
Endorsement of TNBC Biomarkers in Precision Therapy by Nanotechnology
 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
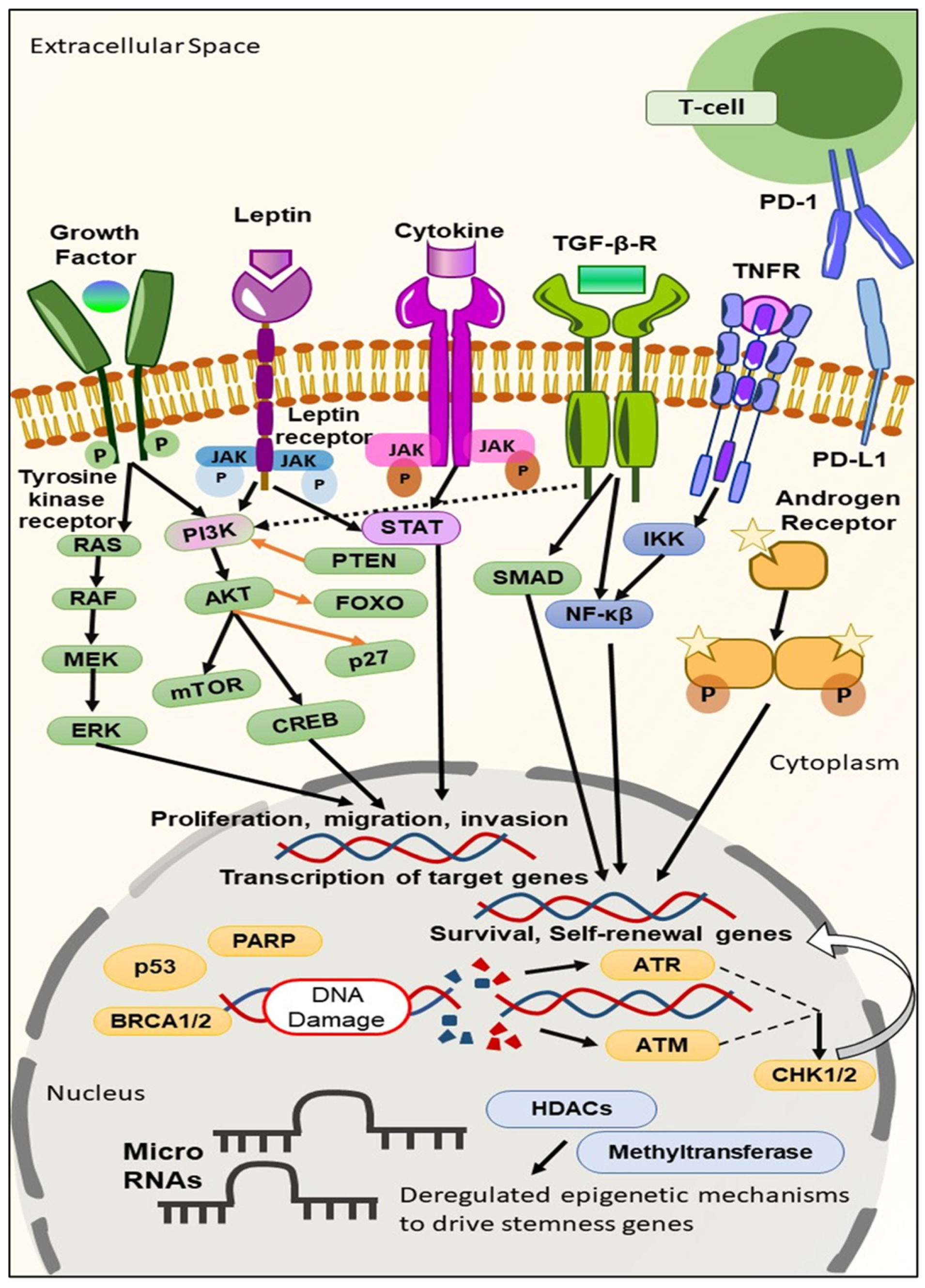
2. Biomarkers Derived from the Molecular Profiling of TNBC
2.1. TNBC Biomarkers on the Cell Surface
2.1.1. Folate Receptor
2.1.2. Epidermal Growth Factor Receptor (EGFR)
2.1.3. Interleukin-3—Receptor α (IL-3Rα)
2.1.4. c-Kit
2.1.5. c-Met
2.1.6. Programmed Cell Death 1 Ligand (PD-L1)
2.1.7. Adenosine 2B Receptor (A2BR)
2.1.8. CD73
2.1.9. GABA Receptor π Subunit (GABRP)
2.1.10. G–Protein-Coupled Receptor 161 (GPR161)
2.1.11. G–Protein-Coupled Kisspeptin Receptor (KISS1R)
2.1.12. Intercellular Adhesion Molecule-1 (ICAM-1)
2.1.13. Leptin Receptor
2.1.14. Monocyte Chemoattractant Protein-1 (MCP-1)
2.1.15. Metabotropic Glutamate Receptor-1 (mGluR1)
2.1.16. MDM2-Binding Protein (MTBP)
2.1.17. Claudin Proteins
2.1.18. Caveolin Proteins
2.1.19. CCR5
2.1.20. Trop 2
2.2. TNBC Biomarkers in the Cytoplasm
2.2.1. PI3K/AKT/mTOR Pathway
2.2.2. Androgen Receptor (AR)
2.2.3. Aldehyde Dehydrogenase 1 (ALDH1)
2.2.4. HOX Genes
2.2.5. Protein Kinase D1 (PKD1)
2.2.6. 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphate-4 (PFKFB4)
2.3. TNBC Biomarkers in the Nucleus
2.3.1. BRCA Genes
2.3.2. TP53
2.3.3. Activating Transcription Factor 4 (ATF4)
2.3.4. ETS Translocation Variant4 (ETV4)
2.3.5. Forkhead Box M1 (FOXM1)
2.3.6. Glucocorticoids
2.4. TNBC Biomarkers in the Blood
2.4.1. Vascular Endothelial Growth Factor (VEGF)
2.4.2. Interleukin-8 (IL-8)
3. Targeted Therapies Based on Biomarker Appraisal
3.1. Signaling Pathway Inhibition
3.1.1. Inhibition of EGFR Signaling Pathway
3.1.2. Inhibition of the PI3K/Akt/mTOR Signaling Pathway
3.1.3. Inhibition of VEGFR
3.2. Immune Checkpoints Inhibition
3.3. Inhibition of Poly (ADP-Ribose) Polymerase (PARP) Enzymes
3.4. Inhibition of Cell Cycle
3.5. Inhibition of Epigenetic Modifications
3.5.1. Inhibition of DNMT
3.5.2. Inhibition of HDAC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Drugs | Phase | Outcome | Refs. |
|---|---|---|---|---|
| PARP | Olaparib | I/II/III |
| [147] |
| Veliparib | II |
| [148] | |
| Iniparib | II |
| [149] | |
| Immune checkpoints | PDL1: Pembrolizumab | FDA-Approved |
| [133,150] |
| PDL1: Atezolizumab, Durvalumab | II/III |
| [134] | |
| Signaling pathways | EGFR: Cetuximab | II |
| [132] |
| EGFR: Erlotinib | Pre-clinical |
| ||
| PI3K: BKM120 | Pre-clinical |
| [123] | |
| Akt: Ipatasertib | II |
| [125] | |
| Angiogenesis | VEGF: Bevacizumab | II/III |
| [127,128] |
| VEGFR: Apatinib | II |
| [132] | |
| Epigenetic modification | DNMT: 5- Azacytidine/AZA, Decitabine/DAC | Pre-clinical |
| [142] |
| HDAC: Suberoylanilide hydroxamic acid (SAHA), Entinostat (ENT) | Pre-clinical |
| [151] | |
| Cell cycle | CDK4/6: Palbociclib | I/II |
| [152] |
| CHK1: MK-8776 | Pre-clinical |
| [153] |
4. TNBC Biomarkers in Cancer Nanotherapeutics
4.1. Significance of Nanotherapeutics in TNBC Therapy
4.2. Correlation of Nanotherapeutics and TNBC Biomarkers
4.2.1. Inorganic Nanoparticles
4.2.2. Polymeric Nanoparticles (PNPs)
4.2.3. Lipid-Based Nanoparticles (LNPs)
5. Clinical Status of TNBC Biomarkers-Based Nanotherapeutics
6. Conclusions and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marra, A.; Trapani, D.; Viale, G.; Criscitiello, C.; Curigliano, G. Practical classification of triple-negative breast cancer: Intratumoral heterogeneity, mechanisms of drug resistance, and novel therapies. NPJ Breast Cancer 2020, 6, 54. [Google Scholar] [CrossRef]
- Shohdy, K.S.; Almeldin, D.S.; Fekry, M.A.; Ismail, M.A.; AboElmaaref, N.A.; ElSadany, E.G.; Hamza, B.M.; El-Shorbagy, F.H.; Ali, A.S.; Attia, H.; et al. Pathological responses and survival outcomes in patients with locally advanced breast cancer after neoadjuvant chemotherapy: A single-institute experience. J. Egypt. Natl. Cancer Inst. 2021, 33, 39. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahmood, S.; Sapiezynski, J.; Garbuzenko, O.B.; Minko, T. Metastatic and triple-negative breast cancer: Challenges and treatment options. Drug Deliv. Transl. Res. 2018, 8, 1483–1507. [Google Scholar] [CrossRef] [PubMed]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef]
- Won, K.; Spruck, C. Triple-negative breast cancer therapy: Current and future perspectives (Review). Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Merkher, Y.; Chen, L.; Liu, N.; Leonov, S.; Chen, Y. Recent advances in therapeutic strategies for triple-negative breast cancer. J. Hematol. Oncol. 2022, 15, 121. [Google Scholar] [CrossRef]
- Mo, H.; Renna, C. Biomarker-Driven Targeted Therapies in Solid Tumor Malignancies. J. Hematol. Oncol. Pharm. 2021, 11, 84–91. [Google Scholar]
- Ye, F.; Zhao, Y.; El-Sayed, R.; Muhammed, M.; Hassan, M. Advances in nanotechnology for cancer biomarkers. Nano Today 2018, 18, 103–123. [Google Scholar] [CrossRef]
- Fleisher, B.; Clarke, C.; Ait-Oudhia, S. Current advances in biomarkers for targeted therapy in triple-negative breast cancer. Breast Cancer Targets Ther. 2016, 8, 183–197. [Google Scholar] [CrossRef]
- da Silva, J.L.; Cardoso Nunes, N.C.; Izetti, P.; de Mesquita, G.G.; de Melo, A.C. Triple negative breast cancer: A thorough review of biomarkers. Crit. Rev. Oncol. Hematol. 2020, 145, 102855. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Zheng, C.; Tang, F.; Rosol, T.J.; Shao, Z.-M. Editorial: Triple-negative breast cancer: Heterogeneity, tumor microenvironment and targeted therapy. Front. Oncol. 2022, 12, 1026566. [Google Scholar] [CrossRef]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar] [CrossRef]
- Sukumar, J.; Gast, K.; Quiroga, D.; Lustberg, M.; Williams, N. Triple-negative breast cancer: Promising prognostic biomarkers currently in development. Expert Rev. Anticancer. Ther. 2021, 21, 135–148. [Google Scholar] [CrossRef]
- Chang, J.Y.H.; Ladame, S. Diagnostic, prognostic, and predictive biomarkers for cancer. In Bioengineering Innovative Solutions for Cancer; Elsevier: Amsterdam, The Netherlands, 2020; pp. 3–21. [Google Scholar]
- DeCarlo, A.; Malardier-Jugroot, C.; Szewczuk, M.R. Next-Generation Multimodality of Nanomedicine Therapy: Folic Acid Conjugated Copolymer and Folate Receptor Interactions Disrupt Receptor Functionality Resulting in Dual Therapeutic AntiCancer Potential in Triple-Negative Breast Cancer. Preprints 2020, 2020080316. [Google Scholar] [CrossRef]
- Norton, N.; Youssef, B.; Hillman, D.W.; Nassar, A.; Geiger, X.J.; Necela, B.M.; Liu, H.; Ruddy, K.J.; Polley, M.-Y.C.; Ingle, J.N.; et al. Folate receptor alpha expression associates with improved disease-free survival in triple negative breast cancer patients. NPJ Breast Cancer 2020, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. VEGF as a Therapeutic Target in Cancer. Oncology 2005, 69, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of theEGFRin cancer. Mol. Oncol. 2017, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Koni, M.; Castellano, I.; Venturelli, E.; Sarcinella, A.; Lopatina, T.; Grange, C.; Cedrino, M.; Femminò, S.; Cossu-Rocca, P.; Orrù, S.; et al. Interleukin-3-Receptor-α in Triple-Negative Breast Cancer (TNBC): An Additional Novel Biomarker of TNBC Aggressiveness and a Therapeutic Target. Cancers 2022, 14, 3918. [Google Scholar] [CrossRef]
- Lopatina, T.; Grange, C.; Cavallari, C.; Navarro-Tableros, V.; Lombardo, G.; Rosso, A.; Cedrino, M.; Pomatto, M.A.C.; Koni, M.; Veneziano, F.; et al. Targeting IL-3Rα on tumor-derived endothelial cells blunts metastatic spread of triple-negative breast cancer via extracellular vesicle reprogramming. Oncogenesis 2020, 9, 90. [Google Scholar] [CrossRef]
- López-Mejía, J.A.; Tallabs-Utrilla, L.F.; Salazar-Sojo, P.; Mantilla-Ollarves, J.C.; Sánchez-Carballido, M.A.; Rocha-Zavaleta, L. c-Kit Induces Migration of Triple-Negative Breast Cancer Cells and Is a Promising Target for Tyrosine Kinase Inhibitor Treatment. Int. J. Mol. Sci. 2022, 23, 8702. [Google Scholar] [CrossRef] [PubMed]
- Nogi; Toriumi, Y.; Fukushima, H.; Uchida, K.; Nogi, H.; Kobayashi, T.; Suzuki, M.; Tabei, I.; Kawase, K. EGFR as paradoxical predictor of chemosensitivity and outcome among triple-negative breast cancer. Oncol. Rep. 1994, 21, 413–417. [Google Scholar] [CrossRef]
- Shams, T.M.; Shams, M.E. Overexpression of c-KIT (CD117) in triple-negative breast cancer. Egypt. J. Pathol. 2011, 31, 113–117. [Google Scholar] [CrossRef]
- Jansson, S.; Bendahl, P.-O.; Grabau, D.A.; Falck, A.-K.; Fernö, M.; Aaltonen, K.; Rydén, L. The Three Receptor Tyrosine Kinases c-KIT, VEGFR2 and PDGFRα, Closely Spaced at 4q12, Show Increased Protein Expression in Triple-Negative Breast Cancer. PLoS ONE 2014, 9, e102176. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, Y.; Guan, B.; Rao, Q.; Wang, J.; Ma, H.; Zhang, Z.; Zhou, X. C-kit and PDGFRA gene mutations in triple negative breast cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 4280–4285. [Google Scholar]
- Johansson, I.; Aaltonen, K.E.; Ebbesson, A.; Grabau, D.; Wigerup, C.; Hedenfalk, I.; Rydén, L. Increased gene copy number of KIT and VEGFR2 at 4q12 in primary breast cancer is related to an aggressive phenotype and impaired prognosis. Genes Chromosom. Cancer 2011, 51, 375–383. [Google Scholar] [CrossRef]
- Gaule, P.B.; Crown, J.; O’Donovan, N.; Duffy, M.J. cMET in triple-negative breast cancer: Is it a therapeutic target for this subset of breast cancer patients? Expert. Opin. Ther. Targets 2014, 18, 999–1009. [Google Scholar] [CrossRef]
- Breen, L.; Gaule, P.B.; Canonici, A.; Walsh, N.; Collins, D.M.; Cremona, M.; Hennessy, B.T.; Duffy, M.J.; Crown, J.; Donovan, N.O.; et al. Targeting c-Met in triple negative breast cancer: Preclinical studies using the c-Met inhibitor, Cpd A. Investig. New Drugs 2020, 38, 1365–1372. [Google Scholar] [CrossRef]
- Ho-Yen, C.M.; Jones, J.L.; Kermorgant, S. The clinical and functional significance of c-Met in breast cancer: A review. Breast Cancer Res. 2015, 17, 52. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Chen, H.; Karuturi, M.S.; Chavez-MacGregor, M.; Tsavachidis, S.; Meric-Bernstam, F.; Do, K.-A.; Hortobagyi, G.N.; Thompson, P.A.; Mills, G.B.; et al. Frequency of mesenchymal-epithelial transition factor gene (MET) and the catalytic subunit of phosphoinositide-3-kinase (PIK3CA) copy number elevation and correlation with outcome in patients with early stage breast cancer. Cancer 2012, 119, 7–15. [Google Scholar] [CrossRef]
- Yi, Y.W.; You, K.; Bae, E.J.; Kwak, S.-J.; Seong, Y.-S.; Bae, I. Dual inhibition of EGFR and MET induces synthetic lethality in triple-negative breast cancer cells through downregulation of ribosomal protein S6. Int. J. Oncol. 2015, 47, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.; Khalil, F.; Antonia, S. PD-L1 Expression Is Increased in a Subset of Basal Type Breast Cancer Cells. PLoS ONE 2014, 9, e88557. [Google Scholar] [CrossRef] [PubMed]
- Muenst, S.; Schaerli, A.R.; Gao, F.; Däster, S.; Trella, E.; Droeser, R.A.; Muraro, M.G.; Zajac, P.; Zanetti, R.; Gillanders, W.E.; et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Cocco, S.; Piezzo, M.; Calabrese, A.; Cianniello, D.; Caputo, R.; Di Lauro, V.; Fusco, G.; Di Gioia, G.; Licenziato, M.; De Laurentiis, M. Biomarkers in Triple-Negative Breast Cancer: State-of-the-Art and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 4579. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.A.; Levine, K.; Oesterreich, S. Targeting adenosine receptor 2B in triple negative breast cancer. J. Cancer Metastasis Treat. 2018, 4, 13. [Google Scholar] [CrossRef]
- Fernandez-Gallardo, M.; González-Ramírez, R.; Sandoval, A.; Felix, R.; Monjaraz, E. Adenosine Stimulate Proliferation and Migration in Triple Negative Breast Cancer Cells. PLoS ONE 2016, 11, e0167445. [Google Scholar] [CrossRef] [PubMed]
- Lafont, V.; Michaud, H.-A.; Bonnefoy, N. CD73: A new biomarker in triple-negative breast cancer. Transl. Cancer Res. 2018, 7, S594–S596. [Google Scholar] [CrossRef]
- Petruk, N.; Tuominen, S.; Åkerfelt, M.; Mattsson, J.; Sandholm, J.; Nees, M.; Yegutkin, G.G.; Jukkola, A.; Tuomela, J.; Selander, K.S. CD73 facilitates EMT progression and promotes lung metastases in triple-negative breast cancer. Sci. Rep. 2021, 11, 6035. [Google Scholar] [CrossRef]
- Sizemore, G.M.; Sizemore, S.T.; Seachrist, D.D.; Keri, R.A. GABA(A) Receptor Pi (GABRP) Stimulates Basal-like Breast Cancer Cell Migration through Activation of Extracellular-regulated Kinase 1/2 (ERK1/2). J. Biol. Chem. 2014, 289, 24102–24113. [Google Scholar] [CrossRef]
- Azizi-Tabesh, G.; Kamaliyan, Z.; Darbeheshti, F.; Omranipour, R.; Soleimani, V.; Alipour, N.; Mirfakhraie, R.; Yassaee, V.R. Overexpression of GABRP Gene in Triple Negative Breast Cancer: Molecular Mechanisms and Interpretation. Int. J. Cancer Manag. 2021, 14, e119130. [Google Scholar] [CrossRef]
- Juvale, I.I.A.; Hassan, Z.; Has, A.T.C. The Emerging Roles of π Subunit-Containing GABAA Receptors in Different Cancers. Int. J. Med Sci. 2021, 18, 3851–3860. [Google Scholar] [CrossRef] [PubMed]
- Feigin, M.E.; Xue, B.; Hammell, M.C.; Muthuswamy, S.K. G-protein–coupled receptor GPR161 is overexpressed in breast cancer and is a promoter of cell proliferation and invasion. Proc. Natl. Acad. Sci. USA 2014, 111, 4191–4196. [Google Scholar] [CrossRef] [PubMed]
- Feigin, M.E.; Xue, B.; Hammell, M.; Muthuswamy, S.K. Abstract A236: The orphan G-protein coupled receptor GPR161 is an oncogene in triple negative breast cancer. Mol. Cancer Ther. 2013, 12, A236. [Google Scholar] [CrossRef]
- Gutierrez, A.N.; McDonald, P.H. GPCRs as Emerging Cancer Drug Targets: Target Validation of oGPCR, GPR161 and its Role in Triple Negative Breast Cancer. FASEB J. 2019, 33, 674.4. [Google Scholar] [CrossRef]
- Blake, A.; Dragan, M.; Tirona, R.G.; Hardy, D.B.; Brackstone, M.; Tuck, A.B.; Babwah, A.V.; Bhattacharya, M. G protein-coupled KISS1 receptor is overexpressed in triple negative breast cancer and promotes drug resistance. Sci. Rep. 2017, 7, srep46525. [Google Scholar] [CrossRef]
- Dragan, M.; Nguyen, M.-U.; Guzman, S.; Goertzen, C.; Brackstone, M.; Dhillo, W.S.; Bech, P.R.; Clarke, S.; Abbara, A.; Tuck, A.B.; et al. G protein-coupled kisspeptin receptor induces metabolic reprograming and tumorigenesis in estrogen receptor-negative breast cancer. Cell Death Dis. 2020, 11, 106. [Google Scholar] [CrossRef]
- Guzman, S.; Brackstone, M.; Radovick, S.; Babwah, A.V.; Bhattacharya, M.M. KISS1/KISS1R in Cancer: Friend or Foe? Front. Endocrinol. 2018, 9, 437. [Google Scholar] [CrossRef]
- Rosette, C.; Roth, R.B.; Oeth, P.; Braun, A.; Kammerer, S.; Ekblom, J.; Denissenko, M.F. Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis 2005, 26, 943–950. [Google Scholar] [CrossRef]
- Guo, P.; Huang, J.; Wang, L.; Jia, D.; Yang, J.; Dillon, D.A.; Zurakowski, D.; Mao, H.; Moses, M.A.; Auguste, D.T. ICAM-1 as a molecular target for triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 14710–14715. [Google Scholar] [CrossRef]
- Taftaf, R.; Liu, X.; Singh, S.; Jia, Y.; Dashzeveg, N.K.; Hoffmann, A.D.; El-Shennawy, L.; Ramos, E.K.; Adorno-Cruz, V.; Schuster, E.J.; et al. ICAM1 initiates CTC cluster formation and trans-endothelial migration in lung metastasis of breast cancer. Nat. Commun. 2021, 12, 4867. [Google Scholar] [CrossRef]
- Otvos, L.; Kovalszky, I.; Riolfi, M.; Ferla, R.; Olah, J.; Sztodola, A.; Nama, K.; Molino, A.; Piubello, Q.; Wade, J.D.; et al. Efficacy of a leptin receptor antagonist peptide in a mouse model of triple-negative breast cancer. Eur. J. Cancer 2011, 47, 1578–1584. [Google Scholar] [CrossRef]
- Lipsey, C.C.; Harbuzariu, A.; Robey, R.W.; Huff, L.M.; Gottesman, M.M.; Gonzalez-Perez, R.R. Leptin Signaling Affects Survival and Chemoresistance of Estrogen Receptor Negative Breast Cancer. Int. J. Mol. Sci. 2020, 21, 3794. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Sarkissyan, M.; Paico, K.; Wu, Y.; Vadgama, J.V. MCP-1 is overexpressed in triple-negative breast cancers and drives cancer invasiveness and metastasis. Breast Cancer Res. Treat. 2018, 170, 477–486. [Google Scholar] [CrossRef]
- Kanga, K.J.; Mendonca, P.; Soliman, K.F.; Ferguson, D.T.; Darling-Reed, S.F. Effect of Diallyl Trisulfide on TNF-α-induced CCL2/MCP-1 Release in Genetically Different Triple-negative Breast Cancer Cells. Anticancer. Res. 2021, 41, 5919–5933. [Google Scholar] [CrossRef] [PubMed]
- Cranford, T.L.; Velázquez, K.T.; Enos, R.T.; Bader, J.E.; Carson, M.S.; Chatzistamou, I.; Nagarkatti, M.; Murphy, E.A. Loss of monocyte chemoattractant protein-1 expression delays mammary tumorigenesis and reduces localized inflammation in the C3(1)/SV40Tag triple negative breast cancer model. Cancer Biol. Ther. 2017, 18, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Banda, M.; Speyer, C.L.; Semma, S.N.; Osuala, K.O.; Kounalakis, N.; Torres, K.E.T.; Barnard, N.J.; Kim, H.J.; Sloane, B.F.; Miller, F.R.; et al. Metabotropic Glutamate Receptor-1 Contributes to Progression in Triple Negative Breast Cancer. PLoS ONE 2014, 9, e81126. [Google Scholar] [CrossRef]
- Speyer, C.L.; Smith, J.S.; Banda, M.; Devries, J.A.; Mekani, T.; Gorski, D.H. Metabotropic glutamate receptor-1: A potential therapeutic target for the treatment of breast cancer. Breast Cancer Res. Treat. 2011, 132, 565–573. [Google Scholar] [CrossRef]
- Bastiaansen, A.E.M.; Timmermans, A.M.; Smid, M.; van Deurzen, C.H.M.; Hulsenboom, E.S.P.; der Smissen, W.J.C.P.-V.; Foekens, R.; Trapman-Jansen, A.M.A.C.; Smitt, P.A.E.S.; Luider, T.M.; et al. Metabotropic glutamate receptor 1 is associated with unfavorable prognosis in ER-negative and triple-negative breast cancer. Sci. Rep. 2020, 10, 22292. [Google Scholar] [CrossRef]
- Sexton, R.E.; Hachem, A.H.; Assi, A.A.; Bukhsh, M.A.; Gorski, D.H.; Speyer, C.L. Metabotropic glutamate receptor-1 regulates inflammation in triple negative breast cancer. Sci. Rep. 2018, 8, 16008. [Google Scholar] [CrossRef]
- Iwakuma, T.; Tochigi, Y.; Van Pelt, C.S.; Caldwell, L.C.; Terzian, T.; Parant, J.M.; Chau, G.P.; Koch, J.G.; Eischen, C.M.; Lozano, G. Mtbp haploinsufficiency in mice increases tumor metastasis. Oncogene 2007, 27, 1813–1820. [Google Scholar] [CrossRef]
- Grieb, B.C.; Eischen, C.M. MTBP and MYC: A Dynamic Duo in Proliferation, Cancer, and Aging. Biology 2022, 11, 881. [Google Scholar] [CrossRef] [PubMed]
- Naimi, A.; Zare, N.; Amjadi, E.; Soltan, M. High claudin-4 antigen expression in triple-negative breast cancer by the immunohistochemistry method. J. Res. Med Sci. 2022, 27, 20. [Google Scholar] [CrossRef] [PubMed]
- Gowrikumar, S.; Singh, A.B.; Dhawan, P. Role of Claudin Proteins in Regulating Cancer Stem Cells and Chemoresistance-Potential Implication in Disease Prognosis and Therapy. Int. J. Mol. Sci. 2019, 21, 53. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, M.; Kleinclauss, A.; Kuntz, S.; Grillier-Vuissoz, I. Claudin 1 inhibits cell migration and increases intercellular adhesion in triple-negative breast cancer cell line. Mol. Biol. Rep. 2020, 47, 7643–7653. [Google Scholar] [CrossRef]
- Qian, X.-L.; Pan, Y.-H.; Huang, Q.-Y.; Shi, Y.-B.; Huang, Q.-Y.; Hu, Z.-Z.; Xiong, L.-X. Caveolin-1: A multifaceted driver of breast cancer progression and its application in clinical treatment. OncoTargets Ther. 2019, 12, 1539–1552. [Google Scholar] [CrossRef]
- Yeong, J.; Thike, A.A.; Ikeda, M.; Lim, J.C.T.; Lee, B.; Nakamura, S.; Iqbal, J.; Tan, P.H. Caveolin-1 expression as a prognostic marker in triple negative breast cancers of Asian women. J. Clin. Pathol. 2017, 71, 161–167. [Google Scholar] [CrossRef]
- Jiao, X.; Nawab, O.; Patel, T.; Kossenkov, A.V.; Halama, N.; Jaeger, D.; Pestell, R.G. Recent Advances Targeting CCR5 for Cancer and Its Role in Immuno-Oncology. Cancer Res. 2019, 79, 4801–4807. [Google Scholar] [CrossRef]
- Wang, X.; Han, Y.; Peng, J.; He, J. CCR5 is a prognostic biomarker and an immune regulator for triple negative breast cancer. Aging 2021, 13, 23810–23830. [Google Scholar] [CrossRef]
- Velasco-Velázquez, M.; Jiao, X.; De La Fuente, M.; Pestell, T.G.; Ertel, A.; Lisanti, M.P.; Pestell, R.G. CCR5 Antagonist Blocks Metastasis of Basal Breast Cancer Cells. Cancer Res. 2012, 72, 3839–3850. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Chittoria, N.; Ehsani, S.; Rui, H.; Dolezal, M.; Stork-Sloots, L.; de Snoo, F.; Recknor, C.; Abramson, V. Abstract P5-17-08: A phase Ib/II study of leronlimab combined with carboplatin in patients with CCR5+ metastatic triple-negative breast cancer (mTNBC). Cancer Res. 2022, 82, P5–P17. [Google Scholar] [CrossRef]
- Stewart, J. Leronlimab FDA Approval Status. 2021. Available online: https://www.drugs.com/history/leronlimab.html (accessed on 4 May 2023).
- Mumal, I. CytoDyn Files for FDA Breakthrough Therapy Status for Leronlimab for Metastatic TNBC. Available online: https://breastcancer-news.com/2020/01/14/cytodyn-files-for-breakthrough-therapy-designation-for-leronlimab-for-mtnbc/ (accessed on 4 May 2023).
- Pelosci, A. Leronlimab Provides a Significant Survival Benefit in Metastatic Triple-Negative Breast Cancer. Available online: https://www.cancernetwork.com/view/leronlimab-provides-a-significant-survival-benefit-in-metastatic-triple-negative-breast-cancer (accessed on 27 April 2023).
- Brett, E.; Duscher, D.; Pagani, A.; Daigeler, A.; Kolbenschlag, J.; Hahn, M. Naming the Barriers between Anti-CCR5 Therapy, Breast Cancer and Its Microenvironment. Int. J. Mol. Sci. 2022, 23, 14159. [Google Scholar] [CrossRef] [PubMed]
- CytoDyn, I. A Compassionate Use Study of Leronlimab in Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/study/NCT04313075 (accessed on 1 May 2023).
- CytoDyn, I. Basket Study of Leronlimab (PRO 140) in Patients With CCR5+ Locally Advanced or Metastatic Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT04504942 (accessed on 1 May 2023).
- Jeon, Y.; Jo, U.; Hong, J.; Gong, G.; Lee, H.J. Trophoblast cell-surface antigen 2 (TROP2) expression in triple-negative breast cancer. BMC Cancer 2022, 22, 1014. [Google Scholar] [CrossRef] [PubMed]
- Izci, H.; Punie, K.; Waumans, L.; Laenen, A.; Wildiers, H.; Verdoodt, F.; Desmedt, C.; Ardui, J.; Smeets, A.; Han, S.N.; et al. TROP-2 expression in triple-negative breast cancer: Correlations with tumor-infiltrating lymphocytes, histological subtypes and survival. Res. Sq. 2022, 1–22. [Google Scholar] [CrossRef]
- Al-Saad, S.; Donnem, T.; Al-Shibli, K.; Persson, M.; Bremnes, R.M.; Busund, L.-T. Diverse prognostic roles of Akt isoforms, PTEN and PI3K in tumor epithelial cells and stromal compartment in non-small cell lung cancer. Anticancer. Res. 2009, 29, 4175–4183. [Google Scholar] [PubMed]
- West, L.; Vidwans, S.J.; Campbell, N.P.; Shrager, J.; Simon, G.R.; Bueno, R.; Dennis, P.A.; Otterson, G.A.; Salgia, R. A Novel Classification of Lung Cancer into Molecular Subtypes. PLoS ONE 2012, 7, e31906. [Google Scholar] [CrossRef]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Xie, Y.; Naizabekov, S.; Chen, Z.; Tokay, T. Power of PTEN/AKT: Molecular switch between tumor suppressors and oncogenes. Oncol. Lett. 2016, 12, 375–378. [Google Scholar] [CrossRef]
- Rampurwala, M.; Wisinski, K.B.; O’Regan, R. Role of the androgen receptor in triple-negative breast cancer. Clin. Adv. Hematol. Oncol. H&O 2016, 14, 186–193. [Google Scholar]
- Gerratana, L.; Basile, D.; Buono, G.; De Placido, S.; Giuliano, M.; Minichillo, S.; Coinu, A.; Martorana, F.; De Santo, I.; Del Mastro, L.; et al. Androgen receptor in triple negative breast cancer: A potential target for the targetless subtype. Cancer Treat. Rev. 2018, 68, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, Y.S.; Jang, E.D.; Seo, K.J.; Kim, J.S. Prognostic Impact and Clinicopathological Correlation of CD133 and ALDH1 Expression in Invasive Breast Cancer. J. Breast Cancer 2015, 18, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef]
- Panigoro, S.S.; Kurnia, D.; Kurnia, A.; Haryono, S.J.; Albar, Z.A. ALDH1 Cancer Stem Cell Marker as a Prognostic Factor in Triple-Negative Breast Cancer. Int. J. Surg. Oncol. 2020, 2020, 7863243. [Google Scholar] [CrossRef]
- Yamada, A.; Suzuki, C.; Shima, H.; Kida, K.; Adachi, S.; Yamamoto, S.; Narui, K.; Tanabe, M.; Shimizu, D.; Taniguchi, R.; et al. Aldehyde Dehydrogenase 1-related Genes in Triple-negative Breast Cancer Investigated Using Network Analysis. Anticancer. Res. 2020, 40, 6733–6742. [Google Scholar] [CrossRef]
- de Bessa Garcia, S.; Araújo, M.; Pereira, T.; Freitas, R. HOXB7 Overexpression Leads Triple-Negative Breast Cancer Cells to a Less Aggressive Phenotype. Biomedicines 2021, 9, 515. [Google Scholar] [CrossRef]
- Paço, A.; Leitão-Castro, J.; Freitas, R. Epigenetic Regulation of CDH1 Is Altered after HOXB7-Silencing in MDA-MB-468 Triple-Negative Breast Cancer Cells. Genes 2021, 12, 1575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, Y.; Su, X.; Lu, Y. HOXD8 inhibits the proliferation and migration of triple-negative breast cancer cells and induces apoptosis in them through regulation of AKT/mTOR pathway. Reprod. Biol. 2021, 21, 100544. [Google Scholar] [CrossRef]
- Spasojevic, C.; Marangoni, E.; Vacher, S.; Assayag, F.; Meseure, D.; Château-Joubert, S.; Humbert, M.; Karam, M.; Ricort, J.M.; Auclair, C.; et al. PKD1 is a potential biomarker and therapeutic target in triple-negative breast cancer. Oncotarget 2018, 9, 23208–23219. [Google Scholar] [CrossRef]
- Merzoug-Larabi, M.; Youssef, I.; Bui, A.T.; Legay, C.; Loiodice, S.; Lognon, S.; Babajko, S.; Ricort, J.-M. Protein Kinase D1 (PKD1) Is a New Functional Non-Genomic Target of Bisphenol A in Breast Cancer Cells. Front. Pharmacol. 2020, 10, 1683. [Google Scholar] [CrossRef]
- Dai, T.; Rosario, S.R.; Katsuta, E.; Dessai, A.S.; Paterson, E.J.; Novickis, A.T.; Gomez, E.C.; Zhu, B.; Liu, S.; Wang, H.; et al. Hypoxic activation of PFKFB4 in breast tumor microenvironment shapes metabolic and cellular plasticity to accentuate metastatic competence. Cell Rep. 2022, 41, 111756. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.-C.; Yang, H.; Shan, H.-B.; Su, H.-F.; Jiang, W.-Q.; Shi, Y.-X. PFKFB4 Overexpression Facilitates Proliferation by Promoting the G1/S Transition and Is Associated with a Poor Prognosis in Triple-Negative Breast Cancer. Dis. Markers 2021, 2021, 8824589S. [Google Scholar] [CrossRef] [PubMed]
- Peshkin, B.N.; Alabek, M.L.; Isaacs, C. BRCA1/2 mutations and triple negative breast cancers. Breast Dis. 2011, 32, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Hahnen, E.; Hauke, J.; Engel, C.; Neidhardt, G.; Rhiem, K.; Schmutzler, R.K. Germline Mutations in Triple-Negative Breast Cancer. Breast Care 2017, 12, 15–19. [Google Scholar] [CrossRef]
- Mitri, Z.I.; Abuhadra, N.; Goodyear, S.M.; Hobbs, E.A.; Kaempf, A.; Thompson, A.M.; Moulder, S.L. Impact of TP53 mutations in Triple Negative Breast Cancer. NPJ Precis. Oncol. 2022, 6, 64. [Google Scholar] [CrossRef]
- Peng, J.; Yang, M.; Bi, R.; Wang, Y.; Wang, C.; Wei, X.; Zhang, Z.; Xie, X.; Wei, W. Targeting Mutated p53 Dependency in Triple-Negative Breast Cancer Cells Through CDK7 Inhibition. Front. Oncol. 2021, 11, 664848. [Google Scholar] [CrossRef]
- González-González, A.; Muñoz-Muela, E.; Marchal, J.A.; Cara, F.E.; Molina, M.P.; Cruz-Lozano, M.; Jiménez, G.; Verma, A.; Ramírez, A.; Qian, W.; et al. Activating Transcription Factor 4 Modulates TGFβ-Induced Aggressiveness in Triple-Negative Breast Cancer via SMAD2/3/4 and mTORC2 Signaling. Clin. Cancer Res. 2018, 24, 5697–5709. [Google Scholar] [CrossRef]
- Wang, M.; Lu, Y.; Wang, H.; Wu, Y.; Xu, X.; Li, Y. High ATF4 Expression Is Associated With Poor Prognosis, Amino Acid Metabolism, and Autophagy in Gastric Cancer. Front. Oncol. 2021, 11, 740120. [Google Scholar] [CrossRef]
- Yuan, Z.-Y.; Dai, T.; Wang, S.-S.; Peng, R.-J.; Li, X.-H.; Qin, T.; Song, L.-B.; Wang, X. Overexpression of ETV4 protein in triple-negative breast cancer is associated with a higher risk of distant metastasis. OncoTargets Ther. 2014, 7, 1733–1742. [Google Scholar] [CrossRef]
- Dumortier, M.; Ladam, F.; Damour, I.; Vacher, S.; Bièche, I.; Marchand, N.; de Launoit, Y.; Tulasne, D.; Chotteau-Lelièvre, A. ETV4 transcription factor and MMP13 metalloprotease are interplaying actors of breast tumorigenesis. Breast Cancer Res. 2018, 20, 73. [Google Scholar] [CrossRef]
- Saba, R.; Alsayed, A.; Zacny, J.P.; Dudek, A.Z. The Role of Forkhead Box Protein M1 in Breast Cancer Progression and Resistance to Therapy. Int. J. Breast Cancer 2016, 2016, 9768183. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Hu, P.; Lakowski, T.M. Bioinformatics driven discovery of small molecule compounds that modulate the FOXM1 and PPARA pathway activities in breast cancer. Pharm. J. 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wang, Q.; Xie, Y.; Qiao, X.; Zhang, S.; Wang, Y.; Yang, Y.; Zhang, B. Identification of FOXM1 as a specific marker for triple-negative breast cancer. Int. J. Oncol. 2018, 54, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Kanai, A.; McNamara, K.M.; Iwabuchi, E.; Miki, Y.; Onodera, Y.; Guestini, F.; Khalid, F.; Sagara, Y.; Ohi, Y.; Rai, Y.; et al. Significance of glucocorticoid signaling in triple-negative breast cancer patients: A newly revealed interaction with androgen signaling. Breast Cancer Res. Treat. 2020, 180, 97–110. [Google Scholar] [CrossRef]
- Vilasco, M.; Bracaps; Communal, L.; Hugon-Rodin, J.; Penault-Llorca, F.; Mourra, N.; Wu, Z.; Forgez, P.; Gompel, A. Loss of glucocorticoid receptor activation is a hallmark of BRCA1-mutated breast tissue. Breast Cancer Res. Treat. 2013, 142, 283–296. [Google Scholar] [CrossRef]
- Buschmann, D.; González, R.; Kirchner, B.; Mazzone, C.; Pfaffl, M.; Schelling, G.; Steinlein, O.; Reithmair, M. Glucocorticoid receptor overexpression slightly shifts microRNA expression patterns in triple-negative breast cancer. Int. J. Oncol. 2018, 52, 1765–1776. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, W. The Emerging Regulation of VEGFR-2 in Triple-Negative Breast Cancer. Front. Endocrinol. 2015, 6, 159. [Google Scholar] [CrossRef]
- Fu, S.; Lin, J. Blocking Interleukin-6 and Interleukin-8 Signaling Inhibits Cell Viability, Colony-forming Activity, and Cell Migration in Human Triple-negative Breast Cancer and Pancreatic Cancer Cells. Anticancer. Res. 2018, 38, 6271–6279. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jeon, M.; Lee, J.E.; Nam, S.J. MEK-dependent IL-8 induction regulates the invasiveness of triple-negative breast cancer cells. Tumor Biol. 2015, 37, 4991–4999. [Google Scholar] [CrossRef]
- Verma, M.; Patel, P.; Verma, M. Biomarkers in Prostate Cancer Epidemiology. Cancers 2011, 3, 3773–3798. [Google Scholar] [CrossRef]
- Gluz, O.; Liedtke, C.; Gottschalk, N.; Pusztai, L.; Nitz, U.; Harbeck, N. Triple-negative breast cancer—Current status and future directions. Ann. Oncol. 2009, 20, 1913–1927. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; O’Donovan, N.; Crown, J. Use of molecular markers for predicting therapy response in cancer patients. Cancer Treat. Rev. 2011, 37, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Gómez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pêgo, A.; Chan, A.; et al. Randomized Phase II Study of the Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Cetuximab With Cisplatin Versus Cisplatin Alone in Patients With Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2013, 31, 2586–2592, Erratum in J. Clin. Oncol. 2018, 36, 98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; LaFortune, T.A.; Krishnamurthy, S.; Esteva, F.J.; Cristofanilli, M.; Liu, P.; Lucci, A.; Singh, B.; Hung, M.-C.; Hortobagyi, G.N.; et al. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Reverses Mesenchymal to Epithelial Phenotype and Inhibits Metastasis in Inflammatory Breast Cancer. Clin. Cancer Res. 2009, 15, 6639–6648. [Google Scholar] [CrossRef]
- Ueno, N.T.; Zhang, D. Targeting EGFR in Triple Negative Breast Cancer. J. Cancer 2011, 2, 324–328. [Google Scholar] [CrossRef]
- Arafeh, R.; Samuels, Y. PIK3CA in cancer: The past 30 years. Semin. Cancer Biol. 2019, 59, 36–49. [Google Scholar] [CrossRef]
- Guo, Z.; Primeau, T.; Luo, J.; Zhang, C.; Sun, H.; Hoog, J.; Gao, F.; Huang, S.; Edwards, D.P.; Davies, S.R.; et al. Proteomic Resistance Biomarkers for PI3K Inhibitor in Triple Negative Breast Cancer Patient-Derived Xenograft Models. Cancers 2020, 12, 3857. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Kuei, C.-H.; Lee, H.-H.; Lin, C.-H.; Zheng, J.-Q.; Chiu, H.-W.; Chen, C.-L.; Lin, Y.-F. The Gαh/phospholipase C-δ1 interaction promotes autophagosome degradation by activating the Akt/mTORC1 pathway in metastatic triple-negative breast cancer. Aging 2020, 12, 13023–13037. [Google Scholar] [CrossRef]
- Kim, S.-B.; Dent, R.; Im, S.-A.; Espié, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.J.; et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017, 18, 1360–1372. [Google Scholar] [CrossRef]
- Linderholm, B.; Hellborg, H.; Johansson, U.; Elmberger, G.; Skoog, L.; Lehtiö, J.; Lewensohn, R. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann. Oncol. 2009, 20, 1639–1646. [Google Scholar] [CrossRef]
- Brufsky, A.; Valero, V.; Tiangco, B.; Dakhil, S.; Brize, A.; Rugo, H.S.; Rivera, R.; Duenne, A.; Bousfoul, N.; Yardley, D.A. Second-line bevacizumab-containing therapy in patients with triple-negative breast cancer: Subgroup analysis of the RIBBON-2 trial. Breast Cancer Res. Treat. 2012, 133, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Gerber, B.; Loibl, S.; Eidtmann, H.; Rezai, M.; Fasching, P.; Tesch, H.; Eggemann, H.; Schrader, I.; Kittel, K.; Hanusch, C.; et al. Neoadjuvant bevacizumab and anthracycline–taxane-based chemotherapy in 678 triple-negative primary breast cancers; results from the geparquinto study (GBG 44). Ann. Oncol. 2013, 24, 2978–2984. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, J.; Xu, B.; Jiang, Z.; Ragaz, J.; Tong, Z.; Zhang, Q.; Wang, X.; Feng, J.; Pang, D.; et al. Multicenter phase II study of apatinib, a novel VEGFR inhibitor in heavily pretreated patients with metastatic triple-negative breast cancer. Int. J. Cancer 2014, 135, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-F.; Hsu, P.-N. Cancer immunotherapy by targeting immune checkpoints: Mechanism of T cell dysfunction in cancer immunity and new therapeutic targets. J. Biomed. Sci. 2017, 24, 35. [Google Scholar] [CrossRef] [PubMed]
- Vikas, P.; Borcherding, N.; Zhang, W. The clinical promise of immunotherapy in triple-negative breast cancer. Cancer Manag. Res. 2018, 10, 6823–6833. [Google Scholar] [CrossRef]
- Li, Y.; Zhan, Z.; Yin, X.; Fu, S.; Deng, X. Targeted Therapeutic Strategies for Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 731535. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef]
- Hu, Y.; Gao, J.; Wang, M.; Li, M. Potential Prospect of CDK4/6 Inhibitors in Triple-Negative Breast Cancer. Cancer Manag. Res. 2021, 13, 5223–5237. [Google Scholar] [CrossRef]
- Teo, Z.L.; Versaci, S.; Dushyanthen, S.; Caramia, F.; Savas, P.; Mintoff, C.P.; Zethoven, M.; Virassamy, B.; Luen, S.J.; McArthur, G.A.; et al. Combined CDK4/6 and PI3Kα Inhibition Is Synergistic and Immunogenic in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 6340–6352. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Tambaro, F.P.; Dell’aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2015, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter Hypermethylation and BRCA1 Inactivation in Sporadic Breast and Ovarian Tumors. Gynecol. Oncol. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef]
- Jhan, J.-R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar] [CrossRef]
- Yang, X.; Ferguson, A.T.; Nass, S.J.; Phillips, D.L.; Butash, K.A.; Wang, S.M.; Herman, J.G.; Davidson, N.E. Transcriptional activation of estrogen receptor alpha in human breast cancer cells by histone deacetylase inhibition. Cancer Res. 2000, 60, 6890–6894. [Google Scholar]
- Sulaiman, A.; McGarry, S.; Lam, K.M.; El-Sahli, S.; Chambers, J.; Kaczmarek, S.; Li, L.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; et al. Co-inhibition of mTORC1, HDAC and ESR1α retards the growth of triple-negative breast cancer and suppresses cancer stem cells. Cell Death Dis. 2018, 9, 815. [Google Scholar] [CrossRef]
- Min, A.; Im, S.-A.; Kim, D.K.; Song, S.-H.; Kim, H.-J.; Lee, K.-H.; Kim, T.-Y.; Han, S.-W.; Oh, D.-Y.; O’Connor, M.J.; et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015, 17, 33. [Google Scholar] [CrossRef]
- Tutt, A.N.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.M.; Yau, C.; Sanil, A.; Glas, A.; Petricoin, E.; Wulfkuhle, J.; Severson, T.M.; Linn, S.; Brown-Swigart, L.; Hirst, G.; et al. DNA repair deficiency biomarkers and the 70-gene ultra-high risk signature as predictors of veliparib/carboplatin response in the I-SPY 2 breast cancer trial. NPJ Breast Cancer 2017, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Jensen, K.C.; Vinayak, S.; Kurian, A.W.; Lipson, J.A.; Flaherty, P.J.; Timms, K.; Abkevich, V.; Schackmann, E.A.; Wapnir, I.L.; et al. Phase II Study of Gemcitabine, Carboplatin, and Iniparib As Neoadjuvant Therapy for Triple-Negative and BRCA1/2 Mutation–Associated Breast Cancer With Assessment of a Tumor-Based Measure of Genomic Instability: PrECOG 0105. J. Clin. Oncol. 2015, 33, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Sabnis, G.J.; Goloubeva, O.; Chumsri, S.; Nguyen, N.; Sukumar, S.; Brodie, A.M.H. Functional Activation of the Estrogen Receptor-α and Aromatase by the HDAC Inhibitor Entinostat Sensitizes ER-Negative Tumors to Letrozole. Cancer Res. 2011, 71, 1893–1903. [Google Scholar] [CrossRef] [PubMed]
- Cretella, D.; Fumarola, C.; Bonelli, M.; Alfieri, R.; La Monica, S.; Digiacomo, G.; Cavazzoni, A.; Galetti, M.; Generali, D.; Petronini, P.G. Pre-treatment with the CDK4/6 inhibitor palbociclib improves the efficacy of paclitaxel in TNBC cells. Sci. Rep. 2019, 9, 13014. [Google Scholar] [CrossRef]
- Zhou, Z.-R.; Yang, Z.-Z.; Wang, S.-J.; Zhang, L.; Luo, J.-R.; Feng, Y.; Yu, X.-L.; Chen, X.-X.; Guo, X.-M. The Chk1 inhibitor MK-8776 increases the radiosensitivity of human triple-negative breast cancer by inhibiting autophagy. Acta Pharmacol. Sin. 2017, 38, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Jung, H.; Li, X. Drug Delivery Approaches in Addressing Clinical Pharmacology-Related Issues: Opportunities and Challenges. AAPS J. 2015, 17, 1327–1340. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Laffleur, F.; Keckeis, V. WITHDRAWN: Advances in drug delivery systems: Work in progress still needed? Int. J. Pharm. X 2020, 2, 100050. [Google Scholar] [CrossRef]
- Vargason, A.M.; Anselmo, A.C.; Mitragotri, S. The evolution of commercial drug delivery technologies. Nat. Biomed. Eng. 2021, 5, 951–967. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, V. Chemotherapy: Managing side effects and safe handling. Can. Vet. J. 2009, 50, 665–668. [Google Scholar]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, M.; Gao, X.; Chen, Y.; Liu, T. Nanotechnology in cancer diagnosis: Progress, challenges and opportunities. J. Hematol. Oncol. 2019, 12, 137. [Google Scholar] [CrossRef]
- Hou, K.; Ning, Z.; Chen, H.; Wu, Y. Nanomaterial Technology and Triple Negative Breast Cancer. Front. Oncol. 2022, 11, 828810. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Zhang, F.; Peng, Y.; Xie, T.; Wang, Y.; Lan, Y. Current Progress in Cancer Treatment Using Nanomaterials. Front. Oncol. 2022, 12, 930125. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Kumar, D.N.; Dehari, D.; Singh, S.; Kumar, P.; Bolla, P.K.; Kumar, D.; Agrawal, A.K. Emergence of Nanotechnology as a Powerful Cavalry against Triple-Negative Breast Cancer (TNBC). Pharmaceuticals 2022, 15, 542. [Google Scholar] [CrossRef]
- Siegler, E.L.; Kim, Y.J.; Wang, P. Nanomedicine targeting the tumor microenvironment: Therapeutic strategies to inhibit angiogenesis, remodel matrix, and modulate immune responses. J. Cell. Immunother. 2016, 2, 69–78. [Google Scholar] [CrossRef]
- Fernandes, C.; Suares, D.; Yergeri, M.C. Tumor Microenvironment Targeted Nanotherapy. Front. Pharmacol. 2018, 9, 1230. [Google Scholar] [CrossRef]
- Mendes, R.; Carreira, B.; Baptista, P.V.; Fernandes, A.R. Non-small cell lung cancer biomarkers and targeted therapy–two faces of the same coin fostered by nanotechnology. Expert Rev. Precis. Med. Drug Dev. 2016, 1, 155–168. [Google Scholar] [CrossRef]
- Webb, J.A.; Ou, Y.-C.; Faley, S.; Paul, E.P.; Hittinger, J.P.; Cutright, C.C.; Lin, E.C.; Bellan, L.M.; Bardhan, R. Theranostic Gold Nanoantennas for Simultaneous Multiplexed Raman Imaging of Immunomarkers and Photothermal Therapy. ACS Omega 2017, 2, 3583–3594. [Google Scholar] [CrossRef] [PubMed]
- Harmon, T.; Harbuzariu, A.; Lanier, V.; Lipsey, C.C.; Kirlin, W.; Yang, L.; Gonzalez-Perez, R.R. Nanoparticle-linked antagonist for leptin signaling inhibition in breast cancer. World J. Clin. Oncol. 2017, 8, 54–66. [Google Scholar] [CrossRef] [PubMed]
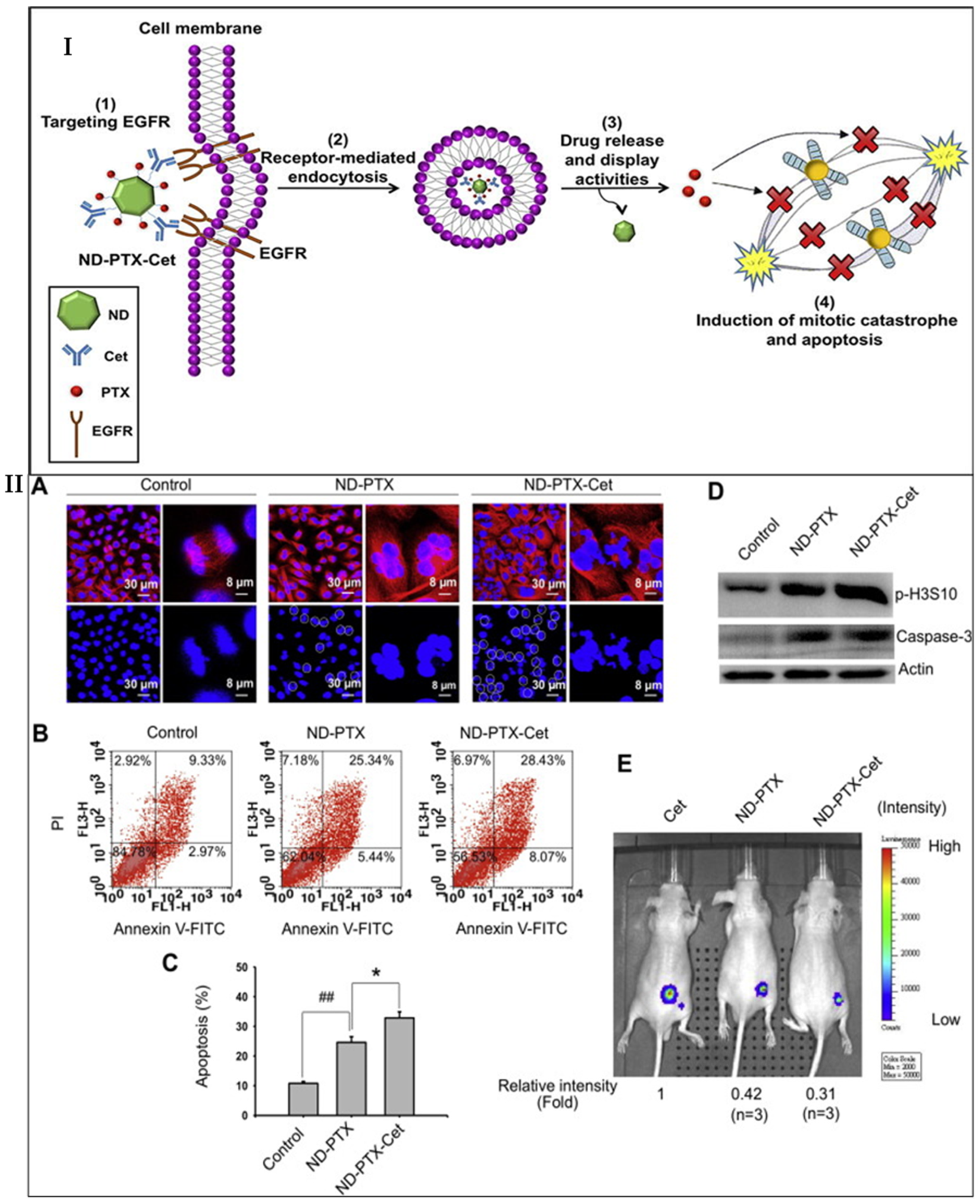
- Liao, W.-S.; Ho, Y.; Lin, Y.-W.; Naveen Raj, E.; Liu, K.K.; Chen, C.; Zhou, X.-Z.; Lu, K.-P.; Chao, J.-I. Targeting EGFR of triple-negative breast cancer enhances the therapeutic efficacy of paclitaxel- and cetuximab-conjugated nanodiamond nanocomposite. Acta Biomater. 2019, 86, 395–405. [Google Scholar] [CrossRef]
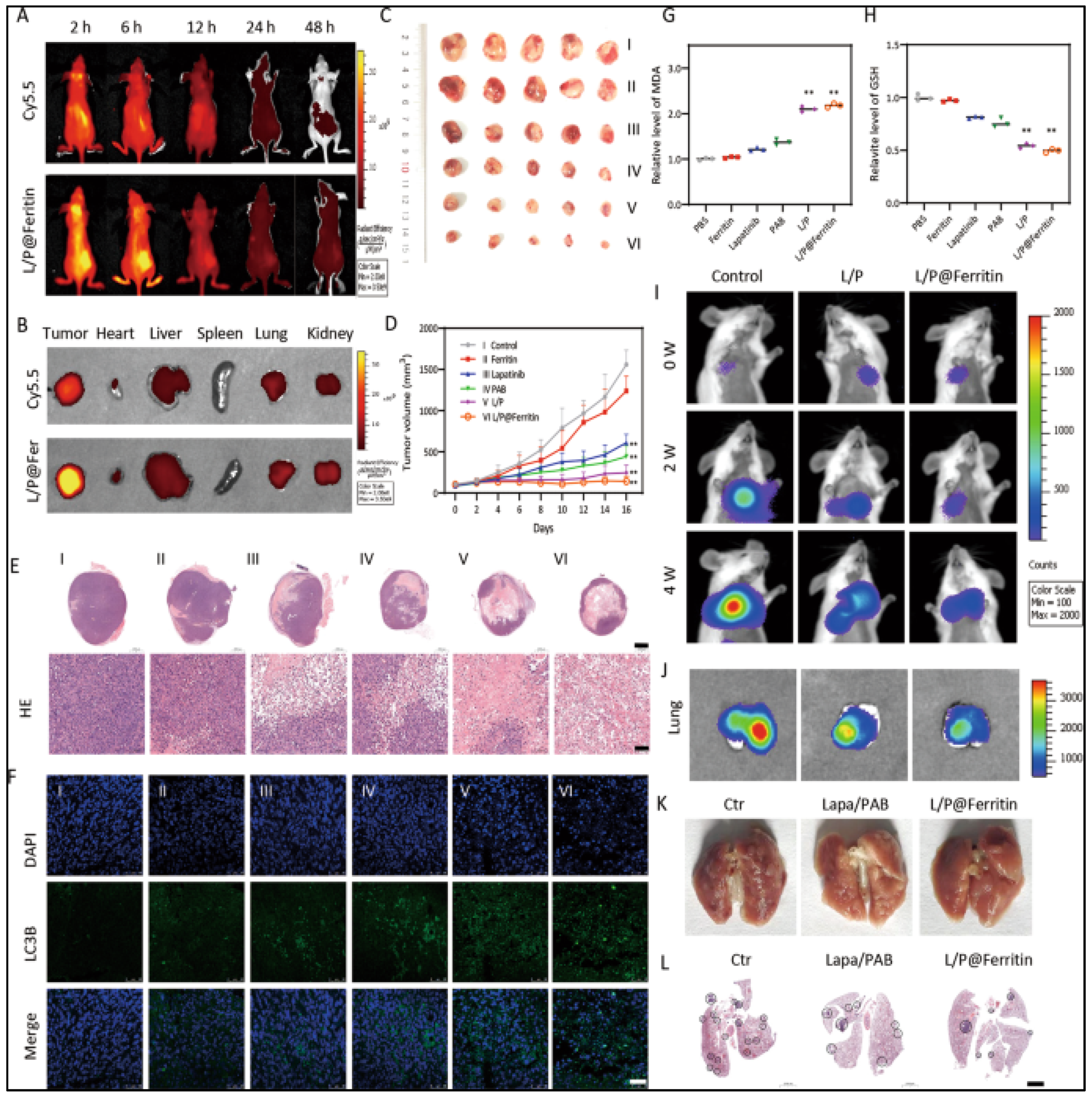
- Wu, X.; Sheng, H.; Zhao, L.; Jiang, M.; Lou, H.; Miao, Y.; Ni Cheng, N.; Zhang, W.; Ding, D.; Li, W. Co-loaded lapatinib/PAB by ferritin nanoparticles eliminated ECM-detached cluster cells via modulating EGFR in triple-negative breast cancer. Cell Death Dis. 2022, 13, 557. [Google Scholar] [CrossRef]
- Malarvizhi, G.L.; Chandran, P.; Retnakumari, A.P.; Ramachandran, R.; Gupta, N.; Nair, S.; Koyakutty, M. A rationally designed photo-chemo core-shell nanomedicine for inhibiting the migration of metastatic breast cancer cells followed by photodynamic killing. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, Q.; Mo, J.; Dai, H. Drug-Loaded Polymeric Nanoparticles for Cancer Stem Cell Targeting. Front. Pharmacol. 2017, 8, 51. [Google Scholar] [CrossRef]
- Babu, A.; Templeton, A.K.; Munshi, A.; Ramesh, R. Nanoparticle-Based Drug Delivery for Therapy of Lung Cancer: Progress and Challenges. J. Nanomater. 2013, 2013, 863951. [Google Scholar] [CrossRef]
- Blanco, E.; Sangai, T.; Wu, S.; Hsiao, A.; Ruiz-Esparza, G.U.; Gonzalez-Delgado, C.A.; Cara, F.E.; Granados-Principal, S.; Evans, K.W.; Akcakanat, A.; et al. Colocalized Delivery of Rapamycin and Paclitaxel to Tumors Enhances Synergistic Targeting of the PI3K/Akt/mTOR Pathway. Mol. Ther. 2014, 22, 1310–1319. [Google Scholar] [CrossRef]
- Li, S.-Y.; Sun, R.; Wang, H.-X.; Shen, S.; Liu, Y.; Du, X.-J.; Zhu, Y.-H.; Jun, W. Combination therapy with epigenetic-targeted and chemotherapeutic drugs delivered by nanoparticles to enhance the chemotherapy response and overcome resistance by breast cancer stem cells. J. Control. Release 2015, 205, 7–14. [Google Scholar] [CrossRef]
- Bakrania, A.K.; Variya, B.C.; Rathod, L.V.; Patel, S.S. DEAE-Dextran coated paclitaxel nanoparticles act as multifunctional nano system for intranuclear delivery to triple negative breast cancer through VEGF and NOTCH1 inhibition. Eur. J. Pharm. Biopharm. 2018, 122, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Sabra, S.A.; Sheweita, S.A.; Haroun, M.; Ragab, D.; Eldemellawy, M.A.; Xia, Y.; Goodale, D.; Allan, A.L.; Elzoghby, A.O.; Rohani, S. Magnetically Guided Self-Assembled Protein Micelles for Enhanced Delivery of Dasatinib to Human Triple-Negative Breast Cancer Cells. J. Pharm. Sci. 2018, 108, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, A.; Ebtekar, M.; Pakravan, N.; Hassan, Z.M. Folate receptor alpha targeted delivery of artemether to breast cancer cells with folate-decorated human serum albumin nanoparticles. Int. J. Biol. Macromol. 2020, 152, 90–101. [Google Scholar] [CrossRef] [PubMed]
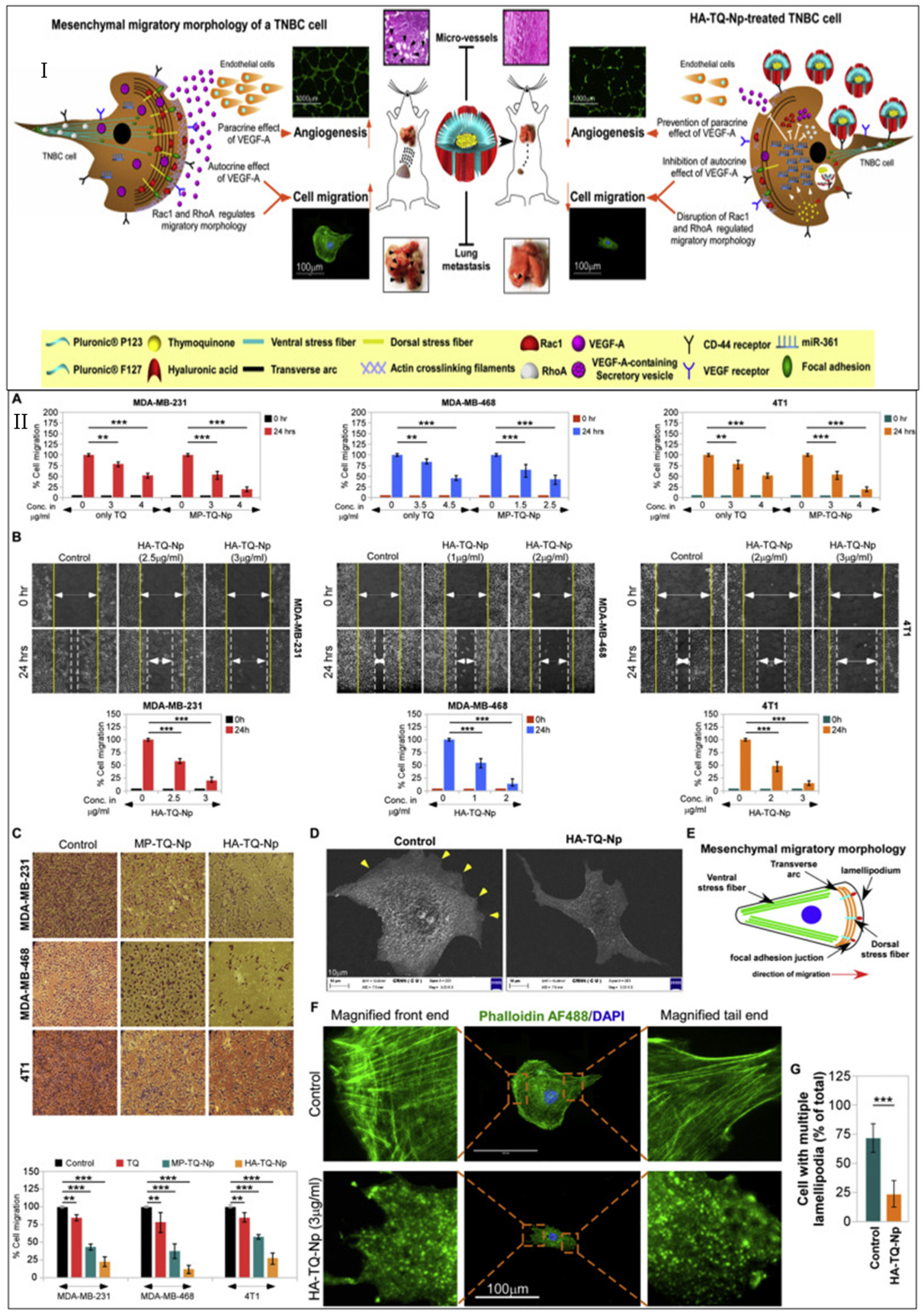
- Bhattacharya, S.; Ghosh, A.; Maiti, S.; Ahir, M.; Debnath, G.H.; Gupta, P.; Bhattacharjee, M.; Ghosh, S.; Chattopadhyay, S.; Mukherjee, P.; et al. Delivery of thymoquinone through hyaluronic acid-decorated mixed Pluronic® nanoparticles to attenuate angiogenesis and metastasis of triple-negative breast cancer. J. Control. Release 2020, 322, 357–374. [Google Scholar] [CrossRef]
- Qin, T.; Xu, X.; Zhang, Z.; Li, J.; You, X.; Guo, H.; Sun, H.; Liu, M.; Dai, Z.; Zhu, H. Paclitaxel/sunitinib-loaded micelles promote an antitumor response in vitro through synergistic immunogenic cell death for triple-negative breast cancer. Nanotechnology 2020, 31, 365101. [Google Scholar] [CrossRef]
- Misra, R.; Patra, B.; Varadharaj, S.; Verma, R.S. Establishing the promising role of novel combination of triple therapeutics delivery using polymeric nanoparticles for Triple negative breast cancer therapy. BioImpacts 2021, 11, 199–207. [Google Scholar] [CrossRef]
- Khesht, A.M.S.; Karpisheh, V.; Gilan, P.S.; Melnikova, L.A.; Zekiy, A.O.; Mohammadi, M.; Hojjat-Farsangi, M.; Zolbanin, N.M.; Mahmoodpoor, A.; Hassannia, H.; et al. Blockade of CD73 using siRNA loaded chitosan lactate nanoparticles functionalized with TAT-hyaluronate enhances doxorubicin mediated cytotoxicity in cancer cells both in vitro and in vivo. Int. J. Biol. Macromol. 2021, 186, 849–863. [Google Scholar] [CrossRef]
- Chen, C.; Guo, Q.; Fu, H.; Yu, J.; Wang, L.; Sun, Y.; Zhang, J.; Duan, Y. Asynchronous blockade of PD-L1 and CD155 by polymeric nanoparticles inhibits triple-negative breast cancer progression and metastasis. Biomaterials 2021, 275, 120988. [Google Scholar] [CrossRef]
- Zeng, X.; Wang, H.; Zhang, Y.; Xu, X.; Yuan, X.; Li, J. pH-Responsive Hyaluronic Acid Nanoparticles for Enhanced Triple Negative Breast Cancer Therapy. Int. J. Nanomed. 2022, 17, 1437–1457. [Google Scholar] [CrossRef]
- Kumar, D.N.; Chaudhuri, A.; Aqil, F.; Dehari, D.; Munagala, R.; Singh, S.; Gupta, R.C.; Agrawal, A.K. Exosomes as Emerging Drug Delivery and Diagnostic Modality for Breast Cancer: Recent Advances in Isolation and Application. Cancers 2022, 14, 1435. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Shrivastava, N.; Kumar, S.; Singh, A.K.; Ali, J.; Baboota, S. Designing and development of omega-3 fatty acid based self-nanoemulsifying drug delivery system (SNEDDS) of docetaxel with enhanced biopharmaceutical attributes for management of breast cancer. J. Drug Deliv. Sci. Technol. 2022, 68, 103117. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Kumar, D.N.; Shaik, R.A.; Eid, B.G.; Abdel-Naim, A.B.; Shadab, M.; Ahmad, A.; Agrawal, A.K. Lipid-Based Nanoparticles as a Pivotal Delivery Approach in Triple Negative Breast Cancer (TNBC) Therapy. Int. J. Mol. Sci. 2022, 23, 10068. [Google Scholar] [CrossRef] [PubMed]
- Sheoran, S.; Arora, S.; Samsonraj, R.; Govindaiah, P.; Vuree, S. Lipid-based nanoparticles for treatment of cancer. Heliyon 2022, 8, e09403. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Yang, J.; Jia, D.; Moses, M.A.; Auguste, D.T. ICAM-1-Targeted, Lcn2 siRNA-Encapsulating Liposomes are Potent Anti-angiogenic Agents for Triple Negative Breast Cancer. Theranostics 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, X.; Zhao, W.; Zeng, C.; Li, W.; Li, B.; Luo, X.; Li, J.; Jiang, J.; Deng, B.; et al. Chemotherapy drugs derived nanoparticles encapsulating mRNA encoding tumor suppressor proteins to treat triple-negative breast cancer. Nano Res. 2019, 12, 855–861. [Google Scholar] [CrossRef]
- Burande, A.S.; Viswanadh, M.K.; Jha, A.; Mehata, A.K.; Shaik, A.; Agrawal, N.; Poddar, S.; Mahto, S.K.; Muthu, M.S. EGFR Targeted Paclitaxel and Piperine Co-loaded Liposomes for the Treatment of Triple Negative Breast Cancer. AAPS PharmSciTech 2020, 21, 151. [Google Scholar] [CrossRef]
- Li, S.; Wu, Y.; Ding, F.; Yang, J.; Li, J.; Gao, X.; Zhang, C.; Feng, J. Engineering macrophage-derived exosomes for targeted chemotherapy of triple-negative breast cancer. Nanoscale 2020, 12, 10854–10862. [Google Scholar] [CrossRef]
- Darabi, F.; Saidijam, M.; Nouri, F.; Mahjub, R.; Soleimani, M. Anti-CD44 and EGFR Dual-Targeted Solid Lipid Nanoparticles for Delivery of Doxorubicin to Triple-Negative Breast Cancer Cell Line: Preparation, Statistical Optimization, and In Vitro Characterization. BioMed Res. Int. 2022, 2022, 6253978. [Google Scholar] [CrossRef]
- Zhu, L.; Mu, Q.; Yu, J.; Griffin, J.I.; Xu, X.; Ho, R.J.Y. ICAM-1 Targeted Drug Combination Nanoparticles Enhanced Gemcitabine-Paclitaxel Exposure and Breast Cancer Suppression in Mouse Models. Pharmaceutics 2021, 14, 89. [Google Scholar] [CrossRef]
- Gao, W.; Zhang, L. Coating nanoparticles with cell membranes for targeted drug delivery. J. Drug Target. 2015, 23, 619–626. [Google Scholar] [CrossRef]
- Yaman, S.; Chintapula, U.; Rodriguez, E.; Ramachandramoorthy, H.; Nguyen, K.T. Cell-mediated and cell membrane-coated nanoparticles for drug delivery and cancer therapy. Cancer Drug Resist. 2020, 3, 879–911. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Qi, J.; Liu, D.; You, Y.; Shu, G.; Du, Y.; Wang, J.; Xu, X.; Ying, X.; Ji, J.; et al. Cancer-cell-biomimetic Upconversion nanoparticles combining chemo-photodynamic therapy and CD73 blockade for metastatic triple-negative breast cancer. J. Control. Release 2021, 337, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Rampado, R.; Crotti, S.; Caliceti, P.; Pucciarelli, S.; Agostini, M. Recent Advances in Understanding the Protein Corona of Nanoparticles and in the Formulation of “Stealthy” Nanomaterials. Front. Bioeng. Biotechnol. 2020, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-M.; Cheng, T.-L.; Roffler, S.R. Polyethylene Glycol Immunogenicity: Theoretical, Clinical, and Practical Aspects of Anti-Polyethylene Glycol Antibodies. ACS Nano 2021, 15, 14022–14048. [Google Scholar] [CrossRef]
- Sushnitha, M.; Evangelopoulos, M.; Tasciotti, E.; Taraballi, F. Cell Membrane-Based Biomimetic Nanoparticles and the Immune System: Immunomodulatory Interactions to Therapeutic Applications. Front. Bioeng. Biotechnol. 2020, 8, 627. [Google Scholar] [CrossRef]
- Ottonelli, I.; Duskey, J.T.; Genovese, F.; Pederzoli, F.; Caraffi, R.; Valenza, M.; Tosi, G.; Vandelli, M.A.; Ruozi, B. Quantitative comparison of the protein corona of nanoparticles with different matrices. Int. J. Pharm. X 2022, 4, 100136. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Lozza, I.; Torres-Suárez, A.I. Actively Targeted Nanomedicines in Breast Cancer: From Pre-Clinal Investigation to Clinic. Cancers 2022, 14, 1198. [Google Scholar] [CrossRef]



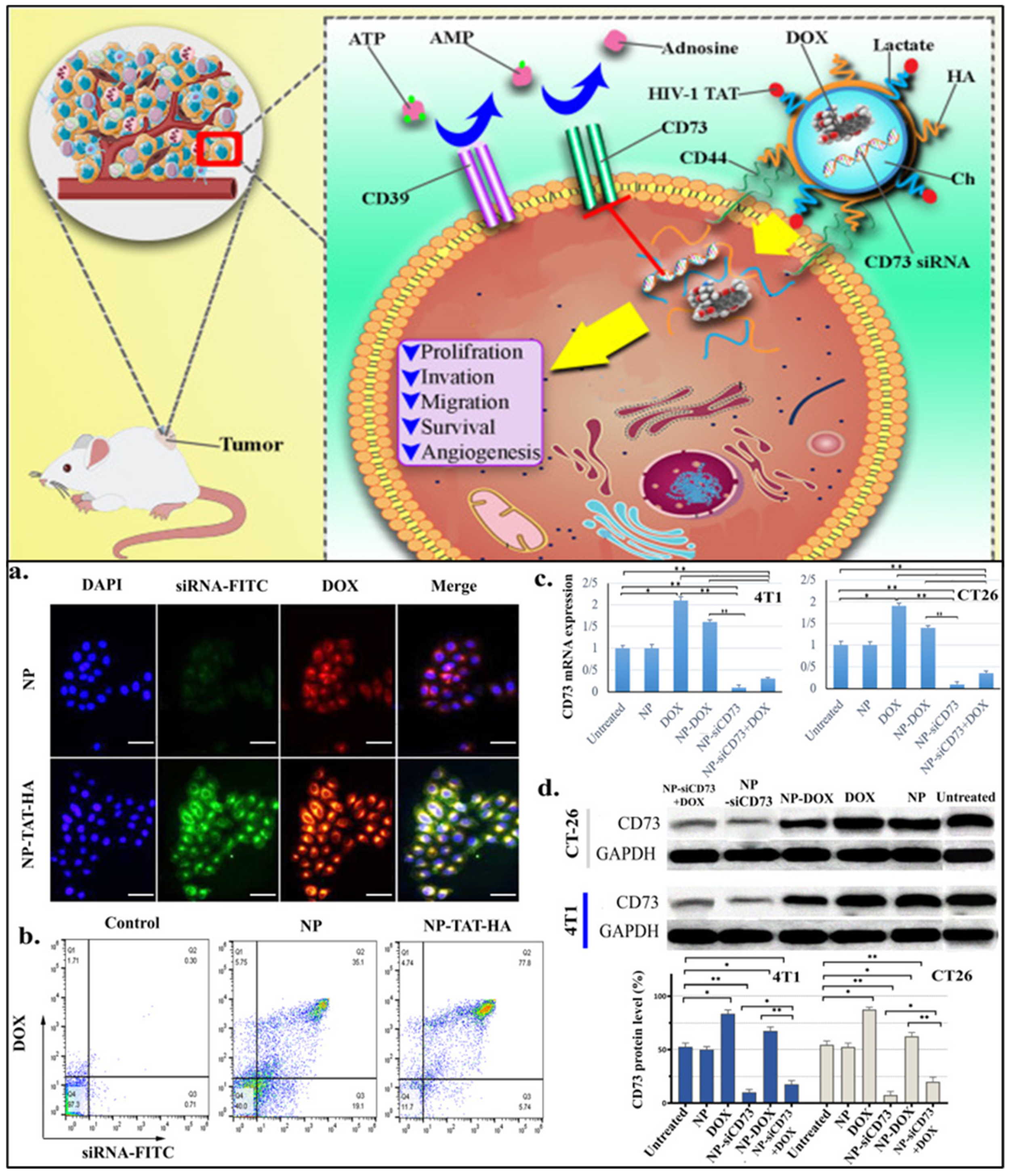

| Agents | Biomarkers (Targeting Moiety) | Clinical Phase | Identifier |
|---|---|---|---|
| APR-246 + Pembrolizumab | TP53 + PD-1 | I/II | NCT04383938 |
| Ribociclib + Bicalutamide | CDK4/6 | I/II | NCT03090165 |
| Taselisib + Enzalutamide | PI3K/AKT/mTOR | I/II | NCT02457910 |
| Alpelisib + Enzalutamide | PI3K/AKT/mTOR | I | NCT03207529 |
| Olaparib + Carboplatin/Paclitaxel | PARP | I | NCT00516724 |
| MEDI4736 + Olaparib and/or Cediranib | PD-L1 + PARP + VEGFR | I/II | NCT02484404 |
| Olaparib + Durvalumab | PARP + PD-L1 | II | NCT03801369 |
| Talazoparib | PARP | II | NCT03901469 |
| Olaparib + Onalespib | PARP + HSP90 | I | NCT02898207 |
| HX008 + Niraparib | PD-1 + PARP | II | NCT04508803 |
| Prexasertib | CHK1 | II | NCT02873975 |
| IDX-1197 | PARP | I/II | NCT04174716 |
| Avelumab | PD-L1 | II | NCT02554812 |
| Nivolumab + Bicalutamide + Ipilimumab | PD-1 + AR + CTLA4 | II | NCT03650894 |
| Avelumab + Binimetinib, Utomilumab, or anti-OX40 antibody | PD-L1 + MEK ½, CD 137 or OX40 | II | NCT03971409 |
| Atezolizumab in different combinations | PD-L1 in different combinations, including chemotherapy, ADC, CD40, IL6R, VEGFA, and AKT | I/II | NCT03424005 |
| Spartalizumab + LAG525 in combination with NIR178, Capmatinib, MCS110, or Canakinumab | PD-1 + LAG-3 in combination with anti- adenosine A2A receptor, Met receptor, CSF-1 or IL1β | I | NCT03742349 |
| Sacituzumab govitecan + Talazoparib | ADC + PARP | I/II | NCT04039230 |
| AMXI-5001 | PARP and a microtubule polymerization inhibitor | I/II | NCT04503265 |
| BKM120/BYL719 + Olaparib | PI3K + PARP | I | NCT01623349 |
| S. No. | Formulation | Targeting Biomarkers | Agent/Drug | Results | Ref. |
|---|---|---|---|---|---|
| 1 | Polymeric micelles | mTOR inhibitor: Rapamycin | Paclitaxel |
| [175] |
| 2 | Polymeric nanoparticles | DNA hypermethylation inhibitor: decitabine | Doxorubicin |
| [176] |
| 3 | PLGA polymeric nanoparticles | tyrosine kinase inhibitor, Dasatinib | Photo-sensitizer: m-tetra (hydroxyphenyl) chlorin (mTHPC), |
| [172] |
| 4 | pH-responsive liposomes | ICAM-1 antibody | Lipocalin 2 (Lcn2) siRNA |
| [190] |
| 5 | Multi-branched gold nanoantennas (MGN) | anti-PDL1 antibodies, and anti-EGFR antibodies | Dithio-bis-(2-nitrobenzoic acid), and pMBA (para mercaptobenzoic acid) |
| [168] |
| 6 | Iron oxide nanoparticles | Leptin antagonist: LPrA2 | Cisplatin, Doxorubicin, Cyclo-phosphamide, and Paclitaxel |
| [169] |
| 7 | Polymeric nanoparticles | VEGFR inhibitor: DEAE-Dextran | Paclitaxel |
| [177] |
| 8 | Oleic acid-coated Magnetite (Fe3O4) based polymeric micelles | Lactoferrin | Dasatinib |
| [178] |
| 9 | Nano-composite | EGFR inhibitor: cetuximab | Paclitaxel |
| [170] |
| 10 | Lipidic nanoparticles | P53 mRNA | Paclitaxel amino lipid |
| [191] |
| 11 | TPGS coated liposomes | EGFR inhibitor: Cetuximab | Paclitaxel and piperine |
| [192] |
| 12 | Albumin nanoparticles | Folic acid | Artemether |
| [179] |
| 13 | Polymeric nanoparticles | hyaluronic acid | Thymoquinone |
| [180] |
| 14 | Polymeric micelles | Inhibitor of tyrosine kinase receptor: sunitinib | Paclitaxel |
| [181] |
| 15 | Macrophage-derived exosomes | c-Met binding peptide | Doxorubicin |
| [193] |
| 16 | Chitosan nanoparticles | PARP inhibitor: olaparib, and FOXM1-siRNA | Paclitaxel |
| [182] |
| 17 | Cancer cell membrane (CM)-cloaked upconversion nanoparticles, | anti-CD73 antibody | ROS-sensitive polymer polyethylene glycol-thioketal-doxorubicin |
| [198] |
| 18 | Chitosan—lactate nanoparticles | HIV-1 derived TAT peptide and CD73 siRNA | Doxorubicin |
| [183] |
| 19 | Polymeric nanoparticles | PD-L1 blocking antibodies | CD155 siRNA |
| [184] |
| 20 | Ferritin nanoparticles | EGFR inhibitor: lapatinib | Pseudolaric acid B |
| [171] |
| 21 | Solid lipid nanoparticles (SLNs) | anti-EGFR/CD44 dual-RNA aptamers, | Doxorubicin |
| [194] |
| 22 | Polymeric micelles | PARP inhibitor; olaparib (OLA) | Dasatinib |
| [185] |
| 23 | Lipid nanoparticle | ICAM—1 binding peptide, LFA1–P | Gemcitabine and Paclitaxel |
| [195] |
| Formulation | Drug | Target/Biomarker | Ligand | Clinical Phase (NCT Number) |
|---|---|---|---|---|
| Glembatumumab—Vedotin-antibody drug conjugate | MMAE (auristatin) | NMB glycoprotein | Glembatumumab (anti NMB glycoprotein monoclonal antibody) | Phase II (NCT01997333) |
| Cofetuzumab—pelidotin (PF-06647020) Albumin nanoparticles | Aur001 (auristatin) | PTK7 (protein tyrosine kinase—7) | Cofetuzumab (anti-PTK7 monoclonal antibody) | Phase I (NCT03243331/NCT02222922) |
| PF-06647263—Albumin nanoparticles | Calicheamicin | Ephrin receptor-4 | Anti-Ephrin receptor-4 monoclonal antibody | Phase I (NCT02078752) |
| Nab-rapamycin—Albumin nanoparticles | Rapamycin | gP 60 receptors | Albumin | Phase I (NCT02646319) |
| C225-ILS-Dox—liposomes | Doxorubicin | EGFR | Antigen-binding fragment of cetuximab | Phase II (NCT02833766) |
| MM310—liposomes | Docetaxel pro-drug | Ephrin A2 | Anti-ephrin A2 monoclonal antibody | Phase I (NCT03076372) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhuri, A.; Kumar, D.N.; Dehari, D.; Patil, R.; Singh, S.; Kumar, D.; Agrawal, A.K. Endorsement of TNBC Biomarkers in Precision Therapy by Nanotechnology. Cancers 2023, 15, 2661. https://doi.org/10.3390/cancers15092661
Chaudhuri A, Kumar DN, Dehari D, Patil R, Singh S, Kumar D, Agrawal AK. Endorsement of TNBC Biomarkers in Precision Therapy by Nanotechnology. Cancers. 2023; 15(9):2661. https://doi.org/10.3390/cancers15092661
Chicago/Turabian StyleChaudhuri, Aiswarya, Dulla Naveen Kumar, Deepa Dehari, Rohit Patil, Sanjay Singh, Dinesh Kumar, and Ashish Kumar Agrawal. 2023. "Endorsement of TNBC Biomarkers in Precision Therapy by Nanotechnology" Cancers 15, no. 9: 2661. https://doi.org/10.3390/cancers15092661