Pentavalent Antimonials: New Perspectives for Old Drugs


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Introduction
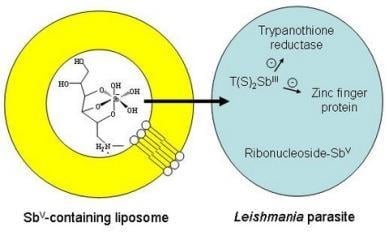
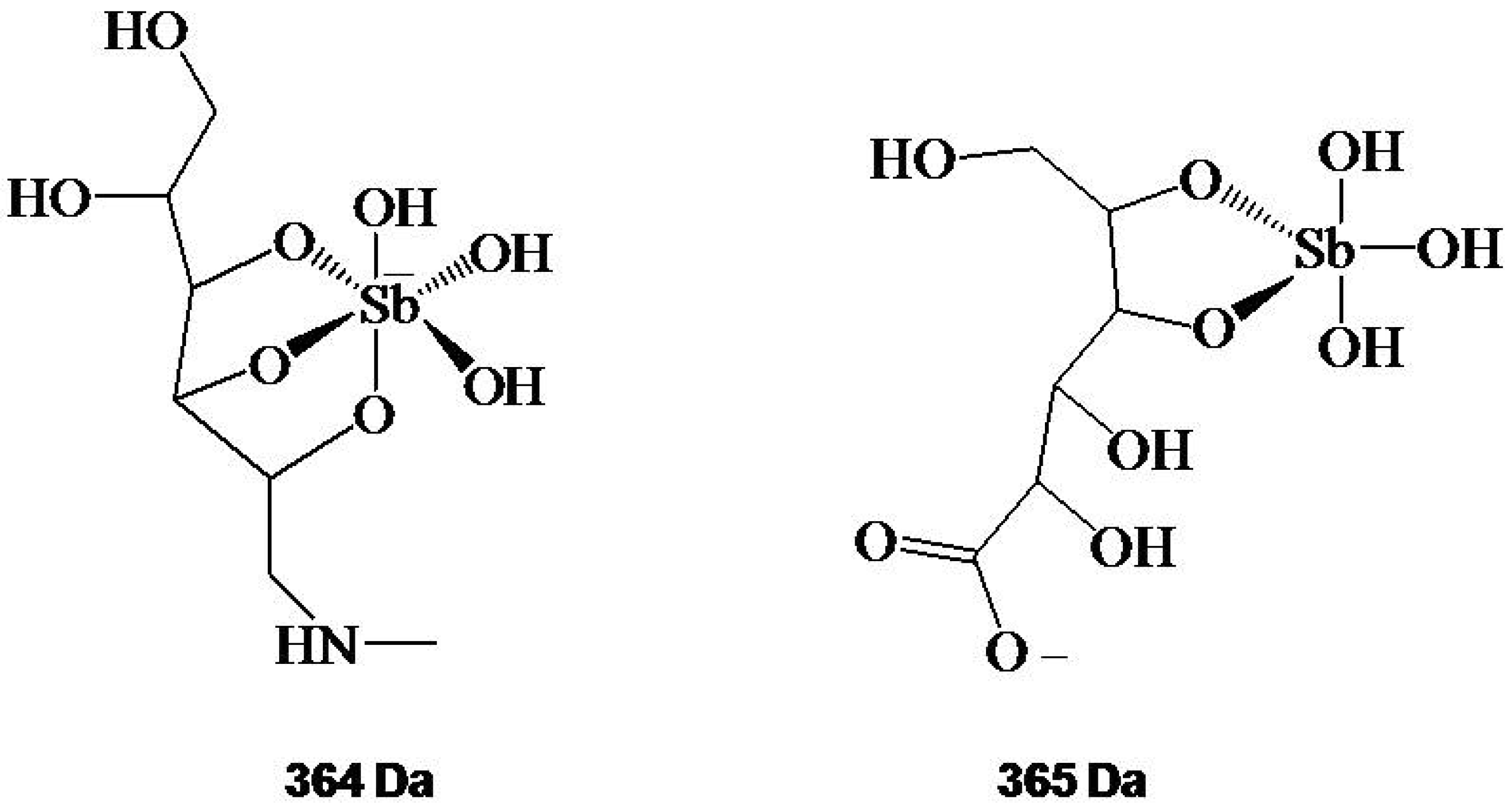
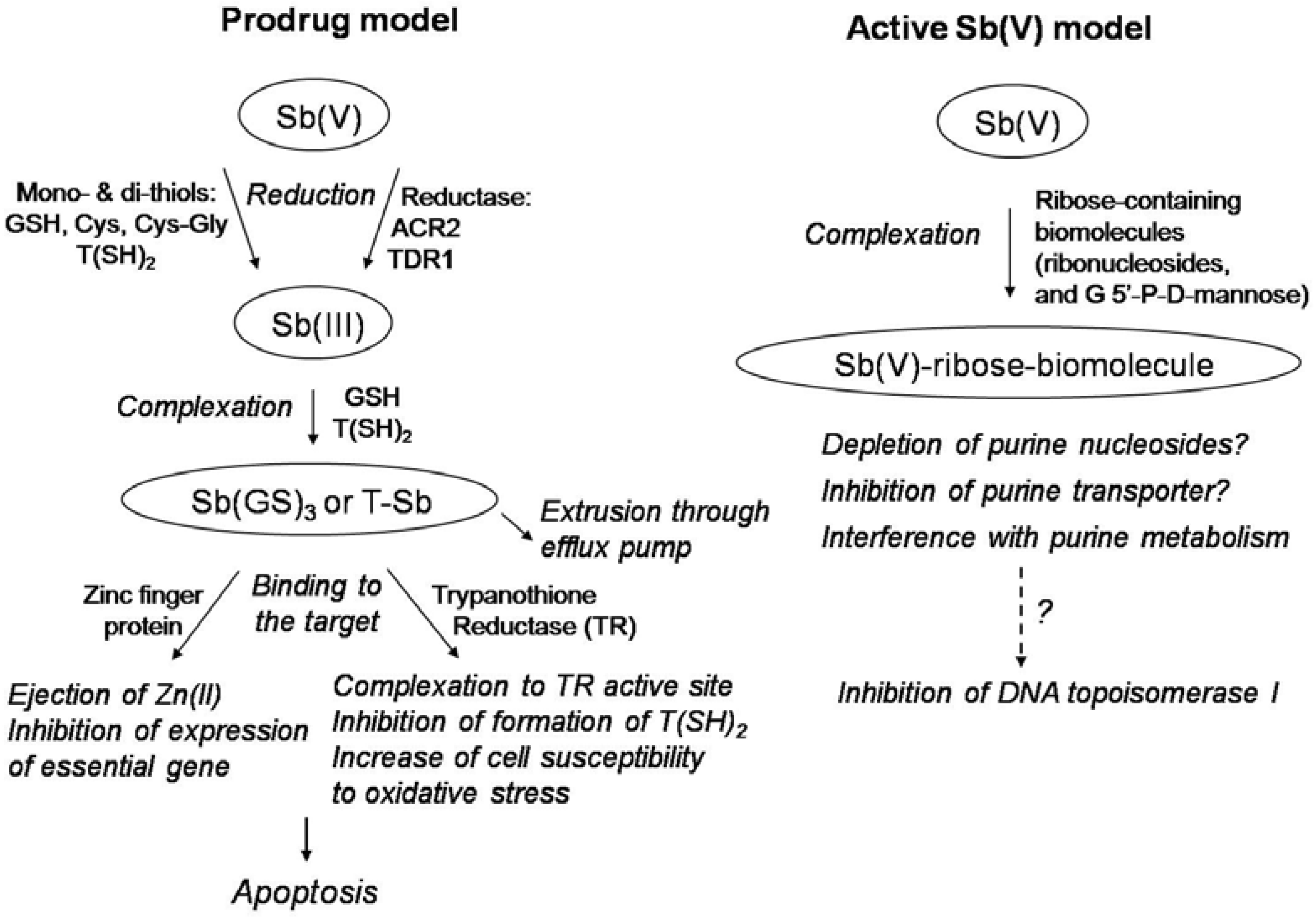
Structure and Mechanism of Action


Synthetic Processes for Pentavalent Antimonials
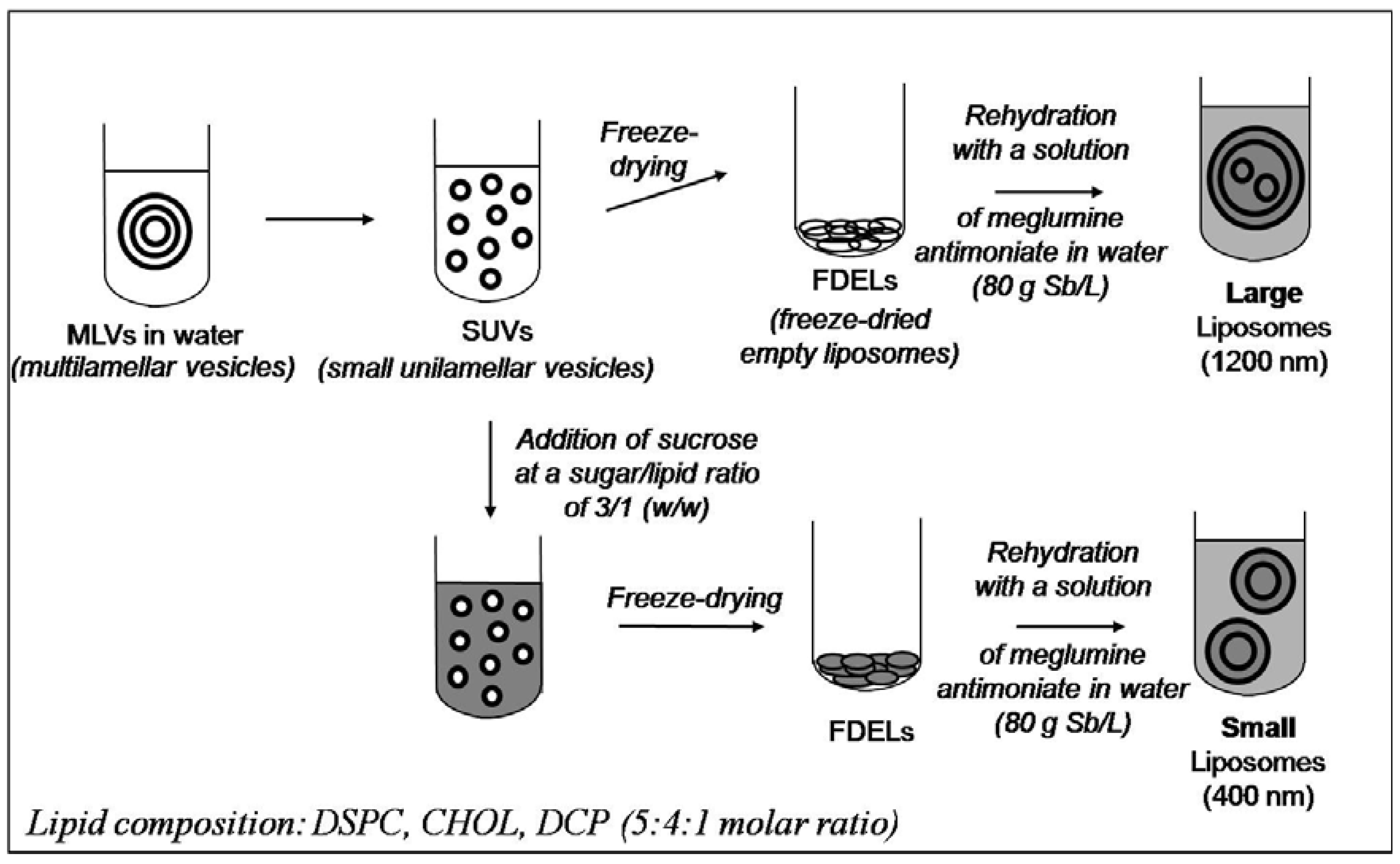
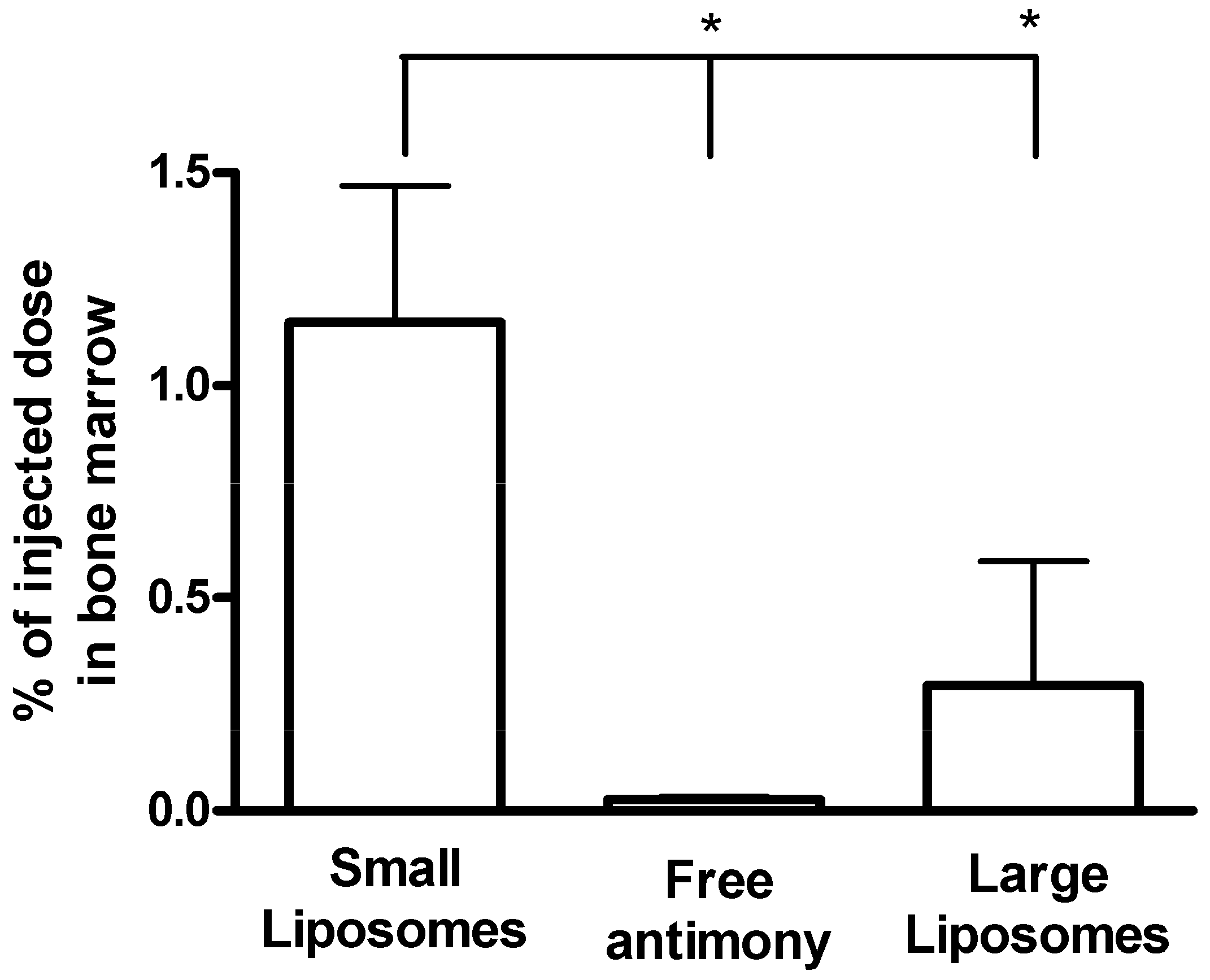
Liposome-based Formulations


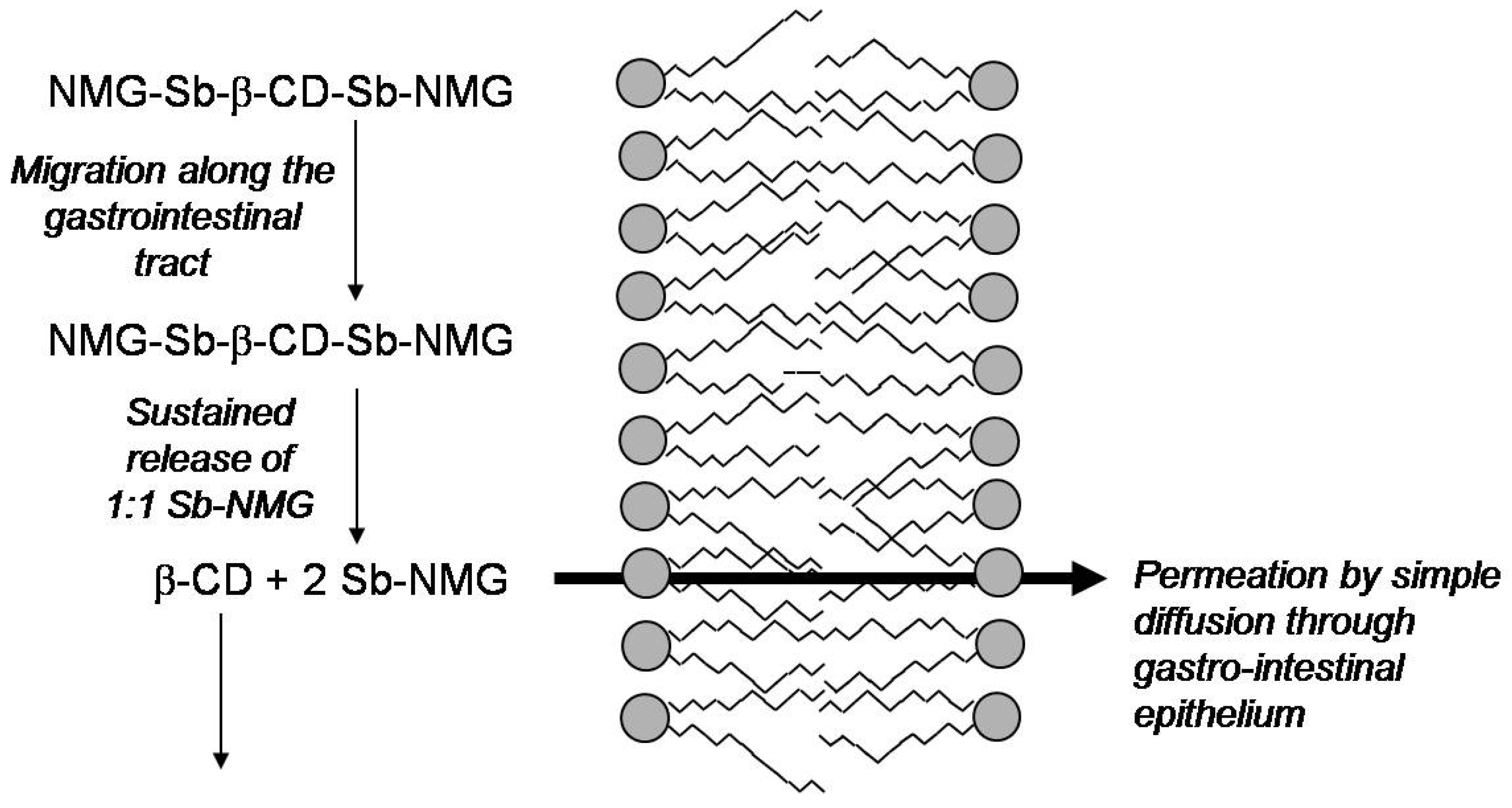
Cyclodextrin-based Formulations

Conclusions
Acknowledgements
References
- Allan, J.W. Persian Metal Technology 700-1300 A.D.; Ithaca Press: London, UK, 1979; pp. 55–59. [Google Scholar]
- Savage-Smith, E. Dioscorides on Pharmacy and Medicine; Riddle, J.M., Ed.; University of Texas Press: Austin, TX, USA, 1985; pp. 96–97. [Google Scholar]
- Estes, J.W. The Medical Skills of Ancient Egypt; Science History Publications: Canton, MA, USA, 1989; p. 155. [Google Scholar]
- Duffin, J.; René, P. 201C;Anti-moine; Anti-biotique”: The public fortunes of the secret properties of antimony potassium tartrate (tartar emetic). J. Hist. Med. Allied Sci. 1991, 46, 440–456. [Google Scholar] [CrossRef]
- World Health Organization. Leishmaniasis. Available online: http://apps.who.int/tdr/svc/diseases/leishmaniasis/.
- Vianna, G. Tratamento da leishmaniose tegumentar por injeções intravenosas de tártaro emético. In 7 Congresso Brasileiro de Medicina Tropical de São Paulo; São Paulo, Brasil, 1912; Volume 4, pp. 426–428. [Google Scholar]
- Di Cristina, G.; Caronia, G. Sulla terapia della leishmaniosi interna. Pathologica 1915, 7, 82–83. [Google Scholar]
- Cole, A.C.E. Kala-azar in east Africa. Trans. R. Soc. Trop. Med. Hyg. 1944, 37, 409–435. [Google Scholar] [CrossRef]
- Cook, C. Leonard Rogers KCSI FRCP FRS (1868-1962) and the founding of the Calcutta School of Tropical Medicine. Notes Rec. R. Soc. 2006, 60, 171–181. [Google Scholar] [CrossRef]
- Marsden, P.D. Pentavalent antimonials: old drugs for new diseases. Rev. Soc. Bras. Med. Trop. 1985, 18, 187–198. [Google Scholar] [CrossRef]
- Berman, J.D. Human leishmaniasis: clinical, diagnostic, and chemotherapeutic developments in the last 10 years. Clin. Infect. Dis. 1997, 24, 684–703. [Google Scholar] [CrossRef]
- Guerin, P.J.; Olliaro, P.; Sundar, S.; Boelaert, M.; Croft, S.L.; Desjeux, P.; Wasunna, M.K.; Bryceson, A.D. Visceral leishmaniasis: current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect. Dis. 2002, 2, 494–501. [Google Scholar] [CrossRef]
- Ridley, R.G. Drug against parasitic diseases; Failamb, A.H., Ridley, R.G., Vial, H.J., Eds.; UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (TDR): Geneva, Switzerland, 2003; pp. 13–21. [Google Scholar]
- Murray, H.W. Progress in the treatment of a neglected infectious disease: Visceral leishmaniasis. Expert Rev. Anti Infect. Ther. 2004, 2, 279–292. [Google Scholar] [CrossRef]
- Meyerhoff, A. U.S. Food and Drug Administration approval of AmBisome (liposomal amphotericin B) for treatment of visceral leishmaniasis. Clin. Infect. Dis. 1999, 28, 42–48. [Google Scholar]
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Engel, J.; Sindermann, H.; Fischer, C.; Junge, K.; Bryceson, A.; Berman, J. Oral Miltefosine for Indian Visceral Leishmaniasis. N. Engl. J. Med. 2002, 347, 1739–1746. [Google Scholar] [CrossRef]
- Steck, E.A. The leishmaniases. Prog. Drug Res. 1974, 18, 289–351. [Google Scholar]
- Sundar, S.; Sinha, P.R.; Agrawal, N.K.; Srivastava, R.; Rainey, P.M.; Berman, J.D.; Murray, H.W.; Singh, V.P. A cluster of cases of severe cardiotoxicity among kala-azar patients treated with a high-osmolarity lot of sodium antimony gluconate. Am. J. Trop. Med. Hyg. 1998, 59, 139–143. [Google Scholar]
- Deps, P.D.; Viana, M.C.; Falqueto, A.; Dietze, R. Avaliação comparativa da eficácia e toxicidade do antimoniato de N-metil-glucamina e do Estibogluconato de Sódio BP88® no tratamento da leishmaniose cutânea localizada. Rev. Soc. Bras. Med. Trop. 2000, 33, 535–543. [Google Scholar] [CrossRef]
- Yan, S.; Jin, L.; Sun, H. The Use of Metals in Medicine. In Metallotherapeutic Drugs and Metal-Based Diagnostic Agents; Gielen, M., Tiekink, E.R.T., Eds.; John Wiley & Sons: New York, NY, USA, 2005; Vol. 10, pp. 441–461. [Google Scholar]
- Headley, J.V.; Yong, M.S.; Brooks, P.W.; Phillips, A. Fast-Atom bombardment mass spectrometry of the organometallic parasiticide, meglumine antimonite. Rapid Comm. Mass Spectrom. 1995, 9, 372–376. [Google Scholar] [CrossRef]
- Roberts, W.L.; McMurray, W.J.; Rainey, P.M. Characterization of the antimonial antileishmanial agent meglumine antimonate (Glucantime). Antimicrob. Agents Chemother. 1998, 42, 1076–1082. [Google Scholar]
- Frézard, F.; Martins, P.S.; Barbosa, M.C.M.; Pimenta, A.M.C.; Ferreira, W.A.; de Melo, J.E.; Mangrum, J.B.; Demicheli, C. New insights into the chemical structure and composition of the pentavalent antimonial drugs, meglumine antimonate and sodium stibogluconate. J. Inorg. Biochem. 2008, 102, 656–665. [Google Scholar] [CrossRef]
- Berman, J.D.; Grogl, M. Leishmania mexicana: chemistry and biochemistry of sodium stibogluconate (Pentostam). Exp. Parasitol. 1988, 67, 96–103. [Google Scholar] [CrossRef]
- Hansen, H.R.; Hansen, C.; Jensen, K.P.; Hansen, S.H.; Sturup, S.; Gammelgaard, B. Characterization of sodium stibogluconate by online liquid separation cell technology monitored by ICPMS and ESMS and computational chemistry. Anal. Chem. 2008, 80, 5993–6000. [Google Scholar] [CrossRef]
- Demicheli, C.; Ochoa, R.; Lula, I.S.; Gozzo, F.C.; Eberlin, M.; Frézard, F. Pentavalent organoantimonial derivatives: two simple and efficient synthetic methods for meglumine antimonate. Applied Organomet. Chem. 2003, 17, 226–231. [Google Scholar] [CrossRef]
- Goodwin, L.C.; Page, J.E. A study of the excretion of organic antimonials using a polarographic procedure. Biochem. J. 1943, 22, 236–240. [Google Scholar]
- Burguera, J.L.; Burguera, M.; Petit de Pena, Y.; Lugo, A.; Anez, N. Selective determination of antimony(III) and antimony(V) in serum and urine and of total antimony in skin biopsies of patients with cutaneous leishmaniasis treated with meglumine antimoniate. Trace Elem. Med. 1993, 10, 66–70. [Google Scholar]
- Shaked-Mishan, P.; Ulrich, N.; Ephros, M.; Zilberstein, D. Novel intracellular Sb(V) reducing activity correlates with antimony susceptibility in Leishmania donovani. J. Biol. Chem. 2001, 276, 3971–3976. [Google Scholar] [CrossRef]
- Ferreira, C.S.; Martins, P.S.; Demicheli, C.; Brochu, C.; Ouellette, M.; Frézard, F. Thiol-induced reduction of antimony(V) into antimony(III): a comparative study with trypanothione, cysteinyl-glycine, cysteine and glutathione. BioMetals 2003, 16, 441–443. [Google Scholar] [CrossRef]
- Frézard, F.; Demicheli, C.; Ferreira, C.S.; Costa, M.A.P. Glutathione-induced conversion of pentavalent antimony to trivalent antimony in meglumine antimoniate. Antimicrob. Agents Chemother. 2001, 45, 913–916. [Google Scholar] [CrossRef]
- Yan, S.C.; Li, F.; Ding, K.Y.; Sun, H. Reduction of pentavalent antimony by trypanothione and formation of a binary and ternary complex of antimony(III) and trypanothione. J. Biol. Inorg. Chem. 2003, 8, 689–697. [Google Scholar] [CrossRef]
- Mego, J.L. Stimulation of intralysosomal proteolysis by cysteinyl-glycine, a product of the action of gamma-glutamyl transpeptidase on glutathione. Biochim. Biophys. Acta 1985, 841, 139–144. [Google Scholar] [CrossRef]
- Gainey, D.; Short, S.; McCoy, K.L. Intracellular location of cysteine transport activity correlates with productive processing of antigen disulfide. J. Cell Physiol. 1996, 168, 248–254. [Google Scholar] [CrossRef]
- Fairlamb, A.H.; Cerami, A. Metabolism and functions of trypanothione in the Kinetoplastida. Annu. Rev. Microbiol. 1992, 46, 695–729. [Google Scholar] [CrossRef]
- Denton, H.; McGregor, J.C.; Coombs, G.H. Reduction of anti-leishmanial pentavalent antimonial drugs by a parasite-specific thiol-dependent reductase, TDR1. Biochem. J. 2004, 381, 405–412. [Google Scholar] [CrossRef]
- Zhou, Y.; Messier, N.; Ouellette, M.; Rosen, B.P.; Mukhopadhyay, R. Leishmania major LmACR2 is a pentavalent antimony reductase that confers sensitivity to the drug Pentostam. J. Biol. Chem. 2004, 279, 37445–37451. [Google Scholar] [CrossRef]
- Gourbal, B.; Sonuc, N.; Bhattacharjee, H.; Legare, D.; Sundar, S.; Ouellette, M.; Rosen, B.P.; Mukhopadhyay, R. Drug uptake and modulation of drug resistance in Leishmania by an aquaglyceroporin. J. Biol. Chem. 2004, 279, 31010–31017. [Google Scholar] [CrossRef]
- Sun, H.; Yan, S.C.; Cheng, W.S. Interaction of antimony tartrate with the tripeptide glutathione. Eur. J. Biochem. 2000, 267, 5450–5457. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Dey, S.; Xu, N.; Gage, D.; Lightbody, J.; Ouellette, M.; Rosen, B.P. Trypanothione overproduction and resistance to antimonials and arsenicals in Leishmania. Proc. Natl. Acad. Sci. USA 1996, 93, 10383–10387. [Google Scholar] [CrossRef]
- Légaré, D.; Richard, D.; Mukhopadhyay, R.; Stierhof, Y.D.; Rosen, B.P.; Haimeur, A.; Papadopoulou, B.; Ouellette, M. The Leishmania ATP-binding cassette protein PGPA is an intracellular metalthiol transporter ATPase. J. Biol. Chem. 2001, 276, 26301–26307. [Google Scholar] [CrossRef]
- Ouellette, M.; Drummelsmith, J.; Papadopoulou, B. Leishmaniasis: drugs in the clinic, resistance and new developments. Drug Resist. Updat. 2004, 7, 257–266. [Google Scholar] [CrossRef]
- Carter, K.C.; Sundar, S.; Spickett, C.; Pereira, O.C.; Mullen, A.B. The in vivo susceptibility of Leishmania donovani to sodium stibogluconate is drug specific and can be reversed by inhibiting glutathione biosynthesis. Antimicrob. Agents Chemother. 2003, 47, 1529–1535. [Google Scholar] [CrossRef]
- Krauth-Siegel, R.L.; Comini, M.A. Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim. Biophys. Acta 2008, 1780, 1236–1248. [Google Scholar]
- Cunningham, M.L.; Fairlamb, A.H. Trypanothione reductase from Leishmania donovani. Purification, characterisation and inhibition by trivalent antimonials. Eur. J. Biochem. 1995, 230, 460–468. [Google Scholar] [CrossRef]
- Wyllie, S.; Cunningham, M.L.; Fairlamb, A.H. Dual action of antimonial drugs on thiol redox metabolism in the human pathogen Leishmania donovani. J. Biol. Chem. 2004, 279, 39925–39993. [Google Scholar] [CrossRef]
- Baiocco, P.; Colotti, G.; Franceschini, S.; Ilari, A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 2009, 52, 2603–2612. [Google Scholar] [CrossRef]
- Demicheli, C.; Frézard, F.; Mangrum, J.B.; Farrell, N.P. Interaction of trivalent antimony with a CCHC zinc finger domain: potential relevance to the mechanism of action of antimonial drugs. Chem. Commun. 2008, 39, 4828–4830. [Google Scholar]
- Leon, O.; Roth, M. Zinc fingers: DNA binding and protein-protein interactions. Biol. Res. 2000, 33, 21–30. [Google Scholar]
- Webb, J.R.; McMaster, W.R. Molecular cloning and expression of a Leishmania major gene encoding a single-stranded DNA-binding protein containing nine "CCHC" zinc finger motifs. J. Biol. Chem. 1993, 268, 13994–14002. [Google Scholar]
- Sereno, D.; Holzmuller, P.; Mangot, I.; Cuny, G.; Ouaissi, A.; Lemesre, J. Antimonial-mediated DNA fragmentation in Leishmania infantum amastigotes. Antimicrob. Agents Chemother. 2001, 45, 2064–2069. [Google Scholar] [CrossRef]
- Sudhandiran, G.; Shaha, C. Antimonial induced increase in intracellular Ca2+ through non-selective cation channels in the host and the parasite is responsible for apoptosis of intracellular Leishmania donovani amastigotes. J. Biol. Chem. 2003, 278, 25120–25132. [Google Scholar] [CrossRef]
- Mukherjee, S.B.; Das, M.; Sudhandiran, G.; Shaha, C. Increase in cytosolic Ca2+ levels through the activation of non-selective cation channels induced by oxidative stress causes mitochondrial depolarization leading to apoptosis-like death in Leishmania donovani promastigotes. J. Biol. Chem. 2002, 277, 24717–24727. [Google Scholar]
- Chakraborty, A.K.; Majumder, H.K. Mode of action of pentavalent antimonials: specific inhibition of type I DNA topoisomerase of Leishmania donovani. Biochem. Biophys. Res. Commun. 1988, 152, 605–611. [Google Scholar] [CrossRef]
- Walker, J.; Saravia, N.G. Inhibition of Leishmania donovani promastigote DNA topoisomerase I and human monocyte DNA topoisomerases I and II by antimonial drugs and classical antitopoisomerase agents. J. Parasitol. 2004, 90, 1155–1162. [Google Scholar] [CrossRef]
- Lucumi, A.; Robledo, S.; Gama, V.; Saravia, N.G. Sensitivity of Leishmania Viannia panamensis to pentavalent antimony is correlated with the formation of cleavable DNA-protein complexes. Antimicrob. Agents Chemother. 1998, 42, 1990–1995. [Google Scholar]
- Demicheli, C.; Frézard, F.; Lecouvey, M.; Garnier-Suillerot, A. Antimony(V) complex formation with adenine nucleosides in aqueous solution. Biochim. Biophys. Acta 2002, 1570, 192–198. [Google Scholar]
- Chai, Y.; Yan, S.; Wong, I.L.K.; Chow, L.M.C.; Sun, H. Complexation of antimony [Sb(V)] with guanosine 5’-monophosphate and guanosine 5’-diphospho-D-mannose: formation of both mono- and bis-adducts. J. Inorg. Biochem. 2005, 99, 2257–2263. [Google Scholar] [CrossRef]
- Demicheli, C.; Santos, L.S.; Ferreira, C.S.; Bouchemal, N.; Hantz, E.; Eberlin, M.N.; Frézard, F. Synthesis and characterization of Sb(V)–adenosine and Sb(V)–guanosine complexes in aqueous solution. Inorganica Chim. Acta 2006, 359, 159–167. [Google Scholar] [CrossRef]
- Ferreira, C.S.; Pimenta, A.M.C.; Demicheli, C.; Frézard, F. Characterization of reactions of antimoniate and meglumine antimoniate with a guanine ribonucleoside at different pH. Biometals 2006, 19, 573–581. [Google Scholar] [CrossRef]
- Hansen, H.R.; Pergantis, S.A. Mass spectrometric identification and characterization of antimony complexes with ribose-containing biomolecules and an RNA oligomer. Anal. Bioanal. Chem. 2006, 385, 821–833. [Google Scholar] [CrossRef]
- Roberts, W.L.; Berman, J.D.; Rainey, P.M. In vitro antileishmanial properties of tri- and pentavalent antimonial preparations. Antimicrob. Agents Chemother. 1995, 39, 1234–1239. [Google Scholar] [CrossRef]
- Marr, J. Purine analogs as chemotherapeutic agents in leishmaniasis and american trypanosomiasis. J. Lab. Clin. Med. 1991, 118, 111–119. [Google Scholar]
- Berman, J.D.; Waddell, D.; Hanson, B.D. Biochemical mechanisms of the antileishmanial activity of sodium stibogluconate. Antimicrob. Agents Chemother. 1985, 27, 916–920. [Google Scholar] [CrossRef]
- Berman, J.D.; Gallalee, J.V.; Best, J.M. Sodium stibogluconate (Pentostam) inhibition of glucose catabolism via the glycolytic pathway, and fatty acid beta-oxidation in Leishmania mexicana amastigotes. Biochem. Pharmacol. 1987, 36, 197–201. [Google Scholar] [CrossRef]
- Murray, H.W. Clinical and experimental advances in treatment of visceral leishmaniasis. Antimicrob. Agents Chemother. 2001, 45, 2185–2197. [Google Scholar] [CrossRef]
- Pathak, M.K.; Yi, T. Sodium stibogluconate is a potent inhibitor of protein tyrosine phosphatases and augments cytokine responses in hemopoietic cell lines. J. Immunol. 2001, 167, 3391–3397. [Google Scholar]
- Muniz-Junqueira, M.I.; Paula-Coelho, V.N. Meglumine antimonate directly increases phagocytosis, superoxide anion and TNF-α production, but only via TNF-α it indirectly increases nitric oxide production by phagocytes of healthy individuals, in vitro. Int. Immunopharmacol. 2008, 8, 1633–1638. [Google Scholar] [CrossRef]
- Murray, H.W.; Berman, J.D.; Wright, S.D. Immunochemotherapy for intracellular Leishmania donovani infection: gamma interferon plus pentavalent antimony. J. Infect. Dis. 1988, 157, 973–978. [Google Scholar] [CrossRef]
- Machado-Pinto, J.; Pinto, J.; Da Costa, C.A.; Genaro, O.; Marques, M.J.; Modabber, F.; Mayrink, W. Immunochemotherapy for cutaneous leishmaniasis: a controlled trial using killed Leishmania (Leishmania) amazonensis vaccine plus antimonial. Int. J. Dermatol. 2002, 41, 73–78. [Google Scholar] [CrossRef]
- Gailliot, P.L. Procédé de préparation de dérivés de l’antimoine. (Societé des usines chimiques de Rhone-Poulenc). FR Patent 8.687.47, 1941. [Google Scholar]
- Joan, F.B.; Concepcio, M.C. New procedure for the preparation of antimonic acid derivatives applicable in the treatment of canine leishmaniosis. ES Patent 2.050.614, 1994. [Google Scholar]
- Demicheli, C. Síntese de derivados de antimônio pentavalente utilizados no tratamento de protozoonoses. Brazil Patent Pending PI 9907575-0, 1999. [Google Scholar]
- Demicheli, C.; Frézard, F. Novo processo para preparação de derivados de antimônio. Brazil Patent Pending PI 0106305-7, 2001. [Google Scholar]
- De Morais-Teixeira, E.; De Carvalho, A.S.; Da Costa, J.C.S.; Duarte, S.L.; Mendonça, J.S.; Boechat, N.; Rabello, A. In vitro and in vivo activity of meglumine antimoniate produced at Farmanguinhos-Fiocruz, Brazil, against Leishmania (Leishmania) amazonensis, L (L.) chagasi and L (Viannia) braziliensis. Mem. Inst. Oswaldo Cruz 2008, 10, 358–362. [Google Scholar]
- Dzamitika, S.A.; Falcão, C.A.; Oliveira, F.B.; Marbeuf, C.; Garnier-Suillerot, A.; Demicheli, C.; Rossi-Bergmann, B.; Frézard, F. Role of residual Sb(III) in meglumine antimoniate cytotoxicity and MRP1-mediated resistance. Chem. Biol. Int. 2006, 160, 217–224. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Alving, C.R.; Steck, E.A.; Chapman, W.L.; Waits, V.B.; Hendricks, L.D.; Swartz, G.M.; Hanson, W.L. Therapy of leishmaniasis: superior efficacies of liposome-encapsulated drugs. Proc. Natl. Acad. Sci. USA 1978, 75, 2959–2963. [Google Scholar]
- New, R.R.; Chance, M.L.; Thomas, S.C.; Peters, W. Antileishmanial activity of antimonials entrapped in liposomes. Nature 1978, 272, 55–56. [Google Scholar] [CrossRef]
- Chapman, W.L.; Hanson, W.L.; Alving, C.R.; Hendricks, L.D. Antileishmanial activity of liposome-encapsulated meglumine antimonate in the dog. Am. J. Vet. Res. 1984, 45, 1028–1030. [Google Scholar]
- Collins, M.; Carter, K.C.; Baillie, A.J.; O´Grady, J. The distribution of free and non-ionic vesicular sodium stibogluconate in the dog. J. Drug Target. 1993, 1, 133–142. [Google Scholar] [CrossRef]
- Alving, C.R.; Swartz, G.M. Liposome technology; Gregoriadis, G., Ed.; CRC Press: Boca Raton, FL, USA, 1984; Vol. 2, pp. 55–68. [Google Scholar]
- Rao, L.S. Anti-leishmanial pharmaceutical formulation. US Patent 4.594.241, 1986. [Google Scholar]
- Schettini, D.A.; Ribeiro, R.R.; Demicheli, C.; Rocha, O.G.F.; Melo, M.N.; Michalick, M.S.M.; Frézard, F. Improved targeting of antimony to the bone marrow of dogs using liposomes of reduced size. Int. J. Pharm. 2006, 315, 140–147. [Google Scholar] [CrossRef]
- Frézard, F.; Michalick, M.S.M.; Soares, C.F.; Demicheli, C. Novel methods for the encapsulation of meglumine antimoniate in liposomes. Braz. J. Med. Biol. Res. 2000, 33, 841–846. [Google Scholar] [CrossRef]
- Frézard, F.; Demicheli, C.; Schettini, D.A.; Ribeiro, R.R.; Melo, M.N.; Michalick, M.S.M. Processo para a preparação de formulações farmacêuticas do antimoniato de meglumina em lipossomas e uso das formulações farmacêuticas em animais acometidos com leishmaniose visceral. Brazil Patent Pending PI0405489-0, 2004. [Google Scholar]
- Demicheli, C.; Rocha, O.G.F.; Schettini, D.A.; Frézard, F. Liposomes: physicochemical and pharmacological properties, applications in antimony-based chemotherapy. Quim. Nova 2005, 28, 511–518. [Google Scholar] [CrossRef]
- Oliva, G.; Gradoni, L.; Ciaramella, P.; De Luna, R.; Cortese, L.; Orsini, S.; Davidson, R.N.; Persechino, A. Activity of liposomal amphotericin B (AmBisome) in dogs naturally infected with Leishmania infantum. J. Antimicrob. Chemother. 1995, 36, 1013–1019. [Google Scholar] [CrossRef]
- Valladares, J.E.; Freixas, J.; Alberola, J.; Franquelo, C.; Cristofol, C.; Arboix, M. Pharmacokinetics of liposome-encapsulated meglumine antimoniate after intramuscular and subcutaneous administration in dogs. Am. J. Med. Hyg. 1997, 57, 403–406. [Google Scholar]
- Valladares, J.E.; Riera, C.; González-Ensenyat, P.; Díez-Cascón, A.; Ramos, G.; Solano-Gallego, L.; Gállego, M.; Gállego, M.; Portús, N.; Arboix, M.; Alberola, J. Long term improvement in the treatment of canine leishmaniosis using antimony liposomal formulation. Vet. Parasitol. 2001, 97, 15–21. [Google Scholar] [CrossRef]
- Nieto, J.; Alvar, J.; Mullen, K.C.; Carter, K.C.; Rodríguez, C.; San Andrés, M.I.; San Andrés, M.D.; Baillie, A.J.; Gonzalez, F. Pharmacokinetics, toxicities, and efficacies of sodium stibogluconate formulations after intravenous administration in animals. Antimicrob. Agents Chemother. 2003, 47, 2781–2787. [Google Scholar] [CrossRef]
- Schettini, D.A.; Costa Val, A.P.; Souza, L.F.; Demicheli, C.; Rocha, O.G.F.; Melo, M.N.; Michalick, M.S.M.; Frézard, F. Distribution of liposome-encapsulated antimony in dogs. Braz. J. Med. Biol. Res. 2003, 36, 269–272. [Google Scholar] [CrossRef]
- Schettini, D.A.; Costa Val, A.P.; Souza, L.F.; Demicheli, C.; Rocha, O.G.F.; Melo, M.N.; Michalick, M.S.M.; Frézard, F. Pharmacokinetic and parasitological evaluation of the bone marrow of dogs with visceral leishmaniasis submitted to multiple dose treatment with liposome-encapsulated meglumine antimoniate. Braz. J. Med. Biol. Res. 2005, 38, 1879–1883. [Google Scholar] [CrossRef]
- Ribeiro, R.R.; Moura, E.P.; Pimentel, V.M.; Sampaio, W.M.; Silva, S.M.; Schettini, D.A.; Alves, C.F.; Melo, F.A.; Tafuri, W.L.; Demicheli, C.; Melo, M.N.; Frezard, F.; Michalick, M.S.M. Reduced tissue parasitic load and infectivity to sand flies in dogs naturally infected by Leishmania (Leishmania) chagasi following treatment with a liposome formulation of meglumine antimoniate. Antimicrob. Agents Chemother. 2008, 52, 2564–2572. [Google Scholar] [CrossRef]
- Costa-val, A.P. Tratamento da leishmaniose visceral canina com antimonial pentavalente encapsulado em lipossomas. Thesis, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil, 2004; p. 125. [Google Scholar]
- Szebeni, J. The interaction of liposomes with the complement system. Crit. Rev. Ther. Drug Carrier Syst. 1998, 15, 57–58. [Google Scholar] [CrossRef]
- Irie, T.; Uekama, K. Pharmaceutical applications of cyclodextrins. III. Toxicological issues and safety evaluation. J. Pharm. Sci. 1997, 86, 147–162. [Google Scholar] [CrossRef]
- Hirayama, F.; Uekama, K. Cyclodextrin-based controlled drug release system. Adv. Drug Deliv. Rev. 1999, 36, 125–141. [Google Scholar] [CrossRef]
- Demicheli, C.; Frézard, F.J.G.; Millán, R.D.S.; Bejarano, R.O.; Ferreira, L.A.M.; da Silva, J.B.B.; de Melo, A.L. Processo de Preparação de Compostos entre as ciclodextrinas ou seus derivados e o antimônio ou seus derivados, de formulações farmacêuticas contendo esses compostos e produtos associados, para tratamento das leishmanioses e da esquitossomose. Brazil Patent Pending PI0304952-3, 2003. [Google Scholar]
- Demicheli, C.; Ochoa, R.; Silva, J.B.B.; de Melo, A.L.; Falcão, C.A.M.; Rossi-Bergmann, B.; Sinisterra, R.D.; Frézard, F. Oral delivery of meglumine antimoniate-beta-cyclodextrin complex for treatment of leishmaniasis. Antimicrob. Agents Chemother. 2004, 48, 100–103. [Google Scholar] [CrossRef]
- Martins, P.S.; Ochoa, R.; Pimenta, A.M.C.; Ferreira, L.A.M.; de Melo, A.L.; da Silva, J.B.B.; Sinisterra, R.D.; Demicheli, C.; Frézard, F. Mode of action of β-cyclodextrin as an absorption enhancer of the water-soluble drug meglumine antimoniate. Int. J. Pharm. 2006, 325, 39–47. [Google Scholar] [CrossRef]
- Frézard, F.; Martins, P.S.; Bahia, A.P.C.O.; Le Moyec, L.; de Melo, A.L.; Pimenta, A.M.C.; Salerno, M.; da Silva, J.B.B.; Demicheli, C. Enhanced oral delivery of antimony from meglumine antimoniate/β-cyclodextrin nanoassemblies. Int. J. Pharm. 2008, 347, 102–108. [Google Scholar] [CrossRef]
- Sample Availability: Samples of meglumine antimoniate and of liposome- and cyclodextrin-based formulations are available from the corresponding author.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Frézard, F.; Demicheli, C.; Ribeiro, R.R. Pentavalent Antimonials: New Perspectives for Old Drugs. Molecules 2009, 14, 2317-2336. https://doi.org/10.3390/molecules14072317
Frézard F, Demicheli C, Ribeiro RR. Pentavalent Antimonials: New Perspectives for Old Drugs. Molecules. 2009; 14(7):2317-2336. https://doi.org/10.3390/molecules14072317
Chicago/Turabian StyleFrézard, Frédéric, Cynthia Demicheli, and Raul R. Ribeiro. 2009. "Pentavalent Antimonials: New Perspectives for Old Drugs" Molecules 14, no. 7: 2317-2336. https://doi.org/10.3390/molecules14072317
APA StyleFrézard, F., Demicheli, C., & Ribeiro, R. R. (2009). Pentavalent Antimonials: New Perspectives for Old Drugs. Molecules, 14(7), 2317-2336. https://doi.org/10.3390/molecules14072317