Modulation of Huntingtin Toxicity by BAG1 is Dependent on an Intact BAG Domain

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
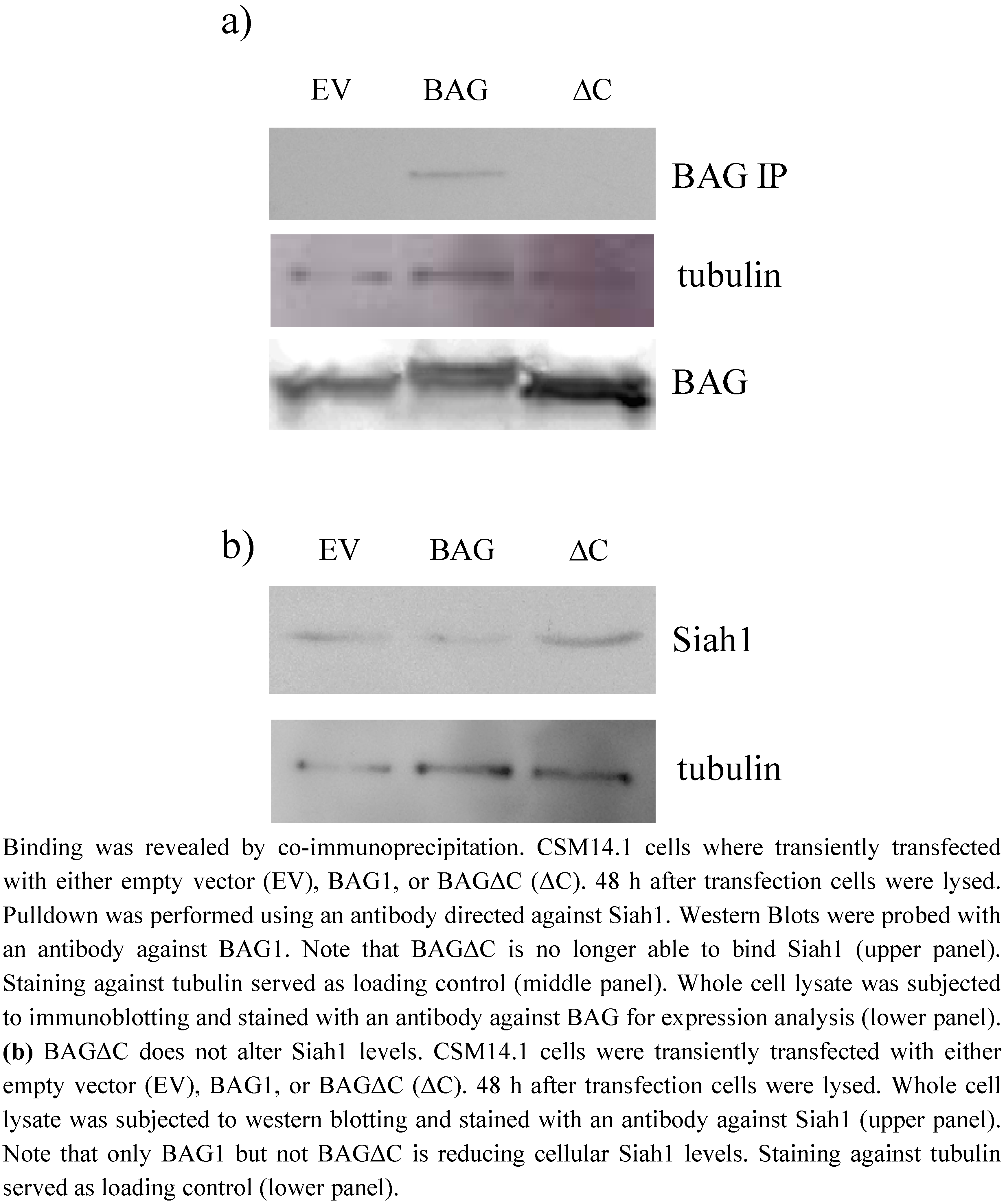
2.1. BAGΔC is not binding and not able to downregulate Siah1

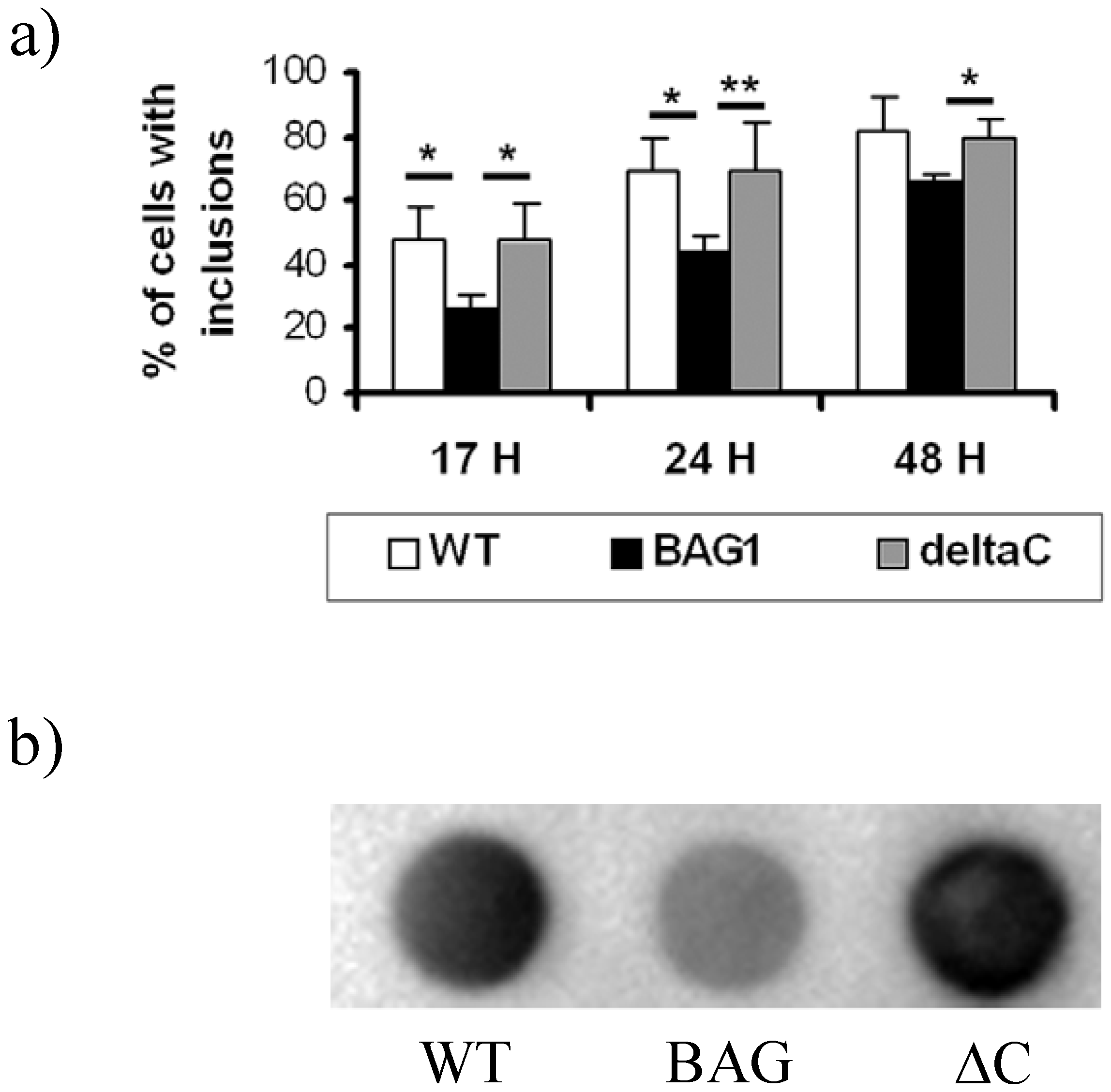
2.2. Reduction of htt inclusions

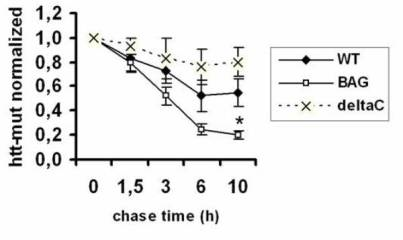
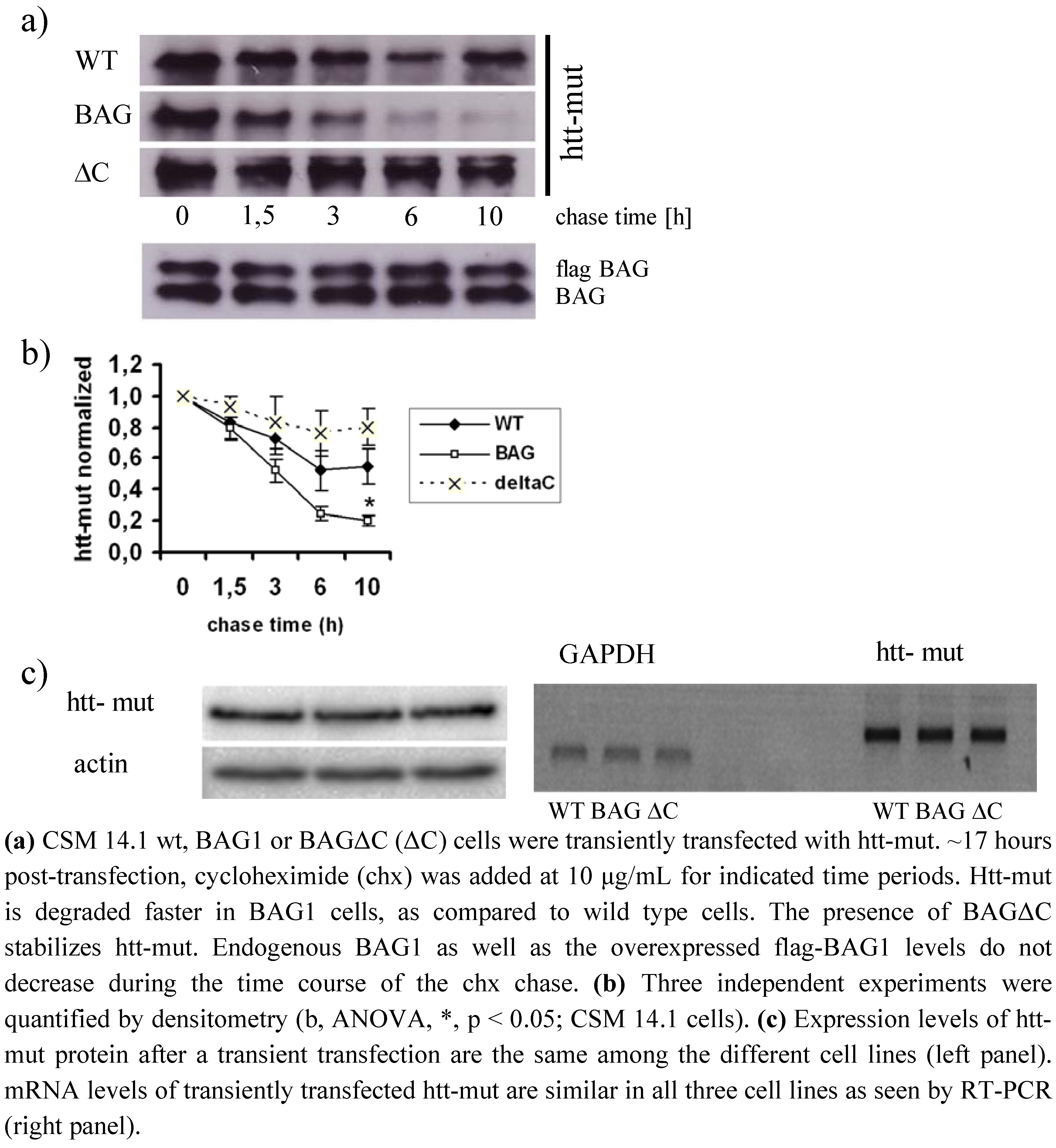
2.3. BAGΔC is no longer able to enhance htt degradation

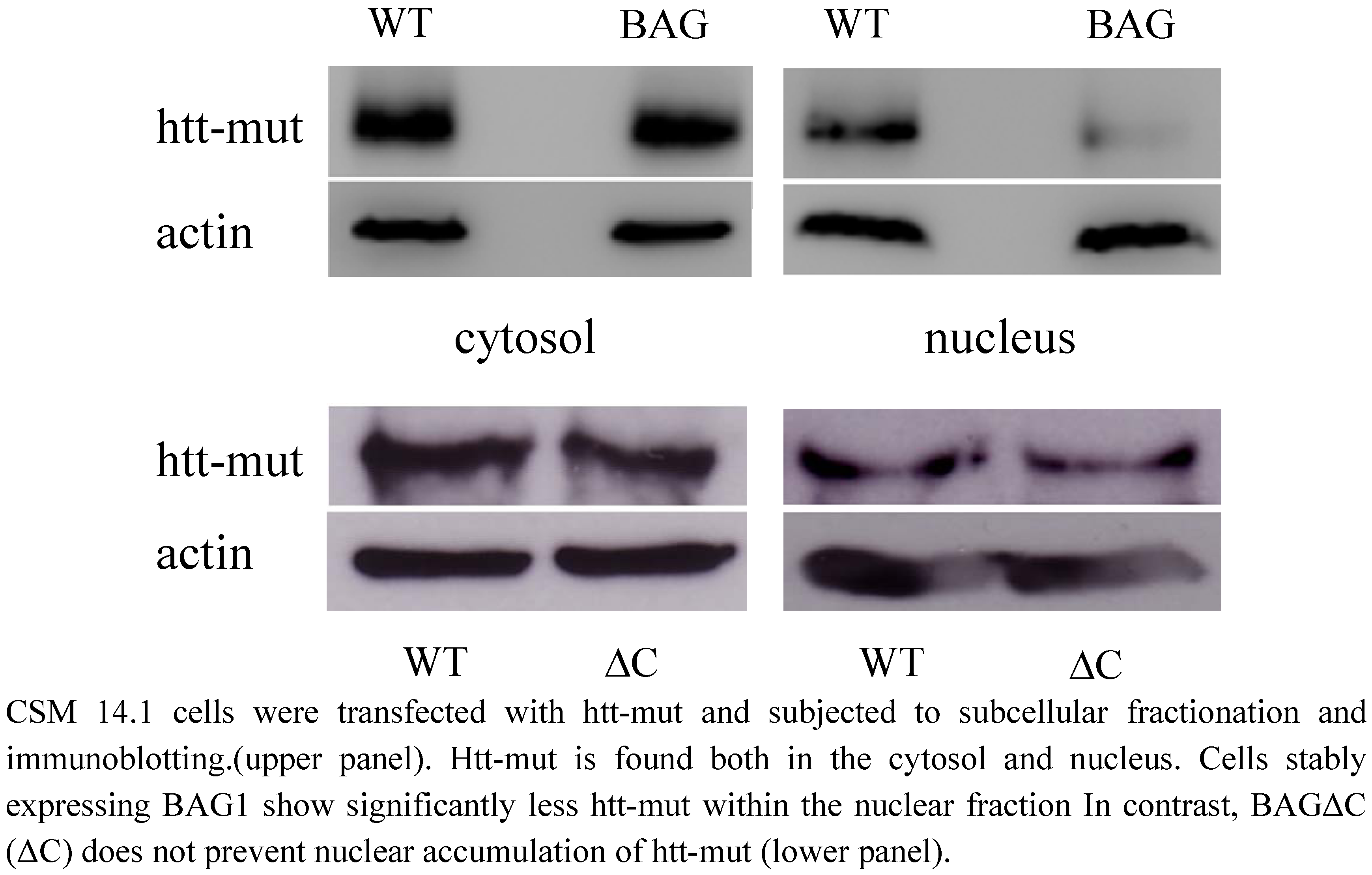
2.4. htt translocation to the nucleus is not modulated by BAGΔC

3. Experimental
3.1. Protein extract preparation
3.2. Filter retardation assay
3.3. Antibodies
3.4. Immunoblotting
3.5. Co-immunoprecipitation
3.6. Cell culture
3.7. Transient transfections
3.8. Calcium phosphate transfection
3.9. Densitometry analysis
4. Conclusions
Acknowledgements
References and Notes
- Takayama, S.; Bimston, D.N.; Matsuzawa, S.; Freeman, B.C.; Aime-Sempe, C.; Xie, Z.; Morimoto, R.I.; Reed, J.C. BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J. 1997, 16, 4887–4896. [Google Scholar] [CrossRef]
- Luders, J.; Demand, J.; Hohfeld, J. The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J. Biol. Chem. 2000, 275, 4613–4617. [Google Scholar]
- Demand, J.; Alberti, S.; Patterson, C.; Hohfeld, J. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 2001, 11, 1569–1577. [Google Scholar] [CrossRef]
- Song, J.; Takeda, M.; Morimoto, R.I. Bag1-Hsp70 mediates a physiological stress signalling pathway that regulates Raf-1/ERK and cell growth. Nat. Cell Biol. 2001, 3, 276–282. [Google Scholar] [CrossRef]
- Planchamp, V.; Bermel, C.; Tonges, L.; Ostendorf, T.; Kugler, S.; Reed, J.C.; Kermer, P.; Bahr, M.; Lingor, P. BAG1 promotes axonal outgrowth and regeneration in vivo via Raf-1 and reduction of ROCK activity. Brain 2008, 131, 2606–2619. [Google Scholar] [CrossRef]
- Kermer, P.; Krajewska, M.; Zapata, J.M.; Takayama, S.; Mai, J.; Krajewski, S.; Reed, J.C. Bag1 is a regulator and marker of neuronal differentiation. Cell Death Differ. 2002, 9, 405–413. [Google Scholar] [CrossRef]
- Gotz, R.; Wiese, S.; Takayama, S.; Camarero, G.C.; Rossoll, W.; Schweizer, U.; Troppmair, J.; Jablonka, S.; Holtmann, B.; Reed, J.C.; Rapp, U.R.; Sendtner, M. Bag1 is essential for differentiation and survival of hematopoietic and neuronal cells. Nat. Neurosci. 2005, 8, 1169–1178. [Google Scholar] [CrossRef]
- Kermer, P.; Digicaylioglu, M.H.; Kaul, M.; Zapata, J.M.; Krajewska, M.; Stenner-Liewen, F.; Takayama, S.; Krajewski, S.; Lipton, S.A.; Reed, J.C. BAG1 over-expression in brain protects against stroke. Brain Pathol. 2003, 13, 495–506. [Google Scholar]
- Matsuzawa, S.; Takayama, S.; Froesch, B.A.; Zapata, J.M.; Reed, J.C. p53-inducible human homologue of Drosophila seven in absentia (Siah) inhibits cell growth: suppression by BAG-1. EMBO J. 1998, 17, 2736–2747. [Google Scholar] [CrossRef]
- Group, H.s.D.C.R. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar]
- Dunah, A.W.; Jeong, H.; Griffin, A.; Kim, Y.M.; Standaert, D.G.; Hersch, S.M.; Mouradian, M.M.; Young, A.B.; Tanese, N.; Krainc, D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 2002, 296, 2238–2243. [Google Scholar]
- Wyttenbach, A.; Swartz, J.; Kita, H.; Thykjaer, T.; Carmichael, J.; Bradley, J.; Brown, R.; Maxwell, M.; Schapira, A.; Orntoft, T.F.; Kato, K.; Rubinsztein, D.C. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum. Mol. Genet. 2001, 10, 1829–1845. [Google Scholar]
- Zhai, W.; Jeong, H.; Cui, L.; Krainc, D.; Tjian, R. In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell 2005, 123, 1241–1253. [Google Scholar]
- Zuccato, C.; Tartari, M.; Crotti, A.; Goffredo, D.; Valenza, M.; Conti, L.; Cataudella, T.; Leavitt, B.R.; Hayden, M.R.; Timmusk, T.; Rigamonti, D.; Cattaneo, E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003, 35, 76–83. [Google Scholar] [CrossRef]
- Gunawardena, S.; Her, L.S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef]
- Szebenyi, G.; Morfini, G.A.; Babcock, A.; Gould, M.; Selkoe, K.; Stenoien, D.L.; Young, M.; Faber, P.W.; MacDonald, M.E.; McPhaul, M.; Brady, S.T. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 2003, 40, 41–52. [Google Scholar] [CrossRef]
- Bennett, E.J.; Shaler, T.A.; Woodman, B.; Ryu, K.Y.; Zaitseva, T.S.; Becker, C.H.; Bates, G.P.; Schulman, H.; Kopito, R.R. Global changes to the ubiquitin system in Huntington's disease. Nature 2007, 448, 704–708. [Google Scholar]
- Verhoef, L.G.; Lindsten, K.; Masucci, M.G.; Dantuma, N.P. Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum. Mol. Genet. 2002, 11, 2689–2700. [Google Scholar] [CrossRef]
- Jana, N.R.; Zemskov, E.A.; Wang, G.; Nukina, N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 2001, 10, 1049–1059. [Google Scholar] [CrossRef]
- Bae, B.I.; Hara, M.R.; Cascio, M.B.; Wellington, C.L.; Hayden, M.R.; Ross, C.A.; Ha, H.C.; Li, X.J.; Snyder, S.H.; Sawa, A. Mutant huntingtin: nuclear translocation and cytotoxicity mediated by GAPDH. Proc. Natl. Acad. Sci. USA 2006, 103, 3405–3409. [Google Scholar]
- Saudou, F.; Finkbeiner, S.; Devys, D.; Greenberg, M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 1998, 95, 55–66. [Google Scholar] [CrossRef]
- Peters, M.F.; Nucifora, F.C., Jr.; Kushi, J.; Seaman, H.C.; Cooper, J.K.; Herring, W.J.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Nuclear targeting of mutant Huntingtin increases toxicity. Mol. Cell Neurosci. 1999, 14, 121–128. [Google Scholar] [CrossRef]
- Sroka, K.; Voigt, A.; Deeg, S.; Reed, J.C.; Schulz, J.B.; Bahr, M.; Kermer, P. BAG1 modulates huntingtin toxicity, aggregation, degradation, and subcellular distribution. J. Neurochem. 2009, 111, 801–807. [Google Scholar] [CrossRef]
- Liman, J.; Ganesan, S.; Dohm, C.P.; Krajewski, S.; Reed, J.C.; Bahr, M.; Wouters, F.S.; Kermer, P. Interaction of BAG1 and Hsp70 mediates neuroprotectivity and increases chaperone activity. Mol. Cell Biol. 2005, 25, 3715–3725. [Google Scholar] [CrossRef]
- Wanker, E.E.; Scherzinger, E.; Heiser, V.; Sittler, A.; Eickhoff, H.; Lehrach, H. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol. 1999, 309, 375–386. [Google Scholar] [CrossRef]
- Jana, N.R.; Dikshit, P.; Goswami, A.; Kotliarova, S.; Murata, S.; Tanaka, K.; Nukina, N. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 2005, 280, 11635–11640. [Google Scholar]
- Sample Availability: Comercially available.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liman, J.; Sroka, K.; Dohm, C.P.; Deeg, S.; Bähr, M.; Kermer, P. Modulation of Huntingtin Toxicity by BAG1 is Dependent on an Intact BAG Domain. Molecules 2010, 15, 6678-6687. https://doi.org/10.3390/molecules15106678
Liman J, Sroka K, Dohm CP, Deeg S, Bähr M, Kermer P. Modulation of Huntingtin Toxicity by BAG1 is Dependent on an Intact BAG Domain. Molecules. 2010; 15(10):6678-6687. https://doi.org/10.3390/molecules15106678
Chicago/Turabian StyleLiman, Jan, Kamila Sroka, Christoph P. Dohm, Sebastian Deeg, Mathias Bähr, and Pawel Kermer. 2010. "Modulation of Huntingtin Toxicity by BAG1 is Dependent on an Intact BAG Domain" Molecules 15, no. 10: 6678-6687. https://doi.org/10.3390/molecules15106678