4.2. Synthetic procedures
4.2.1. General procedure for the synthesis of o-halo(hetero)aroyl chlorides 1b–h
A suspension of the appropriate acid (2 mmol) in toluene (20 mL), DMF (5 drops) and SOCl2 (20 mmol, 2.38 g) was refluxed for 3 h or overnight. The solvent and excess SOCl2 were removed under reduced pressure. Additional toluene (4 × 5 mL) was added, and the solvent was removed under reduced pressure. The remaining acid chloride was immediately used in subsequent reaction steps (with no further purification).
4.2.2. General procedure for the synthesis of ynones 2a-h
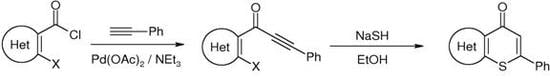
The appropriate acid chloride 1 (2 mmol) was dissolved in dry CH2Cl2 (3 mL). Triethylamine (2 mL), phenylacetylene (1 mmol, 102 mg) and Pd(II) acetate (10 µmol, 2 mg) were added to the solution, which was stirred at room temperature under a N2 atmosphere (N2 balloon) for the indicated time. The solvent was removed under reduced pressure and water (20 mL) was added to the residue. The resulting solution was acidified with 5% HCl and extracted with CH2Cl2 or EtOAc (2 × 15 mL). The combined organic layers were washed with a saturated NaHCO3 solution and a saturated NaCl solution and were then dried over anhydrous Na2SO4. After removal of the solvent, the residue was purified by column chromatography through a silica gel column (eluent given below). An analytically pure sample was obtained by recrystallization from the appropriate solvent, which is indicated below.
1-(2-Chlorophenyl)-3-phenylprop-2-yn-1-one (
2a1). Reaction time: 2.5 h. Eluent: toluene/light petroleum, 1:1 v/v. Yield: 209 mg, 87%; orange oil; [
51]
1H-NMR (300 MHz): δ 8.08 (m, 1H, H-6), 7.65 (m, 2H, Ph H-2,6), 7.48 (m, 2H, H-3,4), 7.46 (m, 1H, Ph H-4), 7.41 (m, 2H, Ph H-3,5), 7.40 (m, 1H, H-5);
13C-NMR (75 MHz): δ 176.7 (C=O,
3J(CO,H6) = 5.3 Hz), 135.9 (C-1), 133.5 (C-2), 133.3 (C-4), 133.1 (Ph C-2,6), 132.4 (C-6), 131.5 (C-3), 130.9 (Ph C-4), 128.7 (Ph C-3,5), 126.8 (C-5), 120.1 (Ph C-1), 93.9 (COC≡
C,
3J(C,Ph H2,6) = 5.5 Hz), 88.3 (CO
C≡C); IR (liquid film): 2196 (C≡C), 1649 (C=O) cm
−1; MS
m/z (%): 242/240 (M
+, 14/40), 214/212 ([M − C=O]
+, 25/72) 141/139 ([COC
6H
4Cl]
+, 12/34), 129 ([COC≡CC
6H
5]
+, 100), 101 ([C≡CC
6H
5]
+, 14).
1-(2-Fluorophenyl)-3-phenylprop-2-yn-1-one (
2a2). Reaction time: 45 min. Eluent: CH
2Cl
2/light petroleum, 1:1 v/v. Yield: 164 mg, 73%; light orange oil; [
52]
1H-NMR (500 MHz): δ 8.11 (m, 1H, H-6), 7.67 (m, 2H, Ph H-2,6), 7.59 (m, 1H, H-4), 7.49 (m, 1H, Ph H-4), 7.42 (m, 2H, Ph H-3,5), 7.28 (m, 1H, H-5), 7.19 (m, 1H, H-3);
13C-NMR (125 MHz): δ 174.2 (C=O), 162.1 (C-2,
1J(C2,F2) = 261.7 Hz), 135.6 (C-4,
3J(C4,F2) = 9.2 Hz), 133.2 (Ph C-2,6), 131.8 (C-6), 130.9 (Ph C-4), 128.6 (Ph C-3,5), 125.6 (C-1,
2J(C1,F2) = 7.7 Hz), 124.2 (C-5,
4J(C5,F2) = 3.9 Hz), 120.1 (Ph C-1), 117.1 (C-3,
2J(C3,F2) = 21.9 Hz), 93.0 (COC≡
C,
J(C,F2) = 3.2 Hz), 88.5 (CO
C≡C);
19F-NMR (470 MHz): δ −111.3 (F-2); IR (liquid film): 2200 (C≡C), 1647 (C=O) cm
−1; MS
m/z (%): 224 (M
+, 17), 196 ([M − C=O]
+, 36), 129 ([COC≡CC
6H
5]
+, 100), 123 ([COC
6H
4F]
+, 84), 101 ([C≡CC
6H
5]
+, 21), 74 (83).
1-(2-Bromothiophen-3-yl)-3-phenylprop-2-yn-1-one (2b). Reaction time: 19 h. Eluent: CH2Cl2/light petroleum, 3:2 v/v. Yield: 147 mg, 67%; whitish needles, mp 66–67 °C (EtOH/H2O); 1H- NMR (500 MHz): δ 7.65 (m, 2H, Ph H-2,6), 7.57 (d, 3J = 5.8 Hz, 1H, H-4), 7.48 (m, 1H, Ph H-4), 7.41 (m, 2H, Ph H-3,5), 7.28 (d, 3J = 5.8 Hz, 1H, H-5); 13C-NMR (125 MHz): δ 170.5 (C=O, 3J(CO,H4) = 2.0 Hz), 138.2 (C-3, 2J(C3,H4) = 4.5 Hz, 3J(C3,H5) = 8.4 Hz), 133.0 (Ph C-2,6), 130.9 (Ph C-4), 129.9 (C-4, 1J(C4,H4) = 173.4 Hz, 2J(C4,H5) = 3.8 Hz), 128.7 (Ph C-3,5), 126.0 (C-5, 1J(C5,H5) = 190.3 Hz, 2J(C5,H4) = 6.0 Hz), 120.3 (C-2, 3J(C2,H4) = 12.1 Hz, 3J(C2,H5) = 6.8 Hz), 120.0 (Ph C-1, 3J(Ph C1,Ph H3,5) = 8.5 Hz, 4J(Ph C1,Ph H4) = 1.3 Hz), 93.1 (COC≡C), 88.0 (COC≡C); IR: 2202 (C≡C), 1630 (C=O) cm−1; MS m/z (%): 292/290 (M+, 22/23), 264/262 ([M − C=O]+, 28/28), 139 (65), 129 ([COC≡CC6H5]+, 100), 101 ([C≡CC6H5]+, 16), 74 (79). Calcd. for C13H7BrOS: C, 53.63; H, 2.42; S, 11.01. Found: C, 53.31; H, 2.39; S, 10.92.
1-(4-Bromothiophen-3-yl)-3-phenylprop-2-yn-1-one (2c). Reaction time: 3 h. Eluent: CH2Cl2/light petroleum, 3:2 v/v. Yield: 148 mg, 25%; brownish oil; 1H-NMR (500 MHz): δ 8.42 (d, 4J = 3.5 Hz, 1H, H-2), 7.65 (m, 2H, Ph H-2,6), 7.49 (m, 1H, Ph H-4), 7.41 (m, 2H, Ph H-3,5), 7.36 (d, 4J = 3.5 Hz, 1H, H-5); 13C-NMR (125 MHz): δ 170.2 (C=O, 3J(CO,H2) = 3.6 Hz, 4J(CO,H5) = 0.6 Hz), 138.6 (C-3, 2J(C3,H2) = 3.2 Hz, 3J(C3,H5) = 7.9 Hz), 137.9 (C-2, 1J(C2,H2) = 189.5 Hz, 3J(C2,H5) = 5.3 Hz), 133.0 (Ph C-2,6), 130.9 (Ph C-4), 128.7 (Ph C-3,5), 126.1 (C-5, 1J(C5,H5) = 192.9 Hz, 3J(C5,H2) = 5.0 Hz), 119.9 (Ph C-1, 3J(Ph C1,Ph H3,5) = 8.6 Hz, 4J(Ph C1,Ph H4) = 1.3 Hz), 109.7 (C-4, 2J(C4,H5) = 0.9 Hz, 3J(C4,H2) = 11.2 Hz), 91.9 (COC≡C), 87.2 (COC≡C); IR (liquid film): 2189 (C≡C), 1640 (C=O) cm−1; MS m/z (%): 292/290 (M+, 2/2), 264/262 ([M − C=O]+, 2/1), 191/189 ([COC4H2SBr]+, 65/61), 129 ([COC≡CC6H5]+, 7), 101 ([C≡CC6H5]+, 1), 81 (100). Calcd. for C13H7BrOS: C, 53.63; H, 2.42; S, 11.01. Found: C, 53.38; H, 2.50; S, 10.67.
1-(3-Bromothiophen-2-yl)-3-phenylprop-2-yn-1-one (2d). Reaction time: 23 h. Eluent: CH2Cl2/light petroleum, 3:2 v/v. Yield: 134 mg, 46%; colorless needles, mp 74–77 °C (EtOH/H2O); 1H- NMR (500 MHz): δ 7.69 (m, 2H, Ph H2,6), 7.62 (d, 3J = 5.2 Hz, 1H, H-5), 7.50 (m, 1H, Ph H-4), 7.42 (m, 2H, Ph H-3,5), 7.18 (d, 3J = 5.2 Hz, 1H, H-4); 13C-NMR (125 MHz): δ 168.3 (C=O), 138.0 (C-2, 3J(C2,H4) = 7.4 Hz, 3J(C2,H5) = 4.8 Hz), 134.0 (C-4, 1J(C4,H4) = 176.5 Hz, 2J(C4,H5) = 4.1 Hz), 133.6 (C-5, 1J(C5,H5) = 188.1 Hz, 2J(C5,H4) = 6.1 Hz), 133.1 (Ph C-2,6), 131.0 (Ph C-4), 128.7 (Ph C-3,5), 119.9 (Ph C-1), 116.4 (C-3, 2J(C3,H4) = 2.4 Hz, 3J(C3,H5) = 12.2 Hz), 94.1 (COC≡C), 87.3 (COC≡C); IR: 2198 (C≡C), 1627 (C=O) cm−1; MS m/z (%): 292/290 (M+, 25/22), 264/262 ([M − C=O]+, 52/47), 139 (95), 129 ([COC≡CC6H5]+, 97), 101 ([C≡CC6H5]+, 22), 84 (73), 74 (77), 43 (100). Calcd. for C13H7BrOS: C, 53.63; H, 2.42; S, 11.01. Found: C, 53.63; H, 4.00; S, 10.85. HRMS Calcd. for C13H7BrOS: 289.9401. Found: 289.9403.
1-(3-Chloro-1-benzo[b]thiophen-2-yl)-3-phenylprop-2-yn-1-one (2e). Reaction time: 2.5 h. Eluent: toluene. Yield: 213 mg, 72%; light tan crystals, mp 117–120 °C (MeOH); 1H-NMR (500 MHz): δ 8.03 (d, 3J = 8.0 Hz, 1H, H-4), 7.85 (d, 3J = 8.1 Hz, 1H, H-7), 7.72 (m, 2H, Ph H-2,6), 7.57 (m, 1H, H-6), 7.52 (m, 2H, H-5, Ph H-4), 7.45 (m, 2H, Ph H-3,5); 13C-NMR (125 MHz): δ 169.3 (C=O), 139.3 (C-7a), 137.6 (C-3a), 136.4 (C-2), 133.2 (Ph C-2,6), 131.2 (Ph C-4), 129.0 (C-6), 128.7 (Ph C-3,5), 126.9 (C-3, 3J(C3,H4) = 7.7 Hz), 125.7 (C-5), 124.4 (C-4), 122.9 (C-7), 119.9 (Ph C-1), 95.1 (COC≡C, 3J(C,Ph H2,6) = 5.4 Hz), 88.0 (COC≡C); IR: 2199 (C≡C), 1601 (C=O) cm−1; MS m/z (%): 298/296 (M+, 27/68), 270/268 ([M − C=O]+, 40/100), 129 ([COC≡CC6H5]+, 100), 101 ([C≡CC6H5]+, 14). Calcd. for C17H9ClOS•0.2 H2O: C, 67.98; H, 3.15. Found: C, 68.11; H, 2.82.
1-(2-Chloropyridin-3-yl)-3-phenylprop-2-yn-1-one (2f). Reaction time: 2.5 h. Eluent: CH2Cl2/EtOAc, 20:1 v/v. Yield: 123 mg, 51%; yellowish-brown crystals, mp 69–71 °C (MeOH); 1H- NMR (500 MHz): δ 8.56 (dd, 3J(H5,H6) = 4.7 Hz, 4J = 1.9 Hz, 1H, H-6), 8.34 (dd, 3J = 7.7 Hz, 4J = 1.9 Hz, 1H, H-4), 7.65 (m, 2H, Ph H-2,6), 7.51 (m, 1H, Ph H-4), 7.42 (m, 3H, H-5, Ph H-3,5); 13C-NMR (125 MHz): δ 175.5 (C=O, 3J(CO,H4) = 5.3 Hz), 152.3 (C-6, 1J(C6,H6) = 183.7 Hz, 2J(C6,H5) = 3.8 Hz, 3J(C6,H4) = 8.2 Hz), 149.5 (C-2, 3J(C2,H4) = 8.8 Hz, 3J(C2,H6) = 13.8 Hz, 4J(C2,H5) = 1.5Hz), 140.7 (C-4, 1J(C4,H4) = 166.2 Hz, 2J(C4,H5) = 1.9 Hz, 3J(C4,H6) = 6.7 Hz), 132.6 (C-3), 133.2 (Ph C-2,6), 131.3 (Ph C-4), 128.8 (Ph C-3,5), 122.4 (C-5, 1J(C5,H5) = 168.2 Hz, 2J(C5,H6) = 8.2 Hz), 119.5 (Ph C-1, 3J(Ph C1,Ph H3,5) = 8.6 Hz, 4J(Ph C1,Ph H4) = 1.4 Hz), 95.7 (COC≡C, 3J(C,Ph H2,6) = 5.3 Hz), 87.9 (COC≡C); 15N-NMR (50 MHz): δ −70.3 (N-1); IR: 2199 (C≡C), 1636 (C=O) cm−1; MS m/z (%): 243/241 (M+, 6/15), 215/213 ([M − C=O]+, 6/21), 129 ([COC≡CC6H5]+, 100), 101 ([C≡CC6H5]+, 13). Calcd. for C14H8ClNO: C, 69.58; H, 3.34; N, 5.80. Found: C, 69.59; H, 3.16; N, 5.67.
1-(3-Chloropyridin-4-yl)-3-phenylprop-2-yn-1-one (2g). Reaction time: 23 h. Eluent: CH2Cl2/EtOAc, 20:1 v/v. Yield: 70 mg, 19%; brown crystals, mp 77–79 °C (MeOH/H2O, 5:1 v/v); 1H- NMR (500 MHz): δ 8.76 (s, 1H, H-2), 8.68 (d, 3J = 4.9 Hz, 1H, H-6), 7.81 (d, 3J = 4.9 Hz, 1H, H-5), 7.65 (m, 2H, Ph H-2,6), 7.52 (m, 1H, Ph H-4), 7.43 (m, 2H, Ph H-3,5); 13C-NMR (125 MHz): δ 175.3 (C=O), 151.6 (C-2, 1J(C2,H2) = 188.0 Hz, 3J(C2,H6) = 11.4 Hz, 4J(C2,H5) = 1.0 Hz), 148.4 (C-6, 1J(C6,H6) = 183.6 Hz, 2J(C6,H5) = 2.3 Hz, 3J(C6,H2) = 11.6 Hz), 141.8 (C-4, 2J(C4,H5) = not resolved, 3J(C4,H2) = 4.9 Hz, 3J(C4,H6) = 6.9 Hz), 133.3 (Ph C-2,6), 131.6 (Ph C-4), 129.3 (C-3), 128.8 (Ph C-3,5), 124.0 (C-5, 1J(C5,H5) = 167.1 Hz, 2J(C5,H6) = 9.7 Hz), 119.3 (Ph C-1, 3J(Ph C1,Ph H3,5) = 8.5 Hz, 4J(Ph C1,Ph H4) = 1.4 Hz), 96.2 (COC≡C, 3J(C,Ph H2,6) = 5.4 Hz), 87.8 (COC≡C); IR: 2197 (C≡C), 1641 (C=O) cm−1; MS m/z (%): 243/241 (M+, 4/12), 215/213 ([M − C=O]+, 4/8), 129 ([COC≡CC6H5]+, 100), 101 ([C≡CC6H5]+, 11). Calcd. for C14H8ClNO•0.2 H2O: C, 68.56; H, 3.45; N, 5.71. Found: C, 68.69; H, 3.25; N, 5.63.
1-(2-Chloroquinolin-3-yl)-3-phenylprop-2-yn-1-one (2h). Reaction time: 200 min. Eluent: light petroleum/EtOAc, 4:1 v/v. Yield: 181 mg, 62%; brownish-yellow crystals, mp 100–101 °C (MeOH); 1H-NMR (500 MHz): δ 8.87 (s, 1H, H-4), 8.08 (dd, 3J = 8.5 Hz, 4J = 1.1 Hz, 1H, H-8), 7.97 (dd, 3J = 8.1 Hz, 4J = 1.4 Hz, 1H, H-5), 7.88 (m, 3J(H7,H6) = 7.0 Hz, 3J(H7,H8) = 8.5 Hz, 4J = 1.4 Hz, 1H, H-7), 7.68 (m, 2H, Ph H-2,6), 7.66 (m, 1H, H-6), 7.51 (m, 1H, Ph H-4), 7.44 (m, 2H, Ph H-3,5); 13C-NMR (125 MHz): δ 175.3 (C=O, 3J(CO,H4) = 5.7 Hz), 148.4 (C-8a), 147.0 (C-2, 3J(C2,H4) = 9.7 Hz), 142.7 (C-4, 1J(C4,H4) = 164.4 Hz, 3J(C4,H5) = 4.6 Hz), 133.2 (C-7, Ph C-2,6), 131.3 (Ph C-4), 130.2 (C-3), 128.9 (C-5), 128.8 (Ph C-3,5), 128.5 (C-8), 128.0 (C-6), 126.0 (C-4a), 119.7 (Ph C-1, 3J(Ph C1,Ph H3,5) = 8.5 Hz, 4J(Ph C1,Ph H4) = 1.3 Hz), 95.2 (COC≡C, 3J(C,Ph H2,6) = 5.3 Hz, 4J(C,Ph H3,5) = 1.0 Hz), 88.0 (COC≡C); 15N-NMR (50 MHz): δ −78.4 (N-1); IR: 2197 (C≡C), 1632 (C=O) cm−1; MS m/z (%): 293/291 (M+, 6/15), 265/263 ([M − C=O]+, 8/24), 239 (41), 129 ([COC≡CC6H5]+, 90), 101 ([C≡CC6H5]+, 45), 43 (100). Calcd. for C18H10ClNO•0.4 H2O: C, 72.32; H, 3.64; N, 4.69. Found: C, 72.27; H, 3.37; N, 5.01.
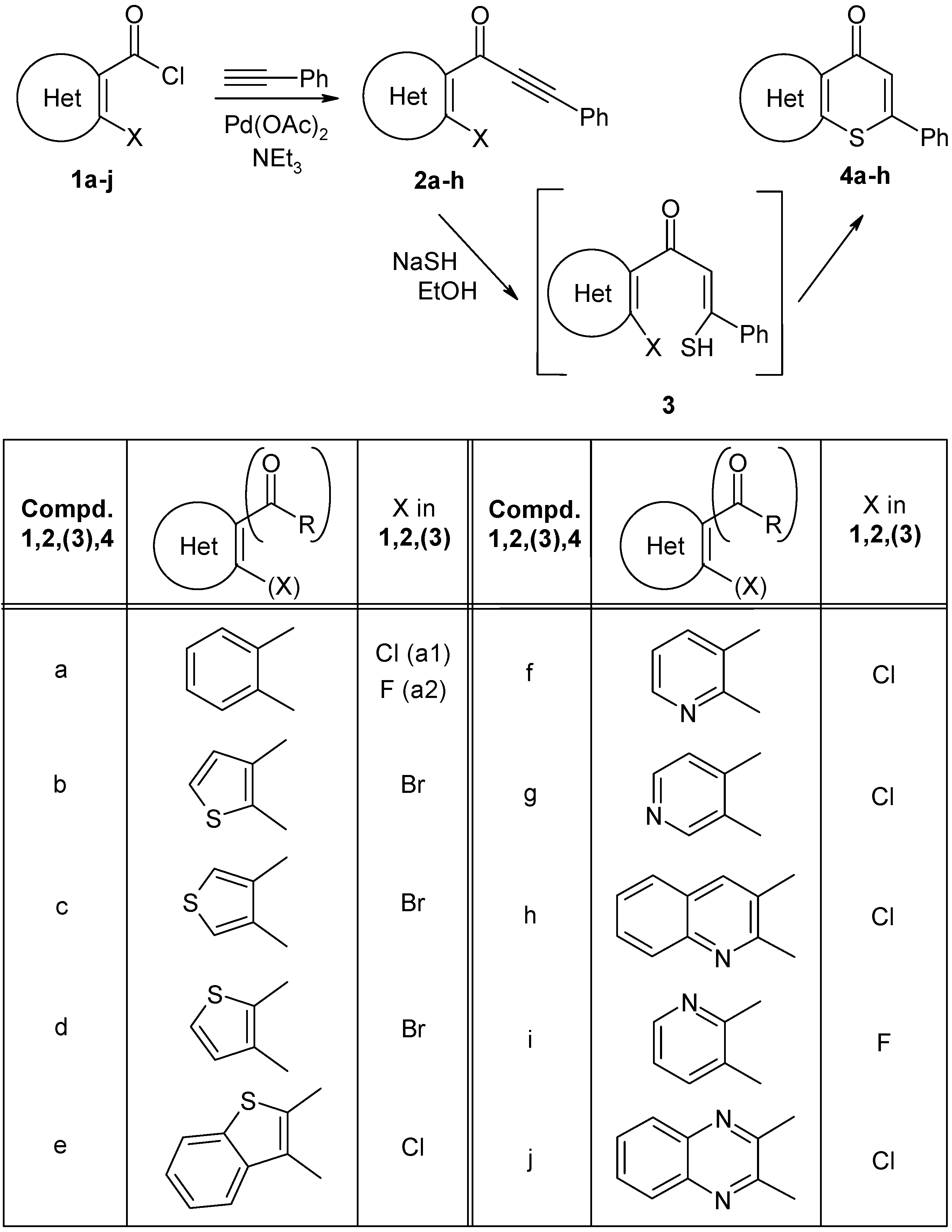
4.2.3. General procedure for the synthesis of thiopyranones 4a–h
A suspension of NaSH hydrate (~60%, 1.2 mmol, 112 mg) was refluxed in EtOH (20 mL) until a cloudy solution was formed. Ynone 2 (0.4 mmol) in EtOH (2 mL) was then added to the solution, which was refluxed for the indicated time. The solvent was removed under reduced pressure and water (20 mL) was added to the residue. The resulting solution was acidified with 5% HCl and extracted with CH2Cl2 (2 × 15 mL). The combined organic layers were washed with a saturated NaHCO3 solution and a saturated NaCl solution. They were then dried over anhydrous Na2SO4. After removal of the solvent, the residue was purified by column chromatography through a silica gel (eluent as indicated below). An analytically pure sample was obtained by recrystallization from an appropriate solvent.
2-Phenyl-4H
-thiochromen-4-one (
4a). Reaction time: 4 h. Eluent: CH
2Cl
2/EtOAc, 15:1 v/v. Yield: 131 mg, 54%; beige needles, mp 121–123 °C (MeOH) (lit. [
18] mp 124–126 °C);
1H= NMR (500 MHz): δ 8.54 (dd, 1H, H-5), 7.68 (m, 2H, Ph H-2,6), 7.65 (dd, 1H, H-8), 7.62 (dt, 1H, H-7), 7.54 (dt, 1H, H-6), 7.51 (m, 1H, Ph H-4), 7.50 (m, 2H, Ph H-3,5), 7.24 (s, 1H, H-3);
13C-NMR (125 MHz): δ 180.8 (C-4), 153.0 (C-2), 137.6 (C-8a), 136.5 (Ph C-1), 131.6 (C-7), 130.8 (C-4a, Ph C-4), 129.2 (Ph C-3,5), 128.5 (C-5), 127.7 (C-6), 126.9 (Ph C-2,6), 126.4 (C-8), 123.4 (C-3,
1J(C3,H3) = 163.6 Hz); IR: 1620 (C=O) cm
−1; MS
m/z (%): 238 (M
+, 92), 210 ([M − C=O]
+, 100), 136 ([COC
6H
4S]
+, 53), 108 ([C
6H
4S]
+, 28).
6-Phenyl-4H-thieno[2,3-b]thiopyran-4-one (4b). Reaction time: 100 min. Eluent: CH2Cl2/EtOAc, 5:1 v/v. Yield: 20 mg, 50%; whitish powder, mp 115–117 °C (MeOH); 1H-NMR (500 MHz): δ 7.78 (d, 3J = 5.3 Hz, 1H, H-3), 7.62 (m, 2H, Ph H-2,6), 7.50 (m, 1H, Ph H-4), 7.49 (m, 2H, Ph H-3,5), 7.46 (d, 3J = 5.3 Hz, 1H, H-2), 7.20 (s, 1H, H-5); 13C-NMR (125 MHz): δ 177.1 (C-4, 2J(C4,H5) = 1.2 Hz, 3J(C4,H3) = 1.3 Hz), 151.5 (C-6, 2J(C6,H5) = 3.4 Hz), 144.3 (C-7a, 3J(C7a,H2) = 6.7 Hz, 3J(C7a,H3) = 10.0 Hz), 137.5 (C-3a, 2J(C3a,H3) = 4.7 Hz, 3J(C3a,H2) = 9.9 Hz, 3J(C3a,H5) = 3.7 Hz), 136.1 (Ph C-1, 3J(Ph C1,H5) = 5.4 Hz), 130.7 (Ph C-4), 129.3 (Ph C-3,5), 127.0 (Ph C-2,6), 125.5 (C-3, 1J(C3,H3) = 174.8 Hz, 2J(C3,H2) = 4.4 Hz, 4J(C3,H5) = 1.0 Hz), 125.3 (C-2, 1J(C2,H2) = 189.6 Hz, 2J(C2,H3) = 7.5 Hz), 125.0 (C-5, 1J(C5,H5) = 163.3 Hz); IR: 1606 (C=O) cm−1; MS m/z (%): 244 (M+, 100), 216 ([M − C=O]+, 31), 142 ([COC4H2S2]+, 97), 120 ([COCH=CC6H5]+, 69), 114 ([C4H2S2]+, 38), 101 ([C=CC6H5]+, 27). Calcd. for C13H8OS2: C, 63.90; H, 3.30. Found: C, 63.57; H, 3.27.
2-Phenyl-4H-thieno[3,4-b]thiopyran-4-one (4c). Reaction time: 22.5 h. Eluent: CH2Cl2/EtOAc, 5:1 v/v. Yield: 28 mg, 34%; brownish needles, mp 160–162 °C (MeOH/H2O); 1H-NMR (500 MHz): δ 8.51 (d, 4J = 3.4 Hz, 1H, H-5), 7.66 (m, 2H, Ph H-2,6), 7.52 (d, 3J = 3.4 Hz, 1H, H-7), 7.49 (m, 1H, Ph H-4), 7.48 (m, 2H, Ph H-3,5), 6.99 (s, 1H, H-3); 13C-NMR (125 MHz): δ 178.2 (C-4, 2J(C4,H3) = 1.1 Hz, 3J(C4,H5) = 2.2 Hz, 4J(C4,H7) = 1.1 Hz), 153.3 (C-2, 2J(C2,H3) = 3.1 Hz), 136.7 (Ph C-1, 3J(Ph C1,H3) = 5.5 Hz), 134.7 (C-4a, 2J(C4a,H5) = 3.2 Hz, 3J(C4a,H3) = 3.8 Hz, 3J(C4a,H7) = 7.8 Hz), 131.4 (C-7a, 2J(C7a,H7) = 2.8 Hz, 3J(C7a,H5) = 9.8 Hz), 130.8 (Ph C-4), 129.7 (C-5, 1J(C5,H5) = 191.8 Hz, 3J(C5,H7) = 4.9 Hz, 4J(C5,H3) = 1.3 Hz), 129.2 (Ph C-3,5), 127.0 (Ph C-2,6), 122.1 (C-3, 1J(C3,H3) = 163.6 Hz), 119.2 (C-7, 1J(C7,H7) = 189.3 Hz, 3J(C7,H5) = 5.7 Hz); IR: 1612 (C=O) cm−1; MS m/z (%): 244 (M+, 48), 216 ([M − C=O]+, 16), 142 ([COC4H2S2]+, 100), 114 ([C4H2S2]+, 36), 82 ([C4H2S]+, 22). Calcd. for C13H8OS2: C, 63.90; H, 3.30. Found: C, 62.82; H 3.28. HRMS (ESI) Calcd. for C13H9OS2 [M+H]: 245.0095. Found: 245.0100.
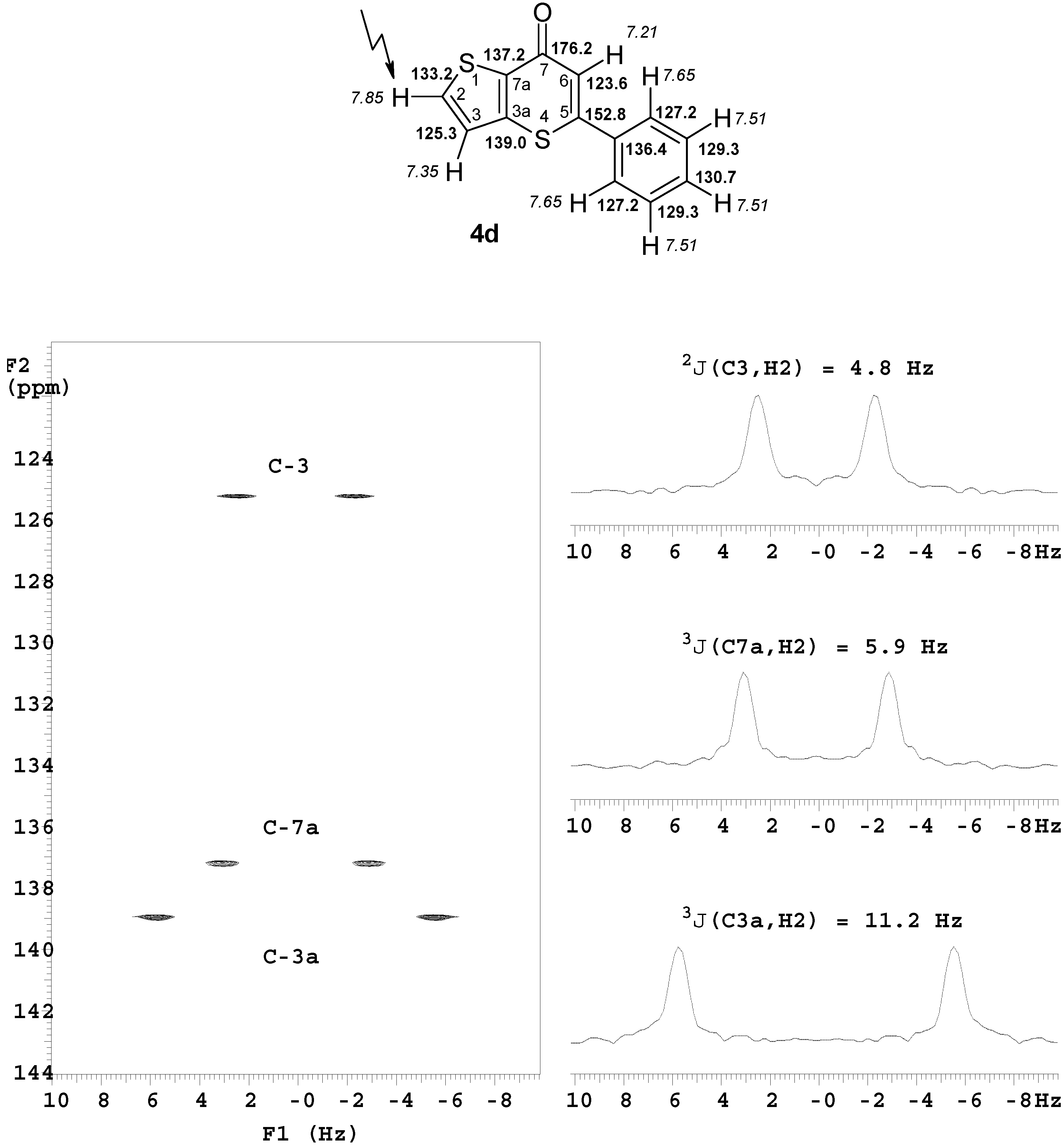
5-Phenyl-7H
-thieno[3,2-b
]thiopyran-7-one (
4d). Reaction time: 1 h. Eluent: CH
2Cl
2/EtOAc, 5:1 v/v. Yield: 80 mg, 87%; brownish powder, mp 82–84 °C (EtOH/H
2O) (lit. [
24] mp 80 °C);
1H-NMR (500 MHz): δ 7.85 (d,
3J = 5.3 Hz, 1H, H-2), 7.65 (m, 2H, Ph H-2,6), 7.51 (m, 3H, Ph H-3,4,5), 7.35 (d,
3J = 5.3 Hz, 1H, H-3), 7.21 (s, 1H, H-6);
13C-NMR (125 MHz): δ 176.2 (C-7,
2J(C7,H6) = 1.1 Hz,
4J(C7,H2) = 1.1 Hz,
4J(C7,H3) = 1.1 Hz), 152.8 (C-5,
2J(C5,H6) = 3.1 Hz), 139.0 (C-3a,
2J(C3a,H3) = 4.4 Hz,
3J(C3a,H2) = 11.2 Hz), 137.2 (C-7a,
3J(C7a,H2) = 5.9 Hz,
3J(C7a,H3) = 7.1 Hz,
3J(C7a,H6) = 4.7 Hz), 136.4 (Ph C-1,
3J(Ph C1,H6) = 5.4 Hz), 133.2 (C-2,
1J(C2,H2) = 186.6 Hz,
2J(C2,H3) = 6.0 Hz), 130.7 (Ph C-4), 129.3 (Ph C-3,5), 127.2 (Ph C-2,6), 125.3 (C-3,
1J(C3,H3) = 173.4 Hz,
2J(C3,H2) = 4.8 Hz), 123.6 (C-6,
1J(C6,H6) = 163.9 Hz); MS
m/z (%): 244 (M
+, 46), 243 ([M − H]
+, 21), 216 ([M − C=O]
+, 16), 142 ([COC
4H
2S
2]
+, 94), 114 ([C
4H
2S
2]
+, 33), 102 ([CH=CC
6H
5]
+, 19), 82 ([C
4H
2S]
+, 20), 69 (100).
2-Phenyl-4H
-thiopyrano[3,2-b
][1]benzothiophen-4-one (
4e). Reaction time: 70 min. Eluent: CH
2Cl
2/EtOAc, 15:1 v/v. Yield: 96 mg, 82%; colorless crystals, mp 166–167 °C (MeOH/EtOAc, 2:1 v/v) (lit. [
25] mp 177 °C);
1H-NMR (500 MHz): δ 8.00 (m, 1H, H-9), 7.99 (m, 1H, H-6), 7.71 (m, 2H, Ph H-2,6), 7.59 (m, 1H, H-7), 7.54 (m, 3H, Ph H-3,4,5), 7.53 (m, 1H, H-8), 7.32 (s, 1H, H-3);
13C-NMR (125 MHz): δ 177.0 (C-4,
2J(C4,H3) = 1.1 Hz), 152.1 (C-2,
2J(C2,H3) = 3.1 Hz), 140.7 (C-5a), 137.3 (C-4a,
3J(C4a,H3) = 4.6 Hz), 136.3 (Ph C-1,
3J(Ph C1,H3) = 5.3 Hz), 136.2 (C-9a), 135.4 (C-9b,
3J(C9b,H9) = 3.9 Hz), 130.8 (Ph C-4), 129.4 (Ph C-3,5), 128.6 (C-7,
1J(C7,H7) = 162.0 Hz,
3J(C7,H9) = 7.8 Hz), 127.2 (Ph C-2,6), 125.3 (C-8,
1J(C8,H8) = 162.5 Hz,
3J(C8,H6) = 7.6 Hz), 124.7 (C-3,
1J(C3,H3) = 163.9 Hz), 123.8 (C-6,
1J(C6,H6) = 164.8 Hz,
2J(C6,H7) = 1.5 Hz,
3J(C6,H8) = 8.0 Hz,
4J(C6,H9) = 1.5 Hz), 122.5 (C-9,
1J(C9,H9) = 161.1 Hz,
2J(C9,H8) = 1.4 Hz,
3J(C9,H7) = 8.1 Hz,
4J(C9,H6) = 1.4 Hz); IR: 1618 (C=O) cm
−1; MS
m/z (%): 294 (M
+, 32), 266 ([M–C=O]
+, 34), 164 ([C
8H
4S
2]
+, 25), 132 ([C
8H
4S]
+, 29), 120 (43), 68 (100). Calcd. for C
17H
10OS
2•0.1 H
2O: C, 68.94; H, 3.47; S, 21.65. Found: C, 68.91; H, 3.45; S, 21.26.
2-Phenyl-4H
-thiopyrano[2,3-b
]pyridin-4-one (
4f). Reaction time: 1.5 h. Eluent: CH
2Cl
2/EtOAc, 15:1 v/v. Yield: 36 mg, 93%; luminous yellow needles, mp 114–117 °C (MeOH) (lit. [
19] mp 113–116 °C);
1H-NMR (300 MHz): δ 8.81 (dd,
3J = 4.5 Hz,
4J = 1.9 Hz, 1H, H-7), 8.77 (dd,
3J = 8.1 Hz,
4J = 1.9 Hz, 1H, H-5), 7.71 (m, 2H, Ph H-2,6), 7.53 (m, 3H, Ph H-3,4,5), 7.50 (dd,
3J(H6,H7) = 4.5 Hz,
3J(H6,H5) = 8.1 Hz, 1H, H-6), 7.26 (s, 1H, H-3);
13C-NMR (75 MHz): δ 181.3 (C-4,
3J(C4,H5) = 3.5 Hz), 159.1 (C-8a,
3J(C8a,H5) = 6.5 Hz,
3J(C8a,H7) = 14.0 Hz), 154.7 (C-2), 152.8 (C-7,
1J(C7,H7) = 181.7 Hz,
2J(C7,H6) = 3.8 Hz,
3J(C7,H5) = 7.8 Hz), 136.7 (C-5,
1J(C5,H5) = 168.2 Hz,
2J(C5,H6) not resolved,
3J(C5,H7) = 6.2 Hz), 136.3 (Ph C-1), 131.1 (Ph C-4), 129.4 (Ph C-3,5), 128.1 (C-4a), 127.0 (Ph C-2,6), 123.6 (C-3,
1J(C3,H3) = 164.7 Hz), 122.9 (C-6,
1J(C6,H6) = 167.0 Hz,
2J(C6,H5) not resolved,
2J(C6,H7) = 8.2 Hz);
15N-NMR (50 MHz): δ −76.8 (N-8); IR: 1630 (C=O) cm
−1; MS
m/z (%): 239 (M
+, 100), 211 ([M − C=O]
+, 99), 137 ([COC
5H
3NS]
+, 33), 109 ([C
5H
3NS]
+, 57), 105 ([COC
5H
3N]
+, 32), 102 ([CH=CC
6H
5]
+, 33). Calcd. for C
14H
9NOS: C, 70.27; H, 3.79; N, 5.85. Found: C, 70.26; H, 4.04; N, 5.75.
2-Phenyl-4H-thiopyran[2,3-c]pyridin-4-one (4g). Reaction time: 1.5 h. Eluent: CH2Cl2/EtOAc, 5:1 v/v. Yield: 21 mg, 44%; flat brownish prisms, mp 153–154 °C (EtOH/H2O); 1H-NMR (500 MHz): δ 9.02 (s, 1H, H-8), 8.73 (d, 3J = 5.3 Hz, 1H, H-6), 8.27 (d, 3J = 5.3 Hz, 1H, H-5), 7.69 (m, 2H, Ph H-2,6), 7.53 (m, 1H, Ph H-4), 7.52 (m, 2H, Ph H-3,5), 7.27 (s, 1H, H-3); 13C-NMR (125 MHz): δ 179.5 (C-4, 2J(C4,H3) = 1.0 Hz, 3J(C4,H5) = 3.6 Hz, 4J(C4,H8) = 1.6 Hz), 154.0 (C-2, 2J(C2,H3) = 2.5 Hz), 148.8 (C-8, 1J(C8,H8) = 183.4 Hz, 3J(C8,H6) = 11.1 Hz, 4J(C8,H5) = 0.8 Hz), 147.8 (C-6, 1J(C6,H6) = 182.7 Hz, 2J(C6,H5) = 3.2 Hz, 3J(C6,H8) = 11.6 Hz), 136.1 (Ph C-1, 3J(Ph C1,H3) = 5.5 Hz), 135.7 (C-4a, 2J(C4a,H5) not resolved, 3J(C4a,H3) = 3.7 Hz, 3J(C4a,H6) = 6.7 Hz, 3J(C4a,H8) = 5.1 Hz), 133.2 (C-8a, 2J(C8a,H8) = 7.9 Hz, 3J(C8a,H5) = 6.5 Hz, 4J(C8a,H6) = 1.8 Hz), 131.3 (Ph C-4), 129.4 (Ph C-3,5), 127.0 (Ph C-2,6), 123.9 (C-3, 1J(C3,H3) = 164.5 Hz), 120.6 (C-5, 1J(C5,H5) = 168.9 Hz, 2J(C5,H6) = 9.0 Hz, 4J(C5,H3) = 0.8 Hz, 4J(C5,H8) = 1.7 Hz); 15N-NMR (50 MHz): δ −60.0 (N-7); IR: 1635 (C=O) cm−1; MS m/z (%): 239 (M+, 100), 211 ([M − C=O]+, 77), 149 (49), 139 (28), 137 ([COC5H3NS]+, 66), 109 ([C5H3NS]+, 53), 105 ([COC5H3N]+, 20), 102 ([CH=CC6H5]+, 41), 81 (98). Calcd. for C14H9NOS: C, 70.27; H, 3.79; N, 5.85; S, 13.40. Found: C, 70.28; H, 4.02; N, 5.69; S, 13.21. HRMS Calcd. for C14H9NOS: 239.0405. Found: 239.0402.
2-Phenyl-4H-thiopyrano[2,3-b]quinolin-4-one (4h). Reaction time: 100 min. Eluent: CH2Cl2/EtOAc, 20:1 v/v. Yield: 77 mg, 67%; off-white needles, mp 208–210 °C (CH2Cl2/EtOAc, 5:1 v/v); 1H-NMR (500 MHz): δ 9.30 (s, 1H, H-5), 8.09 (d, 3J = 8.6 Hz, 1H, H-9), 8.04 (d, 3J = 8.1 Hz, 1H, H-6), 7.88 (m, 1H, H-8), 7.74 (m, 2H, Ph H-2,6), 7.62 (m, 1H, H-7), 7.54 (m, 3H, Ph H-3,4,5), 7.23 (s, 1H, H-3); 13C-NMR (125 MHz): δ 182.0 (C-4, 2J(C4,H3) = 0.7 Hz, 3J(C4,H5) = 4.1 Hz), 157.1 (C-10a, 3J(C10a,H5) = 8.1 Hz), 155.1 (C-2), 149.2 (C-9a), 138.6 (C-5, 1J(C5,H5) = 166.4 Hz, 3J(C5,H6) = 4.7 Hz), 136.4 (Ph C-1), 133.1 (C-8), 131.2 (Ph C-4), 129.7 (C-6), 129.4 (Ph C-3,5), 128.1 (C-9), 127.2 (C-7), 127.0 (Ph C-2,6), 126.9 (C-5a), 125.1 (C-4a), 122.1 (C-3, 1J(C3,H3) = 164.8 Hz); 15N-NMR (50 MHz): δ −83.8 (N-10); IR: 1631 (C=O) cm−1; MS m/z (%): 289 (M+, 91), 261 ([M − C=O]+, 88), 181 (46), 159 ([C9H5NS]+, 39), 130 ([COCH=CC6H5]+, 77), 101 ([C=CC6H5]+, 44), 75 (82), 68 (100). Calcd. for C18H11NOS•0.2 H2O: C, 73.80; H, 3.92; N, 4.78. Found: C, 73.85; H, 3.71; N, 4.77.
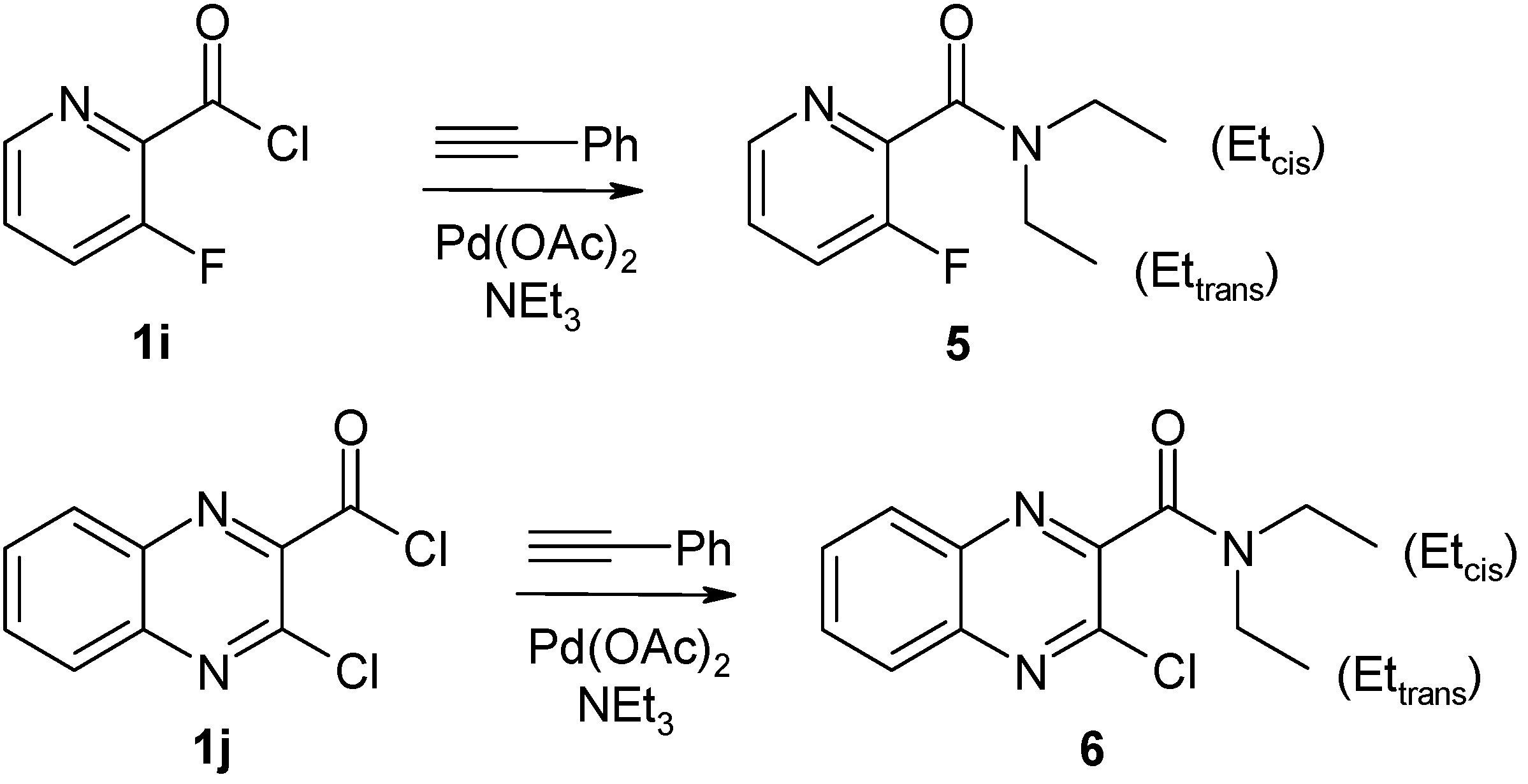
4.2.4. Preparation of N,N-diethyl-3-fluoropyridine-2-carboxamide (5)
Under the conditions given for the synthesis of compounds 2, reaction of 1i (2 mmol, 319 mg) with phenylacetylene (1 mmol, 102 mg), Et3N (2 mL) and Pd(II) acetate (10 µmol, 2 mg) for 26 h resulted in formation of compound 5 after purification by column chromatography (eluent: EtOAc). Yield: 131 mg, 33%; brown oil; 1H-NMR (500 MHz): δ 8.42 (dd, 3J = 4.6 Hz, 4J = 1.1 Hz, 5J(H6,F3) = 1.4 Hz, 1H, H-6), 7.47 (dd, 3J(H4, H5) = 8.5 Hz, 3J(H4,F3) = 8.9 Hz, 4J = 1.1 Hz, 1H, H-4), 7.35 (m, 3J(H5,H6) = 4.6 Hz, 3J(H5,H4) = 8.5 Hz, 4J(H5,F3) = 4.2 Hz, 1H, H-5), 3.58 (q, 3J = 7.1 Hz, 2H, N-CH2 cis), 3.20 (q, 3J = 7.1 Hz, 2H, N-CH2 trans), 1.27 (t, 3J = 7.1 Hz, 3H, CH3 cis), 1.10 (t, 3J = 7.1 Hz, 3H, CH3 trans); 13C-NMR (125 MHz): δ 164.7 (C=O, 3J(CO,F3) = 3.1 Hz), 155.8 (C-3, 1J(C3,F3) = 259.1 Hz, 2J(C3,H4) = 4.8 Hz, 3J(C3,H5) = 9.6 Hz, 4J(C3,H6) = 1.9 Hz), 145.2 (C-6, 1J(C6,H6) = 182.3 Hz, 2J(C6,H5) = 2.7 Hz, 3J(C6,H4) = 7.3 Hz, 4J(C6,F3) = 5.0 Hz), 144.0 (C-2, 2J(C2,F3) = 18.0 Hz, 3J(C2,H4) = 3.8 Hz, 3J(C2,H6) = 12.2 Hz, 4J(C2,H5) = 1.5 Hz), 125.1 (C-5, 1J(C5,H5) = 167.2 Hz, 2J(C5,H4) = 2.0 Hz, 2J(C5,H6) = 9.4 Hz, 3J(C5,F3) = 3.8 Hz), 123.9 (C-4, 1J(C4,H4) = 167.4 Hz, 2J(C4,F3) = 18.4 Hz, 3J(C4,H6) = 7.2 Hz), 42.8 (N-CH2 trans, 1J = 135.2 Hz, 2J(CH2,CH3) = 4.2 Hz), 39.4 (N-CH2 cis, 1J = 136.5 Hz, 2J(CH2,CH3) = 4.2 Hz), 14.0 (CH3 trans, 1J = 127.1 Hz, 2J(CH3,CH2) = 3.1 Hz), 12.8 (CH3 cis, 1J = 127.2 Hz, 2J(CH3,CH2) = 3.4 Hz); 15N-NMR (50 MHz): δ −65.8 (N-1), −248.9 (CONR2); 19F-NMR (470 MHz): δ −124.3 (F-3, 3J(F3,H4) = 8.9 Hz, 4J(F3,H5) = 4.2 Hz, 5J(F3,H6) = 1.4 Hz); IR: 1643 (C=O) cm−1; MS m/z (%): 196 (M+, 7), 124 ([M − N(Et)2]+, 65), 97 ([C5H3NF]+, 55), 72 ([N(Et)2]+, 100), 44 (63). Calcd. for C10H13FN2O•0.2 H2O: C, 60.11; H, 6.76; N, 14.02. Found: C, 60.28; H, 6.59; N, 13.90.
4.2.5. Preparation of 3-chloro-N,N-diethylquinoxaline-2-carboxamide (6)
Under the conditions given for synthesis of compounds 2, reaction of 1j (2 mmol, 455 mg) with phenylacetylene (1 mmol, 102 mg), Et3N (2 mL) and Pd(II) acetate (10 µmol, 2 mg) for 20 h resulted in formation of compound 6 after purification by column chromatography (eluent: CH2Cl2/EtOAc, 5:1 v/v.). Yield: 256 mg, 49%; brownish needles, mp 91–93 °C (MeOH); 1H-NMR (500 MHz): δ 8.10 (m, 1H, H-8), 8.03 (m, 1H, H-5), 7.83 (m, 1H, H-6), 7.80 (m, 1H, H-7), 3.65 (q, 3J = 7.2 Hz, 2H, N-CH2 cis), 3.20 (q, 3J = 7.1 Hz, 2H, N-CH2 trans), 1.34 (t, 3J = 7.2 Hz, 3H, CH3 cis), 1.16 (t, 3J = 7.1 Hz, 3H, CH3 trans); 13C-NMR (125 MHz): δ 164.8 (C=O), 149.1 (C-3), 143.8 (C-2), 141.6 (C-4a), 140.0 (C-8a), 131.6 (C-6), 130.8 (C-7), 129.2 (C-8), 128.3 (C-5), 42.9 (N-CH2 trans), 39.5 (N-CH2 cis), 13.8 (CH3 trans), 12.5 (CH3 cis); 15N-NMR (50 MHz): δ –52.1 (N-1), −63.0 (N-4), −249.2 (CONR2); IR: 1635 (C=O) cm−1; MS m/z (%): 265/263 (M+, 0.5/1.5), 191 ([M − N(Et)2]+, 3), 165/163 ([C8H4N2Cl]+, 7/18), 128 ([C8H4N2]+, 5), 72 ([N(Et)2]+, 100). Calcd. for C13H14ClN3O: C, 59.21; H, 5.35; N, 15.93. Found: C, 59.43; H, 5.42; N, 15.99.
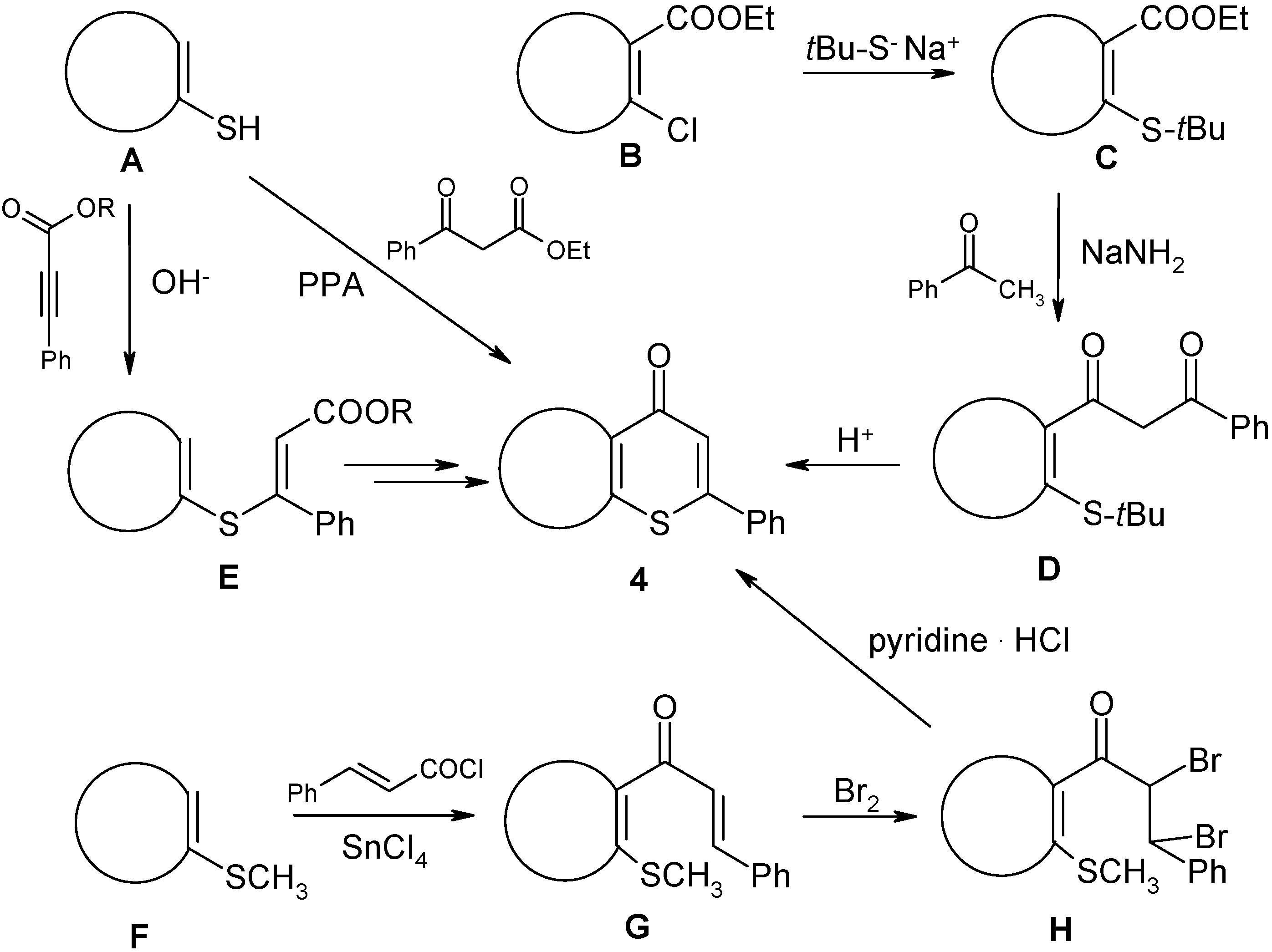
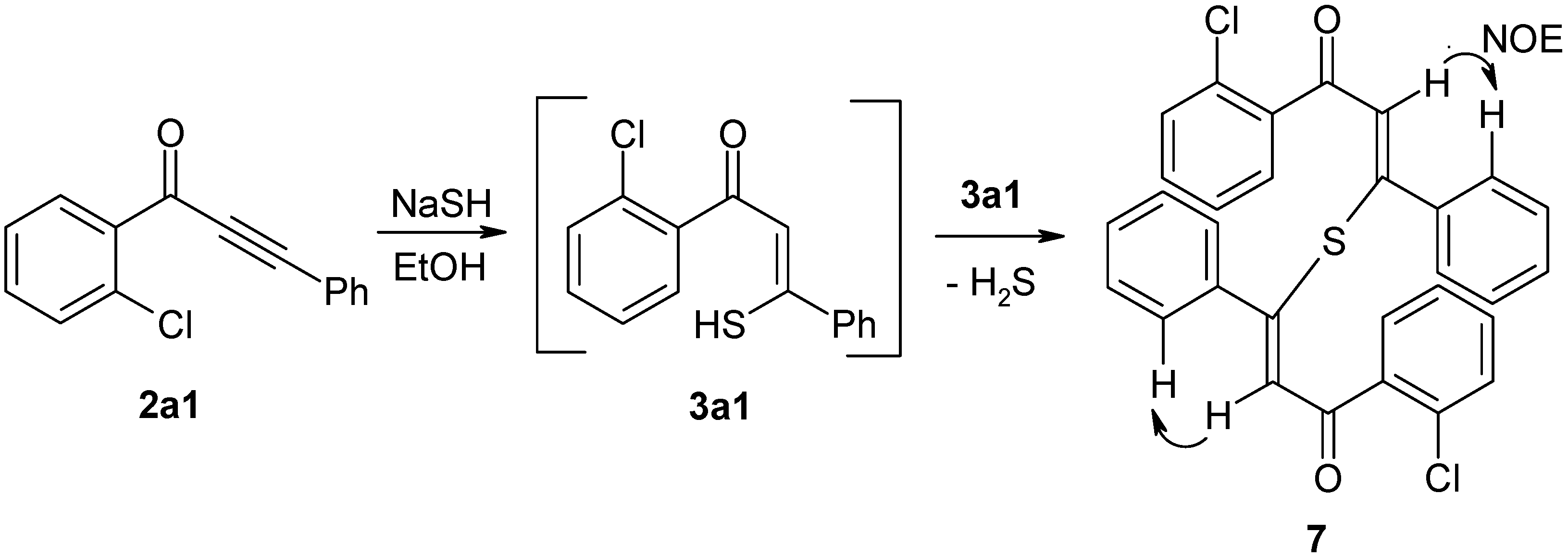
4.2.6. Preparation of (2Z,2'Z)-3,3'-sulfanediylbis[1-(2-chlorophenyl)-3-phenylprop-2-en-1-one] (7)
Ynone 2a1 (0.5 mmol, 120 mg) in EtOH (2 mL) was added to a suspension of NaSH hydrate (~60%, 1.3 mmol, 121 mg) in EtOH (20 mL). The resulting solution was stirred at room temperature for 30 min, and then processed as described in the preparation of compounds 4. The residue was purified by column chromatography through silica gel (eluent: CH2Cl2). Yield: 48 mg, 19%; yellow powder, mp 161–164 °C (EtOH); 1H-NMR (300 MHz): δ 7.65 (m, 2H, H-6), 7.43 (m, 2H, H-3), 7.42 (m, 2H, H-4), 7.38 (m, 2H, H-5), 7.18 (m, 2H, Ph H-4), 7.05 (m, 4H, Ph H-3,5), 6.97 (m, 4H, Ph H-2,6), 6.92 (s, 2H, COCH=C); 13C-NMR (75 MHz): δ 189.7 (C=O), 155.4 (COCH=C), 140.6 (Ph C-1), 139.7 (C-1), 131.9 (C-4), 131.4 (C-2), 130.5 (C-6), 130.2 (C-3), 128.9 (Ph C-4), 128.3 (Ph C-2,6), 128.0 (Ph C-3,5), 127.7 (COCH=C, 1J = 161.5 Hz), 127.1 (C-5); IR: 1638 (C=O) cm−1; MS m/z (%): 516/514 (M+, 0.3/0.4), 275/273 ([M − C15H10ClO]+, 13/33), 238 (19), 149 ([COC(S)C6H5]+, 80), 141/139 ([COC6H4Cl]+, 37/100), 113/111 ([C6H4Cl]+, 29/49), 105 (38), 85 ([COCH=CS]+, 32), 77 ([C6H5]+, 33), 57 (83). HRMS (ESI) Calcd. for C30H21Cl2O2S [M + H]: 515.0639. Found: 515.0642.
4.2.7. Preparation of (2Z)-1-(2-chlorophenyl)-3-(methylamino)-3-phenylprop-2-en-1-one (10)
Ethanolic methylamine-solution (33 wt%, 6 mL, 48 mmol) was added to a solution of ynone 2a1 (0.5 mmol, 120 mg) in pyridine (0.4 mL) and H2O (3 drops). The solution was refluxed for 1 h. The solvent was then removed under reduced pressure and water (20 mL) was added to the residue. The resulting solution was adjusted to pH 5 with 5% HCl and extracted with CH2Cl2 (2 × 15 mL). The combined organic layers were washed with a saturated NaHCO3 solution and a saturated NaCl solution and then dried over anhydrous Na2SO4. After removal of the solvent, the residue was purified by column chromatography through silica gel (eluent: CH2Cl2/EtOAc, 20:1 v/v.), which produced 10 as a slowly crystallizing oil. Yield: 99 mg, 73%; pale orange solid, mp 77–80 °C; 1H-NMR (500 MHz): δ 11.18 (broad, 1H, N-H), 7.49 (m, 1H, H-6), 7.44 (m, 3H, Ph H-3,4,5), 7.42 (m, 2H, Ph H-2,6), 7.37 (m, 1H, H-3), 7.27 (m, 1H, H-4), 7.26 (m, 1H, H-5), 5.44 (s, 1H, COCH=C), 2.96 (d, 3J = 5.4 Hz, 3H, N-CH3);13C-NMR (125 MHz): δ 189.2 (C=O), 167.6 (COCH=C), 141.3 (C-1), 134.8 (Ph C-1), 130.8 (C-2), 130.1 (C-3), 129.9 (C-4), 129.7 (Ph C-4), 129.2 (C-6), 128.5 (Ph C-3,5), 127.7 (Ph C-2,6), 126.5 (C-5), 97.3 (COCH=C, 1J = 164.5 Hz, 3J(C,NH) = 3.6 Hz), 31.6 (N-CH3, 1J = 138.5 Hz, 2J(NCH3,NH) = 3.4 Hz, 4J(NCH3,COCH=C) = 1.1 Hz); 15N-NMR (50 MHz): δ −281.5 (N-CH3); IR: 1595 (C=O) cm−1; MS m/z (%): 273/271 (M+, 12/43), 256/254 (22/45), 236 ([M − Cl]+, 100), 160 ([M–C6H4Cl]+, 57), 141/139 ([COC6H4Cl]+, 14/33), 117 ([CH=C(NH)C6H5]+, 66), 113/111 ([C6H4Cl]+, 13/38), 104 ([HNCC6H5]+, 25), 102 ([CH=CC6H5]+, 97), 77 ([C6H5]+, 74). Calcd. for C16H14ClNO: C, 70.72; H, 5.19; N, 5.15. Found: C, 70.32; H, 5.22; N, 5.32.
4.2.8. Preparation of 1-methyl-2-phenylquinolin-4(1H)-one (11)
Potassium carbonate (2 mmol, 277 mg) was added to a solution of
10 (220 µmol, 60 mg) in DMF (5 mL). The resulting mixture was refluxed for 52 h under a N
2 atmosphere (N
2 balloon). The solvent was then removed under reduced pressure and water (20 mL) was added to the residue. The resulting solution was adjusted to a slightly basic pH with 5% HCl and extracted with CH
2Cl
2 (2 ° 15 mL). The combined organic layers were washed with a saturated NaHCO
3 solution and a saturated NaCl solution and then dried over anhydrous Na
2SO
4. After removal of the solvent, the oily residue was purified by column chromatography through silica gel (eluent: EtOAc/NEt
3, 20:1 v/v.) to generate
11. Yield: 37 mg, 72%; yellowish crystals, mp 137–140 °C (EtOH/H
2O) (lit. [
53] mp 142–145 °C);
1H-NMR (500 MHz): δ 8.50 (dd,
3J = 8.1 Hz,
4J = 1.7 Hz, 1H, H-5), 7.71 (ddd,
3J(H7,H6) = 7.0 Hz,
3J(H7,H8) = 8.6 Hz,
4J = 1.7 Hz, 1H, H-7), 7.55 (d,
3J = 8.6 Hz, 1H, H-8), 7.50 (m, 3H, Ph H-3,4,5), 7.42 (m,
3J(H6,H7) = 7.0 Hz,
3J(H6,H5) = 8.1 Hz,
4J = 1.0 Hz, 1H, H-6), 7.41 (m, 2H, Ph H-2,6), 6.29 (s, 1H, H-3), 3.60 (s, 3H, N-CH
3);
13C-NMR (125 MHz): δ 177.6 (C-4,
2J(C4,H3) = 1.4 Hz,
3J(C4,H5) = 3.9 Hz), 154.7 (C-2,
2J(C2,H3) = 3.7 Hz,
3J(C2,NCH
3) = 3.1 Hz), 141.9 (C-8a,
3J(C8a,H5) = 7.6 Hz,
3J(C8a,H7) = 9.5 Hz,
3J(C8a,NCH
3) = 2.9 Hz), 135.9 (Ph C-1,
3J(Ph C1,H3) = 3.9 Hz), 132.3 (C-7), 129.6 (Ph C-4), 128.8 (Ph C-3,5), 128.5 (Ph C-2,6), 126.8 (C-4a,
3J(C4a,H3) = 4.8 Hz), 126.7 (C-5,
3J(C5,H7) = 7.8 Hz), 123.6 (C-6), 115.9 (C-8), 112.7 (C-3,
1J(C3,H3) = 165.8 Hz), 37.2 (N-CH
3,
1J = 140.1 Hz);
15N-NMR (50 MHz): δ −262.1 (N-1); MS
m/z (%): 235 (M
+, 100), 207 ([M–C=O]
+, 100), 206 ([M–H,–C=O]
+, 55), 102 (40), 77 ([C
6H
5]
+, 47).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






