathway enhances the antitumor effect of cabazitaxel by downregulating Bcl-2 in pancreatic cancer. Int. J. Oncol. 2020, 57, 161–170. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, Z.; Zhang, M.; Gross, N.; Gong, L.; Zhang, S.; Lei, D.; Zeng, Q.; Luo, X.; Li, G.; et al. High Notch1 expression affects chemosensitivity of head and neck squamous cell carcinoma to paclitaxel and cisplatin treatment. Biomed. Pharmacother. 2019, 118, 109306. [Google Scholar] [CrossRef]
- Argirion, I.; Zarins, K.R.; Defever, K.; Suwanrungruang, K.; Chang, J.T.; Pongnikorn, D.; Chitapanarux, I.; Sriplung, H.; Vatanasapt, P.; Rozek, L.S. Temporal changes in head and neck cancer incidence in thailand suggest changing oropharyngeal epidemiology in the region. J. Glob. Oncol. 2019, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Posner, M.R.; Haddad, R.I.; Wirth, L.; Norris, C.M.; Goguen, L.A.; Mahadevan, A.; Sullivan, C.; Tishler, R.B. Induction chemotherapy in locally advanced squamous cell cancer of the head and neck: Evolution of the sequential treatment approach. Semin. Oncol. 2004, 31, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.L.; Zhang, L.; Huang, C.F.; Ma, S.R.; Bu, L.L.; Liu, J.F.; Yu, G.T.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; et al. NOTCH1 inhibition enhances the efficacy of conventional chemotherapeutic agents by targeting head neck cancer stem cell. Sci. Rep. 2016, 6, 24704. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Liu, Y.; Gao, Y.; Yuan, B.; Qi, X.; Fu, Y.; Zhu, Q.; Cao, T.; Zhang, S.; Yin, L.; et al. USP7 is a novel Deubiquitinase sustaining PLK1 protein stability and regulating chromosome alignment in mitosis. J. Exp. Clin. Cancer Res. 2019, 38, 468. [Google Scholar] [CrossRef]
- Hou, Y.; Zhu, Q.; Li, Z.; Peng, Y.; Yu, X.; Yuan, B.; Liu, Y.; Liu, Y.; Yin, L.; Peng, Y.; et al. The FOXM1-ABCC5 axis contributes to paclitaxel resistance in nasopharyngeal carcinoma cells. Cell Death Dis. 2017, 8, e2659. [Google Scholar] [CrossRef]
- Chew, S.C.; Singh, O.; Chen, X.; Ramasamy, R.D.; Kulkarni, T.; Lee, E.J.; Tan, E.H.; Lim, W.T.; Chowbay, B. The effects of CYP3A4, CYP3A5, ABCB1, ABCC2, ABCG2 and SLCO1B3 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of docetaxel in nasopharyngeal carcinoma patients. Cancer Chemother. Pharmacol. 2011, 67, 1471–1478. [Google Scholar] [CrossRef]
- Lin, X.; Yu, T.; Zhang, L.; Chen, S.; Chen, X.; Liao, Y.; Long, D.; Shen, F. Silencing Op18/stathmin by RNA interference promotes the sensitivity of nasopharyngeal carcinoma cells to taxol and high-grade differentiation of xenografted tumours in nude mice. Basic Clin. Pharmacol. Toxicol. 2016, 119, 611–620. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Li, G.; Su, Z.; Ren, S.; Tan, P.; Zhang, X.; Qiu, Y.; Tian, Y. Ephrin typeA receptor 2 regulates sensitivity to paclitaxel in nasopharyngeal carcinoma via the phosphoinositide 3kinase/Akt signalling pathway. Mol. Med. Rep. 2015, 11, 924–930. [Google Scholar] [CrossRef]
- Song, Y.; Peng, X.; Wang, M.; Xie, J.; Tan, G. Gene expression profiling of taxol-resistant nasopharyngeal carcinoma cells with siRNA-mediated FOLR1 downregulation. Int. J. Clin. Exp. Pathol. 2015, 8, 11314–11322. [Google Scholar]
- Li, W.; Tan, G.; Ma, Y.; Li, H.; He, G. Inhibition of alpha folate receptor resulting in a reversal of taxol resistance in nasopharyngeal carcinoma. Otolaryngol. Head Neck Surg. 2012, 146, 250–258. [Google Scholar] [CrossRef]
- Liu, Y.; Li, G.; Liu, C.; Tang, Y.; Zhang, S. RSF1 regulates the proliferation and paclitaxel resistance via modulating NF-kappaB signaling pathway in nasopharyngeal carcinoma. J. Cancer 2017, 8, 354–362. [Google Scholar] [CrossRef][Green Version]
- Huang, L.; Hu, C.; Chao, H.; Wang, R.; Lu, H.; Li, H.; Chen, H. miR-29c regulates resistance to paclitaxel in nasopharyngeal cancer by targeting ITGB1. Exp. Cell Res. 2019, 378, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Cao, P.; Li, J.; He, D.; Han, S.; Zhou, J.; Tan, G.; Li, W.; Yu, F.; Yu, J.; et al. MiR-1204 sensitizes nasopharyngeal carcinoma cells to paclitaxel both in vitro and in vivo. Cancer Biol. Ther. 2015, 16, 261–267. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peng, X.; Cao, P.; He, D.; Han, S.; Zhou, J.; Tan, G.; Li, W.; Yu, F.; Yu, J.; Li, Z.; et al. MiR-634 sensitizes nasopharyngeal carcinoma cells to paclitaxel and inhibits cell growth both in vitro and in vivo. Int. J. Clin. Exp. Pathol. 2014, 7, 6784–6791. [Google Scholar] [PubMed]
- Wang, Q.; Zhang, W.; Hao, S. LncRNA CCAT1 modulates the sensitivity of paclitaxel in nasopharynx cancers cells via miR-181a/CPEB2 axis. Cell Cycle 2017, 16, 795–801. [Google Scholar] [CrossRef]
- Zhu, Y.; He, D.; Bo, H.; Liu, Z.; Xiao, M.; Xiang, L.; Zhou, J.; Liu, Y.; Liu, X.; Gong, L.; et al. The MRVI1-AS1/ATF3 signaling loop sensitizes nasopharyngeal cancer cells to paclitaxel by regulating the Hippo-TAZ pathway. Oncogene 2019, 38, 6065–6081. [Google Scholar] [CrossRef]
- Yardley, D.A.; Burris, H.A., 3rd; Markus, T.; Spigel, D.R.; Greco, F.A.; Mainwaring, M.; Waterhouse, D.M.; Webb, C.D.; Hainsworth, J.D. Phase II trial of docetaxal plus imatinib mesylate in the treatment of patients with metastatic breast cancer. Clin. Breast Cancer 2009, 9, 237–242. [Google Scholar] [CrossRef]
- Bardia, A.; Baselga, J. Neoadjuvant therapy as a platform for drug development and approval in breast cancer. Clin. Cancer Res. 2013, 19, 6360–6370. [Google Scholar] [CrossRef]


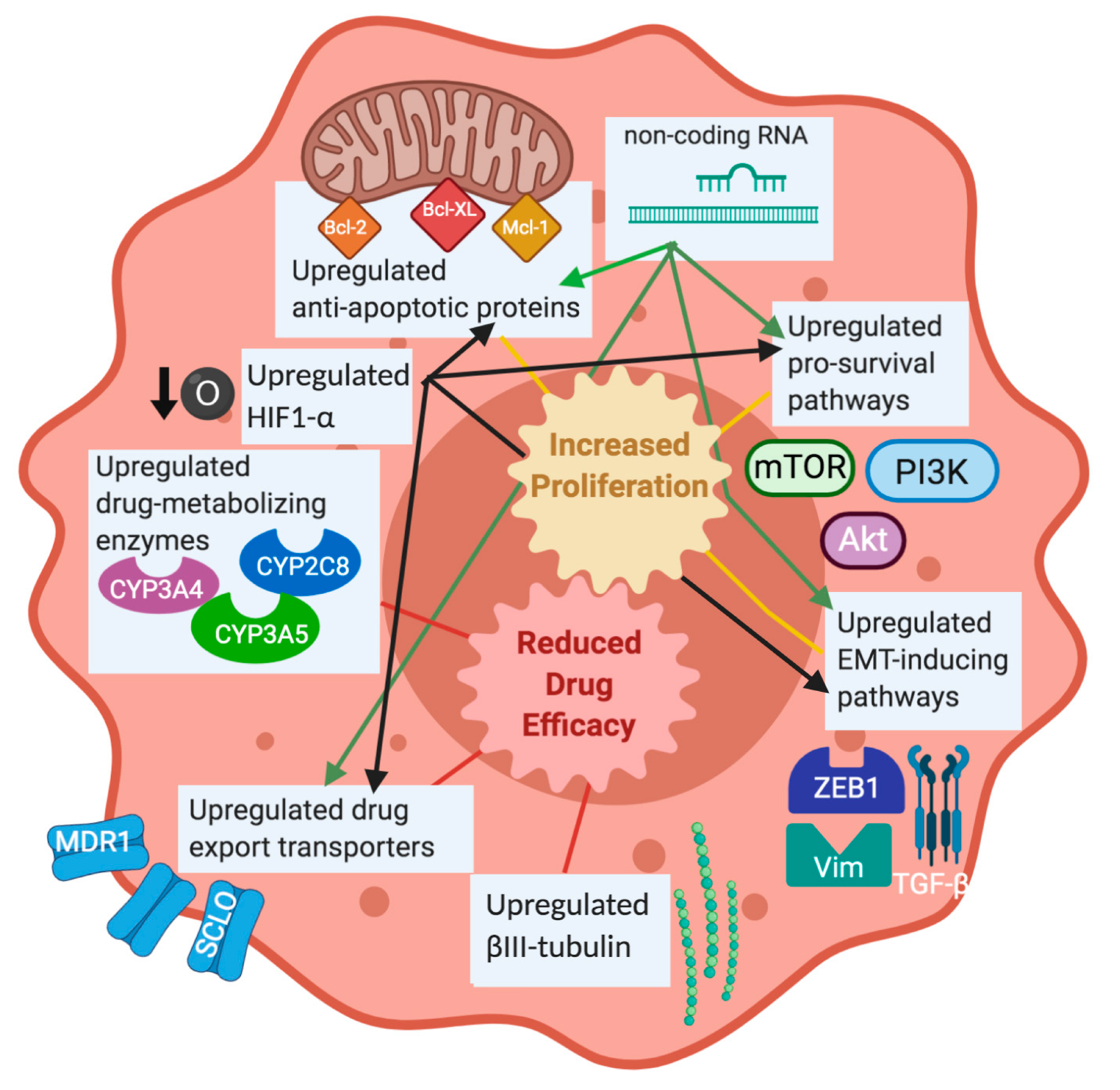{kind=link}
| Mechanism | Cancer Types | Taxane |
|---|---|---|
| Taxane-Metabolizing Enzymes (Cytochrome p450 (CYP) family) | ||
| CYP3A4 | Breast, Ovarian | PTX and DTX |
| CYP2C8 | Ovarian | PTX |
| CYP1 enzymes (CYP1A1 and CYP1B1) | Ovarian | PTX |
| Drug efflux/influx | ||
| MDR1 | Breast, Ovarian, Prostate, Non-small cell lung cancer (NSCLC), Nasopharyngeal | PTX, DTX, and CBZ |
| ABCC10 | Breast | PTX and DTX |
| ABCC5 | Nasopharyngeal | PTX |
| Solute carrier of organic anions (SLCO) | Prostate, Nasopharyngeal | DTX |
| Tubulin subunit and related protein expression | ||
| βIII-tubulin | Breast, Ovarian, Prostate, NSCLC, Pancreatic | PTX, DTX, and CBZ |
| Tau | Breast, Ovarian | PTX |
| Stathmin | Breast, Nasopharyngeal | PTX |
| Signaling molecules | ||
| Polo-like kinase (PLK1) | Ovarian, Prostate, Nasopharyngeal | PTX |
| Bcl-XL/Bcl-2 | Ovarian, Prostate, NSCLC, Pancreatic | PTX, DTX, and CBZ |
| Mcl-1 | Ovarian, Prostate | PTX |
| PI3K/AKT pathway | Ovarian, Prostate, Cervical, Nasopharyngeal | PTX and DTX |
| G Protein-Coupled Receptor Kinase 5 (GRK5) | Breast, Cervical | PTX |
| Survivin | Ovarian, Nasopharyngeal | PTX |
| Cancer Type | Mechanism | Taxane |
|---|---|---|
| Breast | MAP4 | PTX |
| Septin | PTX | |
| Tubulin Binding Cofactor C (TBCC) | PTX | |
| NIMA-related Kinase 2 (NEK2) | PTX | |
| G Protein Signaling Modulator 2 (GPSM2/LGN) | PTX | |
| BRCA1 | PTX | |
| Adenomatous Polyposis Coli (APC) | PTX | |
| p16 | PTX | |
| human Expanded (hEX) | PTX | |
| Yes-Associated Protein (YAP) | PTX | |
| Leucine Zipper Tumor Suppressor 1 (LZTS1) | PTX | |
| Ovarian | Spleen Tyrosine Kinase (SYK) | PTX |
| Cyclin E1 and CDK2 | PTX | |
| Cyclin A1 and CDK1/CDK2 | PTX | |
| BUB1 | PTX | |
| BUBR1 (BUB1-related protein kinase) | PTX | |
| MAD2 (mitotic arrest deficiency 2) | PTX | |
| c-IAP1 and XIAP (X-linked IAP) overexpression | PTX | |
| Src | PTX | |
| Prostate | Androgen Receptor (AR) | DTX and CBZ |
| Pancreatic | NF-kB | CBZ |
| Head and Neck squamous cell carcinoma | Notch signaling | PTX and DTX |
| miRNAs | Cancer Type | Taxane |
|---|---|---|
| miR-451 | NSCLC, Breast | PTX |
| miR-200 family (miR-141, miR-200c, and miR-200a) | Ovarian, Prostate, Breast | PTX, DTX |
| miR-17 | Breast | PTX |
| miR-18a-5p | Breast | PTX |
| miR-18a | Breast | PTX |
| miR-20b | Breast | PTX |
| miR-21 | Prostate | DTX |
| miR-29c | Nasopharyngeal | PTX |
| miR-34a | Prostate | PTX |
| miR-106a | Ovarian | PTX |
| miR-125b | Breast | PTX |
| miR-133b | Ovarian | PTX |
| miR-143 | Prostate | DTX |
| miR-146 | Ovarian | PTX |
| miR-148 | Prostate | PTX |
| miR-194 | Ovarian | PTX |
| miR-433 | Ovarian | PTX |
| miR-520h | Breast | PTX |
| miR-591 | Ovarian | PTX |
| miR-630 | Ovarian | PTX |
| miR-634 | Nasopharyngeal | PTX |
| miR-663 | Breast | PTX |
| miR-1204 | Nasopharyngeal | PTX |
| miR-1307 | Ovarian | PTX |
| miR-3646 | Breast | DTX |
| lncRNAs | Cancer Type | Taxane |
| Colon cancer-associated transcript 1 (CCAT1) | Prostate, Nasopharyngeal | PTX |
| Nuclear paraspeckle assembly transcript 1 (NEAT1) | Prostate, Ovarian | DTX |
| Urothelial carcinoma-associated 1 (UCA1) | Prostate, Ovarian | PTX |
| Ferritin like lnRNAs (FER1L4, FTH1P3) | Breast, Ovarian | PTX |
| AK124454 | Breast | PTX |
| HIF1A-AS2 | Breast | PTX |
| HORAS5 | Prostate | CBZ |
| HOXD-AS1 | Prostate | PTX |
| Long intergenic non-protein coding RNA 00518 (linc00518) | Prostate | PTX |
| LINC01118 | Ovarian | PTX |
| lncRNA H19 | Breast | PTX |
| Long intergenic non-coding RNA, Regulator of Reprogramming (Linc-ROR) | Breast | PTX |
| MT-associated protein tau antisense RNA 1 (MAPT)-AS1 | Breast | PTX |
| MA-linc1 | Breast | PTX |
| Murine retrovirus integration site 1 homolog antisense RNA 1 (MRVI1-AS1) | Nasopharyngeal | PTX |
| NONHSAT141924 | Breast | PTX |
| Small nucleolar RNA host gene 6 (SNHG6) | Prostate | PTX |
| Suppressor of cytokine signaling 2-antisense transcript 1 (SOCS2-AS1) | Prostate | DTX |
| Ref | Phase | Treatment | Prior Taxane | Cancer Type | Primary Outcomes |
|---|---|---|---|---|---|
| [265] | I | Dasatinib+ PTX OR carboplatin | N/A | Advanced and recurrent EOC | Recommended phase II dasatinib dose of 150mg daily with PTX and carboplatin |
| N/A | I | Pembrolizumab+ DTX OR gemcitabine hydrochloride | N/A | Urothelial cancer | Ongoing (NCT02437370) |
| [412] | I/II | Everolimus+ DTX | N/A | mCRPC | Recommended Everolimus dose of 10mg daily and DTX 60 mg/m2 |
| [365] | I/II | CBZ+ abiraterone | DTX (I) and DTX+abiraterone (II) | mCRPC | Manageable safety profile and shows antitumor activity |
| [12] | II | Nab-PTX+ gemcitabine OR simplified LV5FU2 | N/A | Metastatic pancreatic | N/A |
| [262] | II | Dasatinib | One or two regimens of platinum+taxane | EOC or primary peritoneal | Dastinib has minimal activity as a single agent in these cancers |
| [487] | II | DTX+ imatinib | N/A | metastatic BC | Weekly DTX is not enhanced by concurrent imatinib |
| [488] | II | LCL161+ PTX | N/A | TNBC | Toxicity concerns with combination |
| [351] | II | DTX OR CBZ+ prednisone | N/A | mCRPC | Improved prostate-specific antigen response rates |
| [399] | II | DTX+prednisone +placebo OR AT-101 | N/A | mCRPC | Combination did not extend OS |
| [19] | III | CBZ+prednisone OR mitoxantrone+ prednisone | DTX-containing regimen | mCRPC | CBZ + prednisone improves OS |
| [465] | III | Nab-PTX+ gemcitabine | N/A | Metastatic pancreatic | Nab-PTX + gemcitabine improved OS |
| Biomarker | Cancer Types | Biomarker Status | Reference |
|---|---|---|---|
| Tau expression | Breast, Ovarian | Human Specimens | [80,181] |
| miRNAs and lncRNAs | Breast, Ovarian, Prostate | See text for individual references | |
| βIII-tubulin | Ovarian, Prostate | Human Specimens | [193,338] |
| MDR1 and other ABC transporter proteins | Ovarian, Prostate | In vitro | [42,307,324,325] reviewed in [20] |
| Expression of CYP2C8, 3A4, 3A5 | Ovarian | Human Specimens | reviewed in [20] |
| MAPs and MAPKs (IKBKB/STK39 and EDN2/TBK1) | Ovarian | In vitro | [185,186,187] |
| KIF14 | Ovarian | In vitro, Human Specimens | [188] |
| CCNE1 | Ovarian | In vitro | [201] |
| MAD2 (mitotic arrest deficiency 2) | Ovarian | In vitro, Human Specimens | [211,212,213] |
| PLK1 (Polo-like kinase 1) | Ovarian | In vitro | [201,219,220] |
| Inhibitor of apoptosis (IAP) family of proteins | Ovarian | Human Specimens, In vitro | [234,235,236,237,238] |
| Solute carrier of organic anions (SLCO) | Prostate | In vitro, Human Specimens | [326,327,328,331,332,334] |
| Increased Notch1 expression | HNSCC | Human Specimens | [470] |
| HIF1-α | Prostate | In vitro | [449] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maloney, S.M.; Hoover, C.A.; Morejon-Lasso, L.V.; Prosperi, J.R. Mechanisms of Taxane Resistance. Cancers 2020, 12, 3323. https://doi.org/10.3390/cancers12113323
Maloney SM, Hoover CA, Morejon-Lasso LV, Prosperi JR. Mechanisms of Taxane Resistance. Cancers. 2020; 12(11):3323. https://doi.org/10.3390/cancers12113323
Chicago/Turabian StyleMaloney, Sara M., Camden A. Hoover, Lorena V. Morejon-Lasso, and Jenifer R. Prosperi. 2020. "Mechanisms of Taxane Resistance" Cancers 12, no. 11: 3323. https://doi.org/10.3390/cancers12113323
APA StyleMaloney, S. M., Hoover, C. A., Morejon-Lasso, L. V., & Prosperi, J. R. (2020). Mechanisms of Taxane Resistance. Cancers, 12(11), 3323. https://doi.org/10.3390/cancers12113323