
Idiopathic Multicentric Castleman Disease with Cutaneous Manifestation: Case Report

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
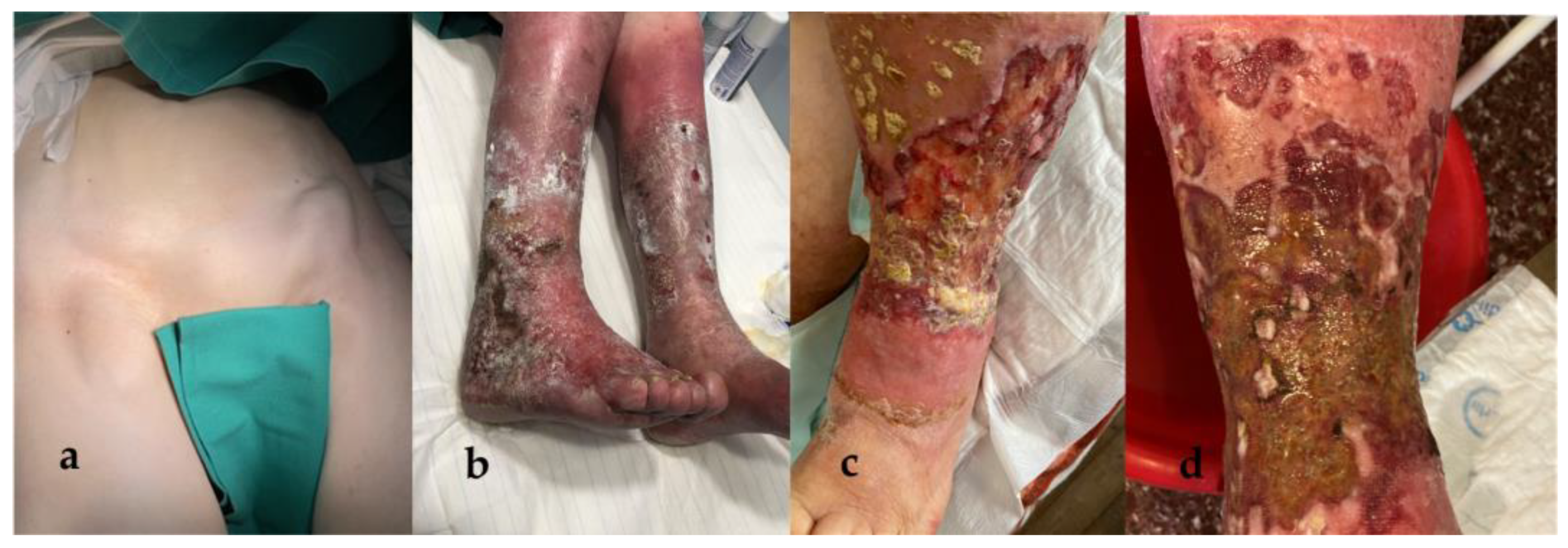
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Castleman, B.; Iverson, L.; Menendez, V.P. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer 1956, 9, 822–830. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Fajgenbaum, D.C. Overview of Castleman disease. Blood 2020, 135, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D. Epidemiology of Castleman Disease. Hematol. Oncol. Clin. N. Am. 2018, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Borok, M.; Damania, B.; Gloghini, A.; Polizzotto, M.N.; Jayanthan, R.K.; Fajgenbaum, D.C.; Bower, M. Castleman disease. Nat. Rev. Dis. Primers 2021, 7, 84. [Google Scholar] [CrossRef]
- Liu, A.Y.; Nabel, C.S.; Finkelman, B.S.; Ruth, J.R.; Kurzrock, R.; van Rhee, F.; Krymskaya, V.P.; Kelleher, D.; Rubenstein, A.H.; Fajgenbaum, D.C. Idiopathic multicentric Castleman’s disease: A systematic literature review. Lancet Haematol. 2016, 3, 163–175. [Google Scholar] [CrossRef]
- Chen, H.; Ba, W.; Chen, H.; Yang, H. Cutaneous involvement as initial presentation of multicentric plasmacytic Castleman disease. J. Cutan. Pathol. 2021, 48, 701–705. [Google Scholar] [CrossRef]
- Cervantes, C.E.; Correa, R. Castleman Disease: A Rare Condition with Endocrine Manifestations. Cureus 2015, 7, e380. [Google Scholar] [CrossRef]
- Oksenhendler, E.; Boutboul, D.; Fajgenbaum, D.; Mirouse, A.; Fieschi, C.; Malphettes, M.; Vercellino, L.; Meignin, V.; Gérard, L.; Galicier, L. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br. J. Haematol. 2018, 180, 206–216. [Google Scholar] [CrossRef]
- Cronin, D.M.; Warnke, R.A. Castleman disease: An update on classification and the spectrum of associated lesions. Adv. Anat. Pathol. 2009, 16, 236–246. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, H.W.; Pittaluga, S.; Jaffe, E.S. Multicentric Castleman disease and the evolution of the concept. Pathologica 2021, 113, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.X.; Ji, Y.; Wu, S. A CARE-compliant article: Unicentric Castleman disease presenting as a retroperitoneal mass of the upper edge of the pancreas: A case report. Medicine 2020, 99, e19515. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tang, D.; Sun, F. Clinical Analysis of Castleman’s Disease of the Lacrimal Gland. J. Ophthalmol. 2020, 2020, 3718305. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Quillin, K.; Rezaee, R. Pediatric Unicentric Castleman Disease of the Temporalis Muscle. JAMA Otolaryngol. Head Neck Surg. 2020, 147, 103–104. [Google Scholar] [CrossRef]
- Pribyl, K.; Vakayil, V.; Farooqi, N.; Arora, N.; Kreitz, B.; Ikramuddin, S.; Linden, M.A.; Harmon, J. Castleman disease: A single-center case series. Int. J. Surg. Case Rep. 2021, 80, 105650. [Google Scholar] [CrossRef]
- Dispenzieri, A. Castleman disease. Cancer Treat. Res. 2008, 142, 293–330. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A. POEMS Syndrome: 2019 Update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2019, 94, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Modolin, M.L.A.; Camargo, C.P.; Milcheski, D.A.; Cintra, W., Jr.; Rocha, R.I.; Clivatti, G.M.; Nascimento, B.; Gemperli, R. Castleman disease. Interaction with dermatopathy: Case report. Int. J. Surg. Case Rep. 2020, 73, 332–337. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sato, Y.; Takata, K.; Kobayashi, K.; Ohno, K.; Iwaki, N.; Orita, Y.; Yoshino, T. Cutaneous multicentric Castleman’s disease mimicking IgG4-related disease. Pathol. Res. Pract. 2012, 208, 746–749. [Google Scholar] [CrossRef]
- Park, H.Y.; Lee, J.J.; Lee, J.B.; Kim, S.J.; Lee, S.C.; Won, Y.H.; Yun, S.J. Castleman’s Disease with Cutaneous Involvement Manifestating as Multiple Violaceous Plaques on Entire Body. Ann. Dermatol. 2011, 23, 169–174. [Google Scholar] [CrossRef]
- Chavez-Alvarez, S.; Villarreal-Martinez, A.; Ocampo-Candiani, J.; Gomez-Flores, M.; Vazquez-Martinez, O.; Gonzalez-Saldivar, G.; Herz-Ruelas, M.E. Cutaneous manifestations of Castleman disease. Int. J. Dermatol. 2020, 59, 1226–1240. [Google Scholar] [CrossRef]
- Aita, T.; Hamaguchi, S.; Shimotani, Y.; Nakamoto, Y. Idiopathic multicentric Castleman disease preceded by cutaneous plasmacytosis successfully treated by tocilizumab. BMJ Case Rep. 2020, 13, e236283. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, B.; Tschen, J.A.; Cohen, P.R.; Zaki, M.H.; Rady, P.L.; Tyring, S.K.; Corringham, R.E.; Kurzrock, R. Cutaneous castleman’s disease responds to anti interleukin-6 treatment Mol. Cancer Ther. 2007, 6, 2386–2390. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Pittaluga, S.; Jaffe, E.S. Multicentric Castleman disease: Where are we now? Semin. Diagn. Pathol. 2016, 33, 294–306. [Google Scholar] [CrossRef]
- Larroche, C.; Cacoub, P.; Soulier, J.; Oksenhendler, E.; Clauvel, J.P.; Piette, J.C.; Raphael, M. Castleman’s disease and lymphoma: Report of eight cases in HIV-negative patients and literature review. Am. J. Hematol. 2002, 69, 119–126. [Google Scholar] [CrossRef]
- Bonekamp, D.; Horton, K.M.; Hruban, R.H.; Fishman, E.K. Castleman disease: The great mimic. Radiographics 2011, 31, 1793–1807. [Google Scholar] [CrossRef] [PubMed]
- Kligerman, S.J.; Auerbach, A.; Franks, T.J.; Galvin, J.R. Castleman Disease of the Thorax: Clinical, Radiologic, and Pathologic Correlation: From the Radiologic Pathology Archives. Radiographics 2016, 36, 1309–1332. [Google Scholar] [CrossRef]
- Lee, E.S.; Paeng, J.C.; Park, C.M.; Chang, W.; Lee, W.W.; Kang, K.W.; Chung, J.K.; Lee, D.S. Metabolic characteristics of Castleman disease on 18F-FDG PET in relation to clinical implication. Clin. Nucl. Med. 2013, 38, 339–342. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; Uldrick, T.S.; Bagg, A.; Frank, D.; Wu, D.; Srkalovic, G.; Simpson, D.; Liu, A.Y.; Menke, D.; Chandrakasan, S.; et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 2017, 129, 1646–1657. [Google Scholar] [CrossRef]
- Wu, D.; Lim, M.S.; Jaffe, E.S. Pathology of Castleman Disease. Hematol. Oncol. Clin. N. Am. 2018, 32, 37–52. [Google Scholar] [CrossRef]
- van Rhee, F.; Oksenhendler, E.; Srkalovic, G.; Voorhees, P.; Lim, M.; Dispenzieri, A.; Ide, M.; Parente, S.; Schey, S.; Streetly, M.; et al. International evidence-based consensus diagnostic and treatment guidelines for unicentric Castleman disease. Blood Adv. 2020, 4, 6039–6050. [Google Scholar] [CrossRef]
- van Rhee, F.; Voorhees, P.; Dispenzieri, A.; Fosså, A.; Srkalovic, G.; Ide, M.; Munshi, N.; Schey, S.; Streetly, M.; Pierson, S.K.; et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018, 132, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Talat, N.; Belgaumkar, A.P.; Schulte, K.M. Surgery in Castleman’s disease: A systematic review of 404 published cases. Ann. Surg. 2012, 255, 677–684. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosmidis, C.S.; Mystakidou, C.M.; Koimtzis, G.; Papadopoulou, E.; Theodorou, V.; Katsios, N.I.; Georgakoudi, E.; Sevva, C.; Charalampous, I.; Varsamis, N.; et al. Idiopathic Multicentric Castleman Disease with Cutaneous Manifestation: Case Report. Medicina 2022, 58, 1222. https://doi.org/10.3390/medicina58091222
Kosmidis CS, Mystakidou CM, Koimtzis G, Papadopoulou E, Theodorou V, Katsios NI, Georgakoudi E, Sevva C, Charalampous I, Varsamis N, et al. Idiopathic Multicentric Castleman Disease with Cutaneous Manifestation: Case Report. Medicina. 2022; 58(9):1222. https://doi.org/10.3390/medicina58091222
Chicago/Turabian StyleKosmidis, Christoforos S., Chrysi Maria Mystakidou, Georgios Koimtzis, Evanthia Papadopoulou, Vasiliki Theodorou, Nikolaos Iason Katsios, Eleni Georgakoudi, Christina Sevva, Ioannis Charalampous, Nikolaos Varsamis, and et al. 2022. "Idiopathic Multicentric Castleman Disease with Cutaneous Manifestation: Case Report" Medicina 58, no. 9: 1222. https://doi.org/10.3390/medicina58091222
APA StyleKosmidis, C. S., Mystakidou, C. M., Koimtzis, G., Papadopoulou, E., Theodorou, V., Katsios, N. I., Georgakoudi, E., Sevva, C., Charalampous, I., Varsamis, N., Koulouris, C., Michael, C., Papadopoulos, K., Anthimidis, G., & Baka, S. (2022). Idiopathic Multicentric Castleman Disease with Cutaneous Manifestation: Case Report. Medicina, 58(9), 1222. https://doi.org/10.3390/medicina58091222