Towards the Development of Synthetic Routes Using Theoretical Calculations: An Application of In Silico Screening to 2,6-Dimethylchroman-4-one

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
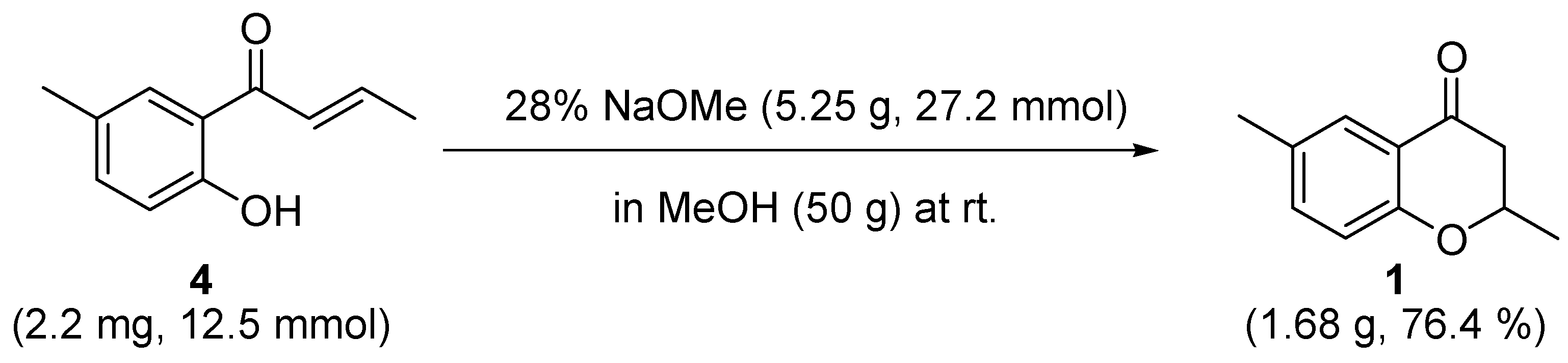{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
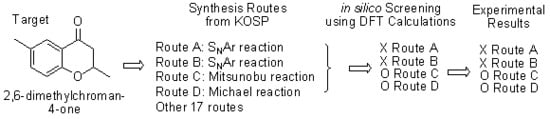
1. Introduction

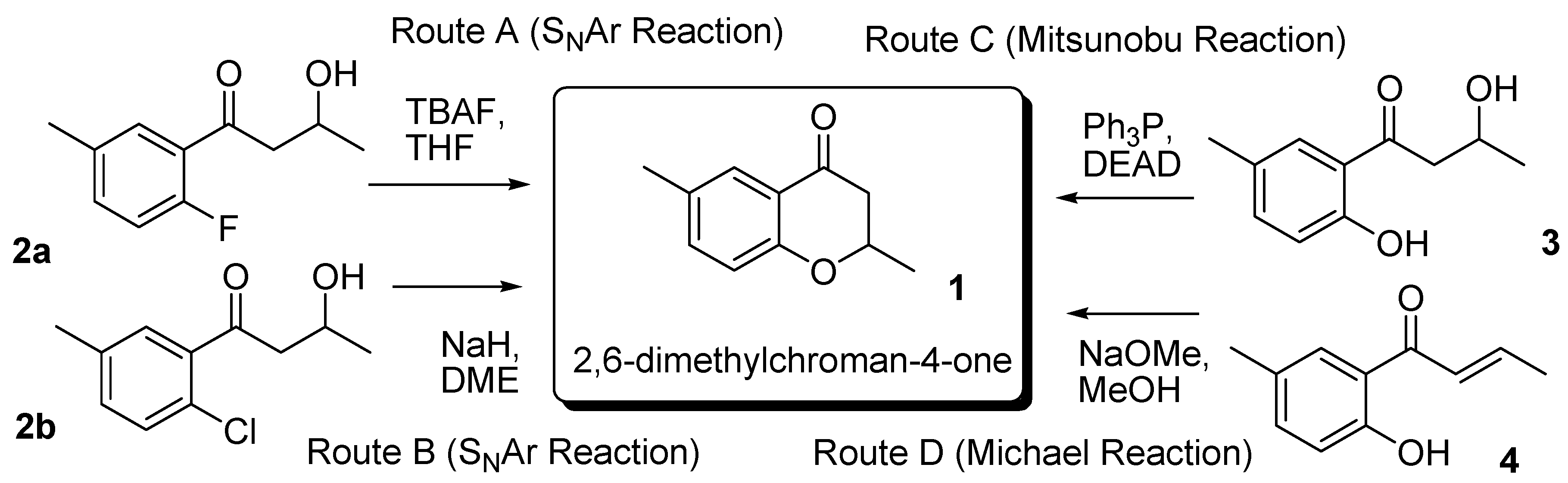
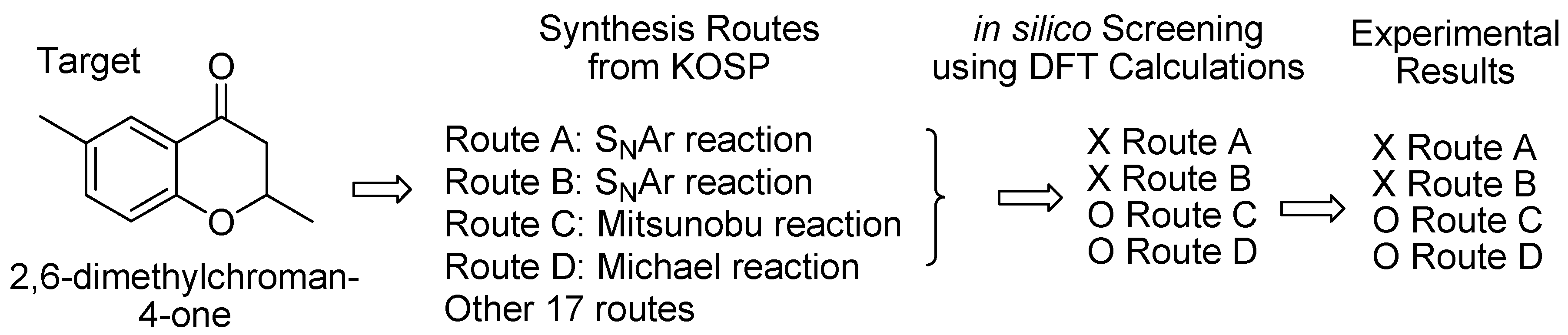
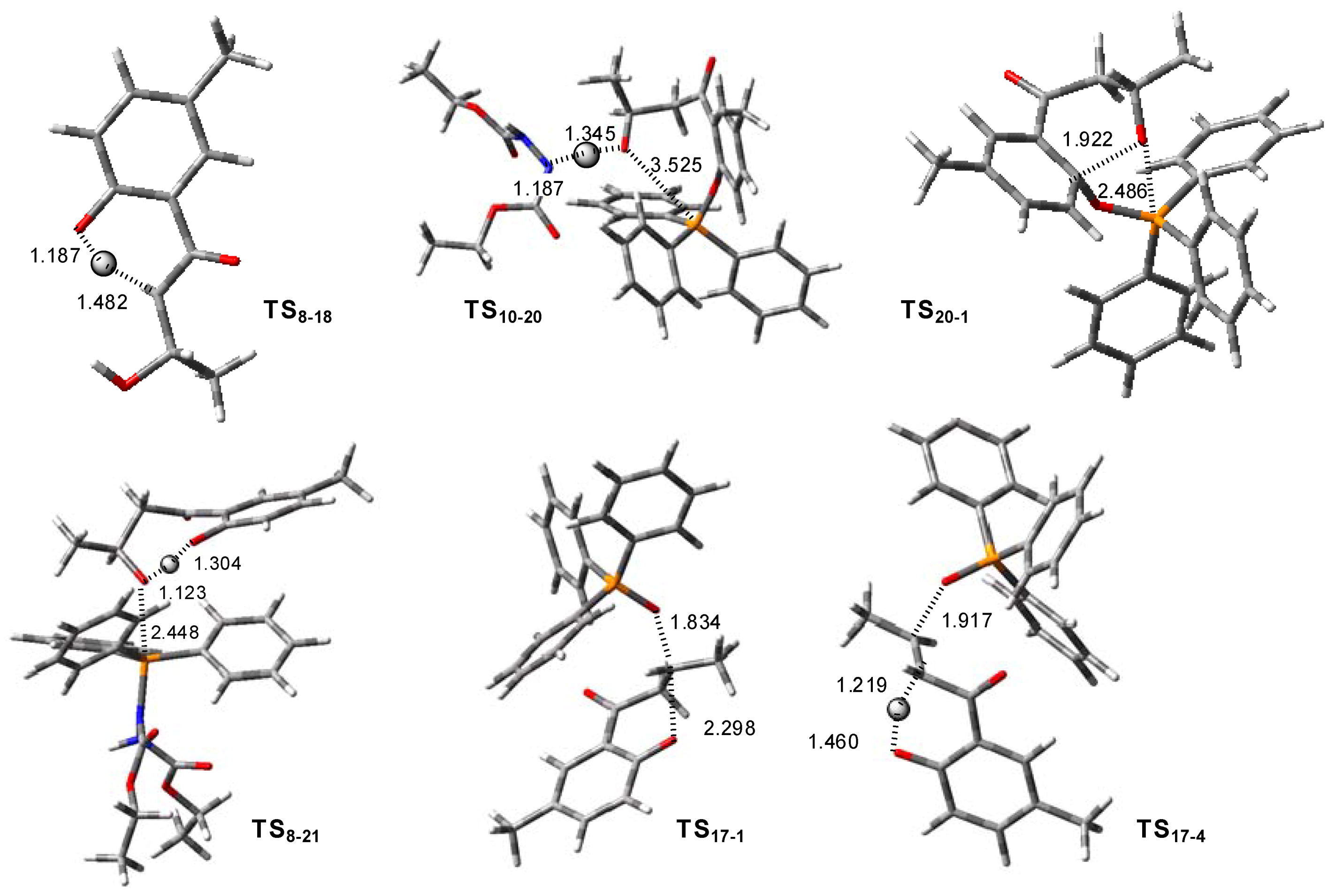
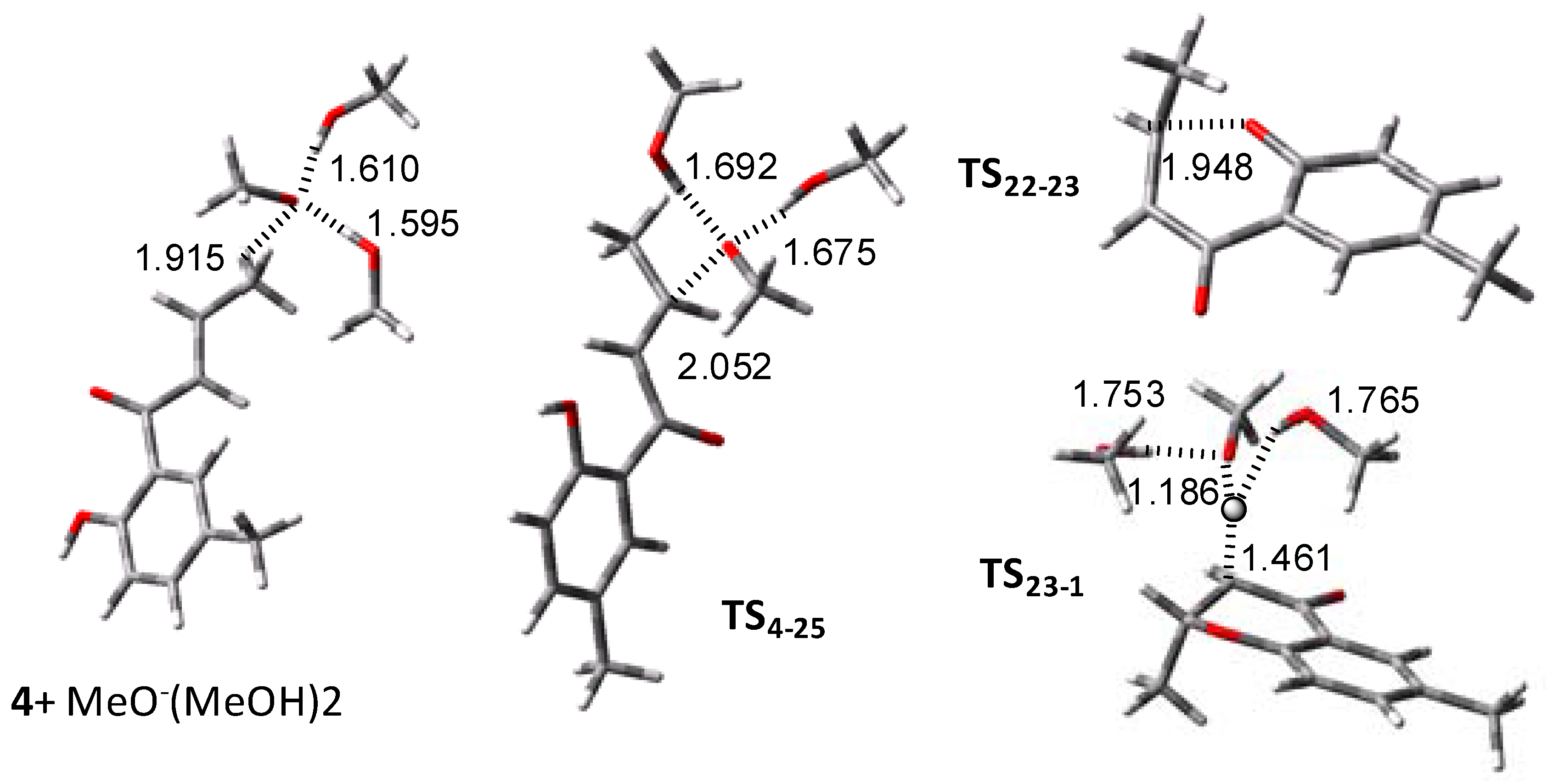
2. Results and Discussion
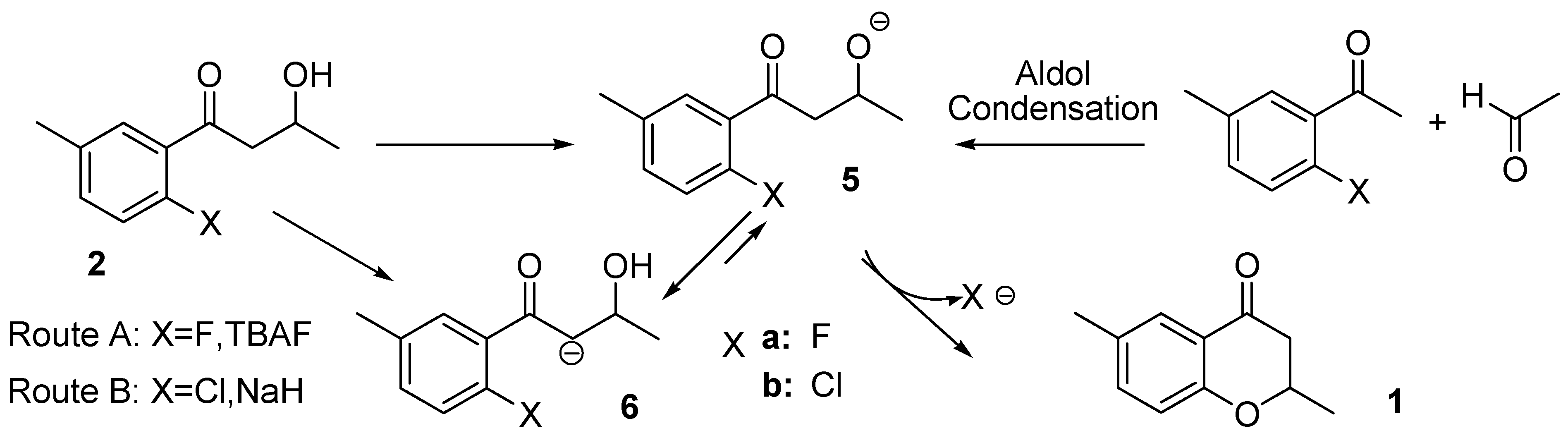
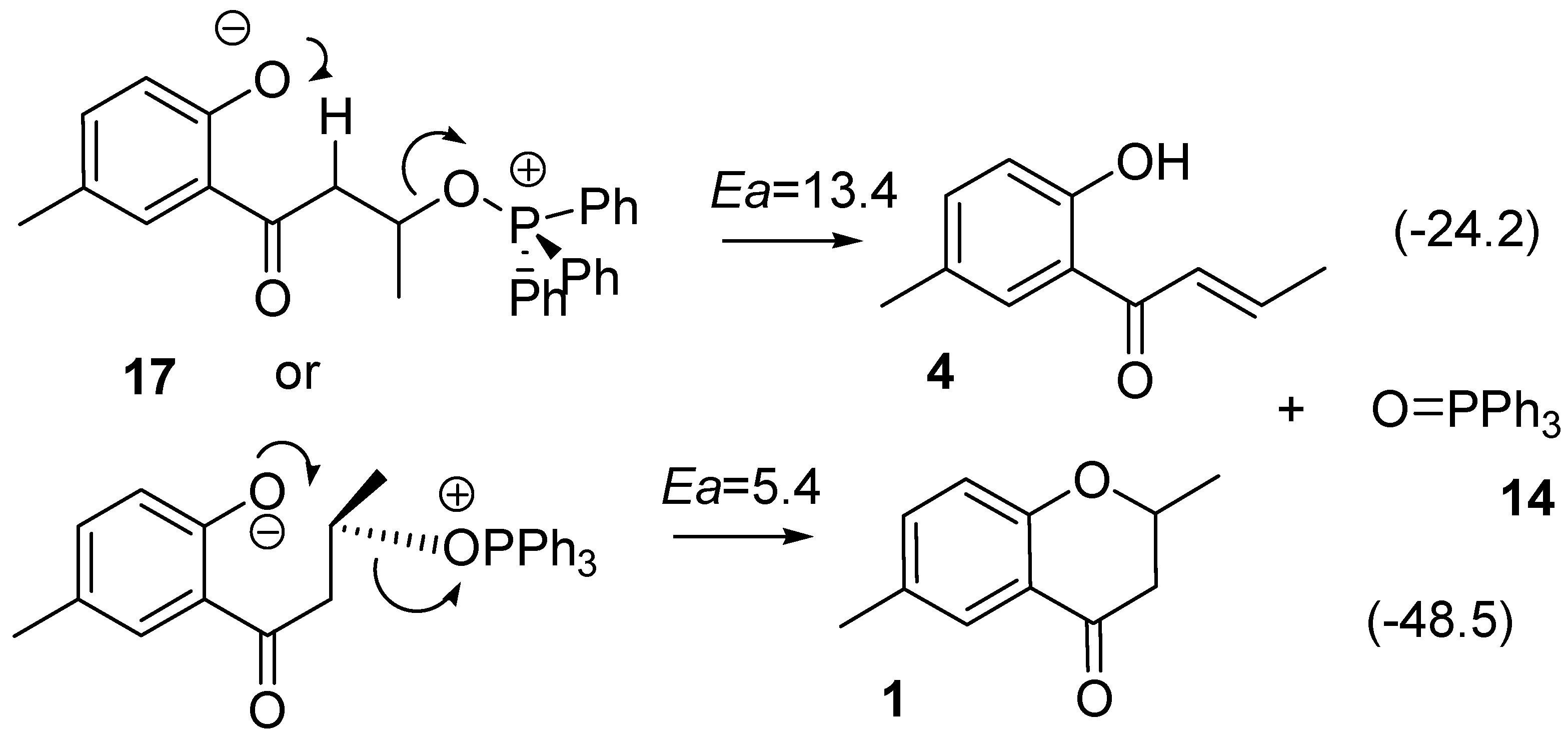
2.1. Routes A and B


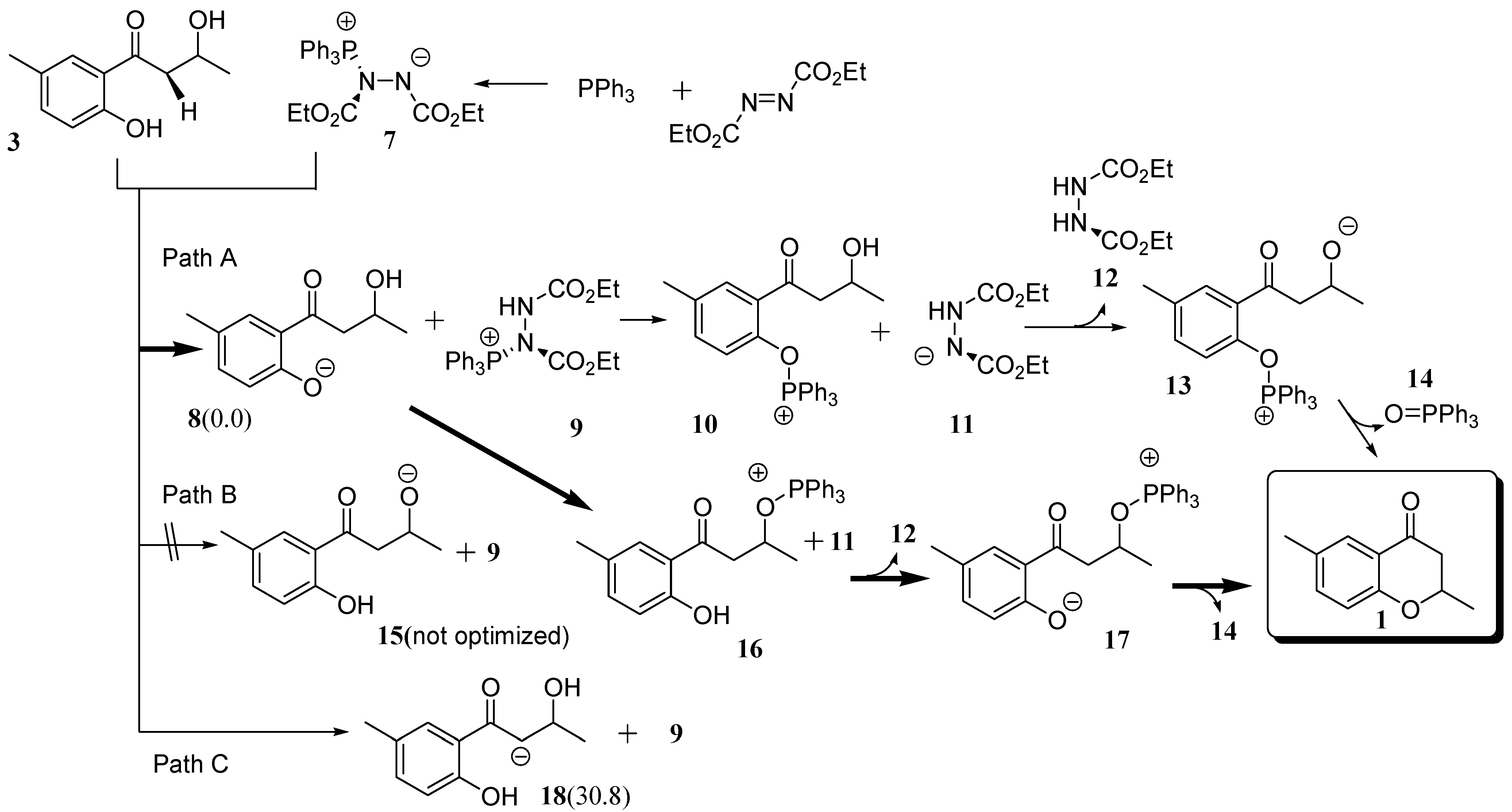
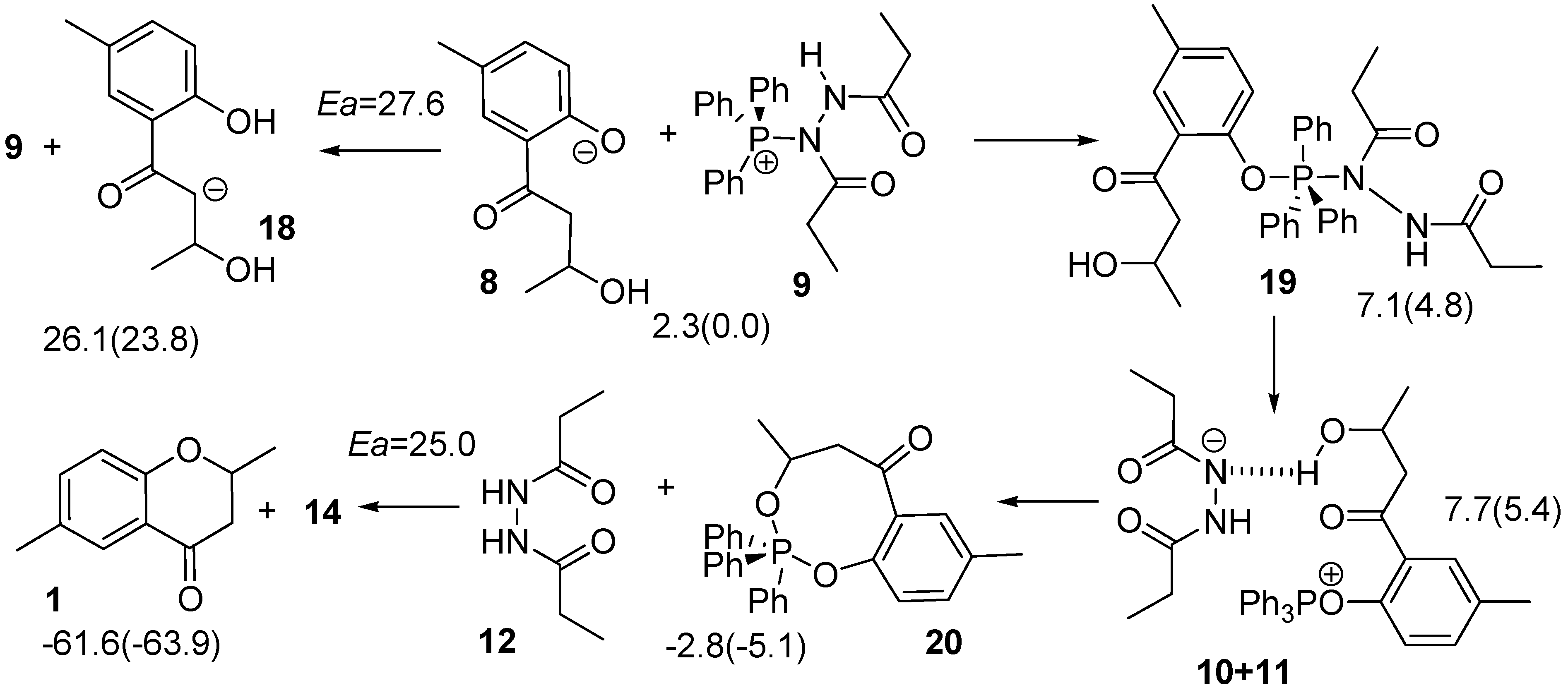
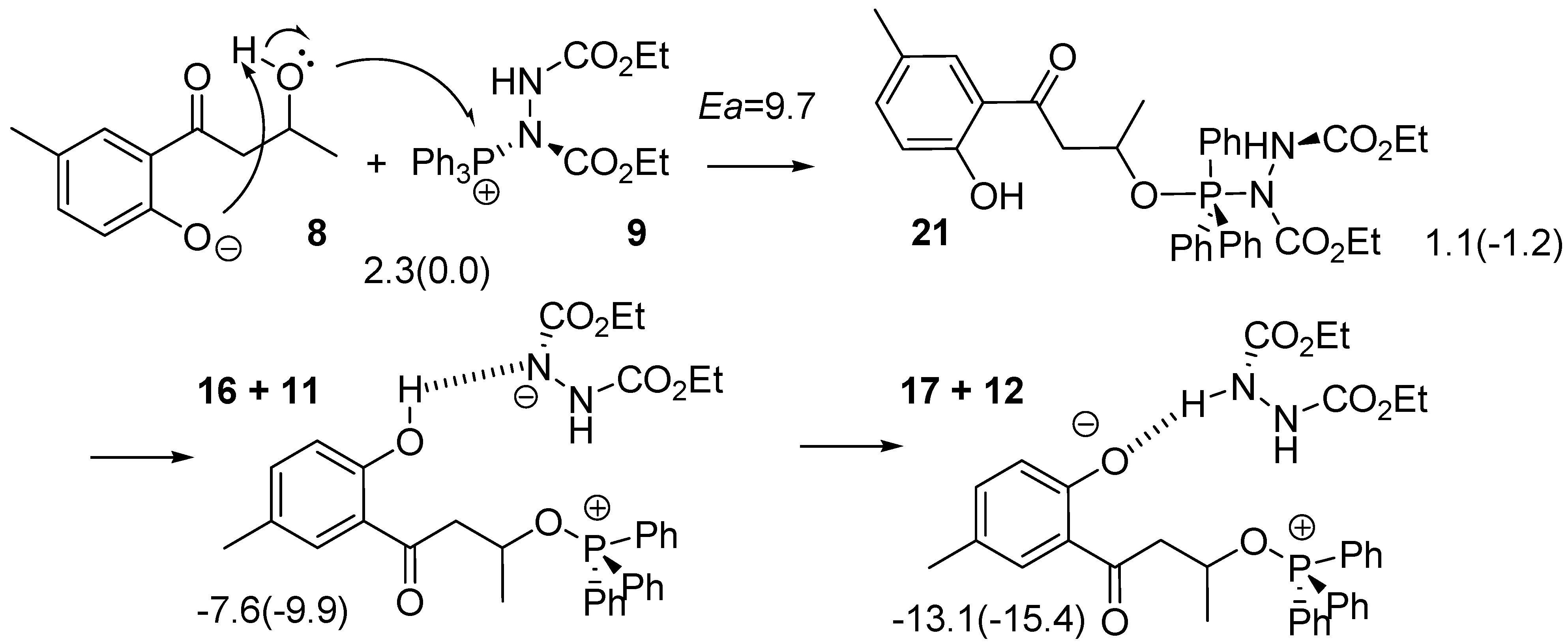
2.2. Route C using the Mitsunobu Reaction






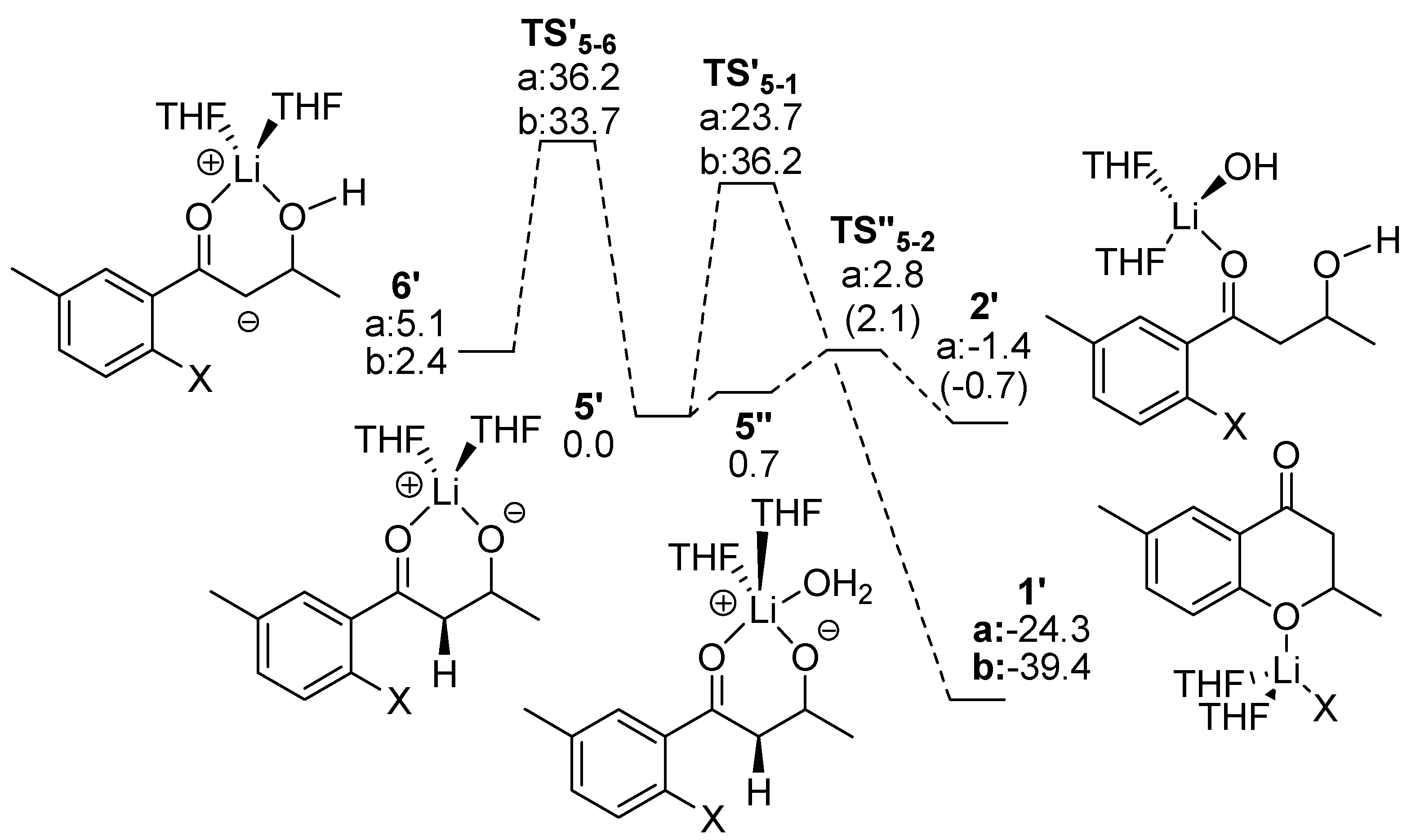
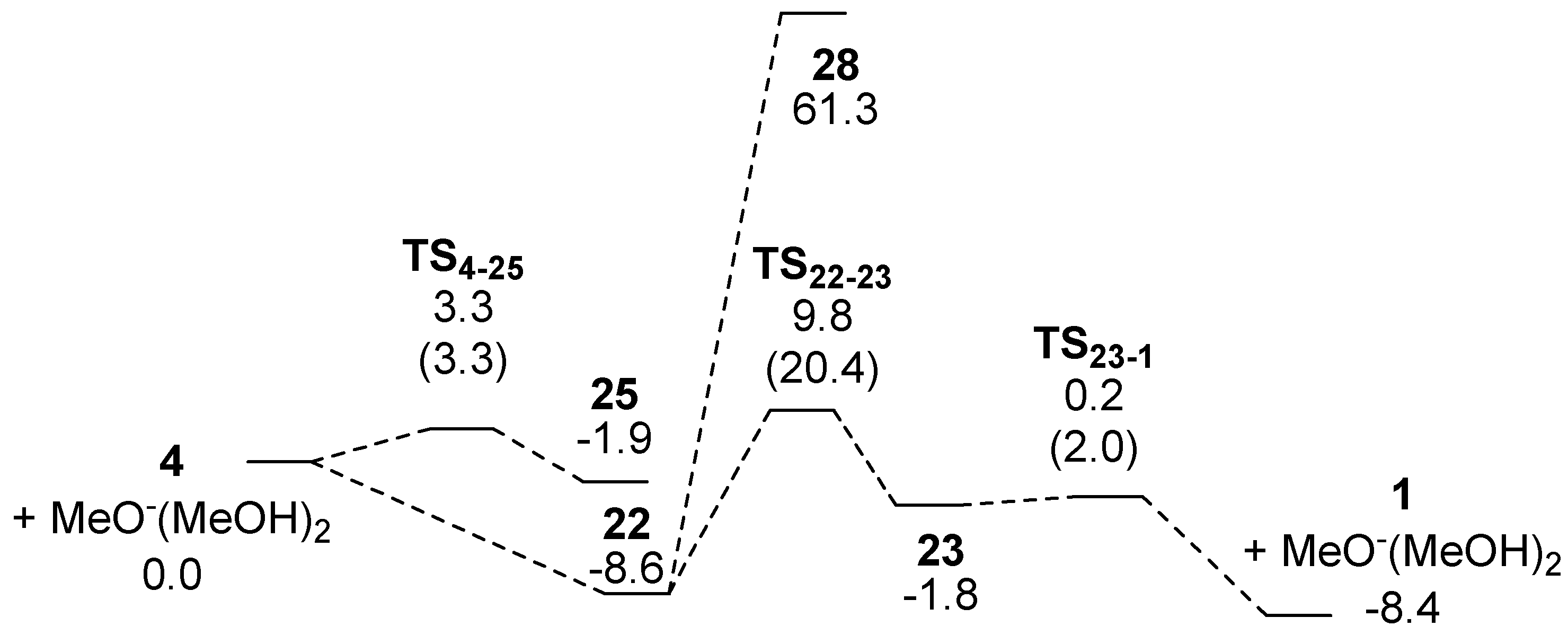
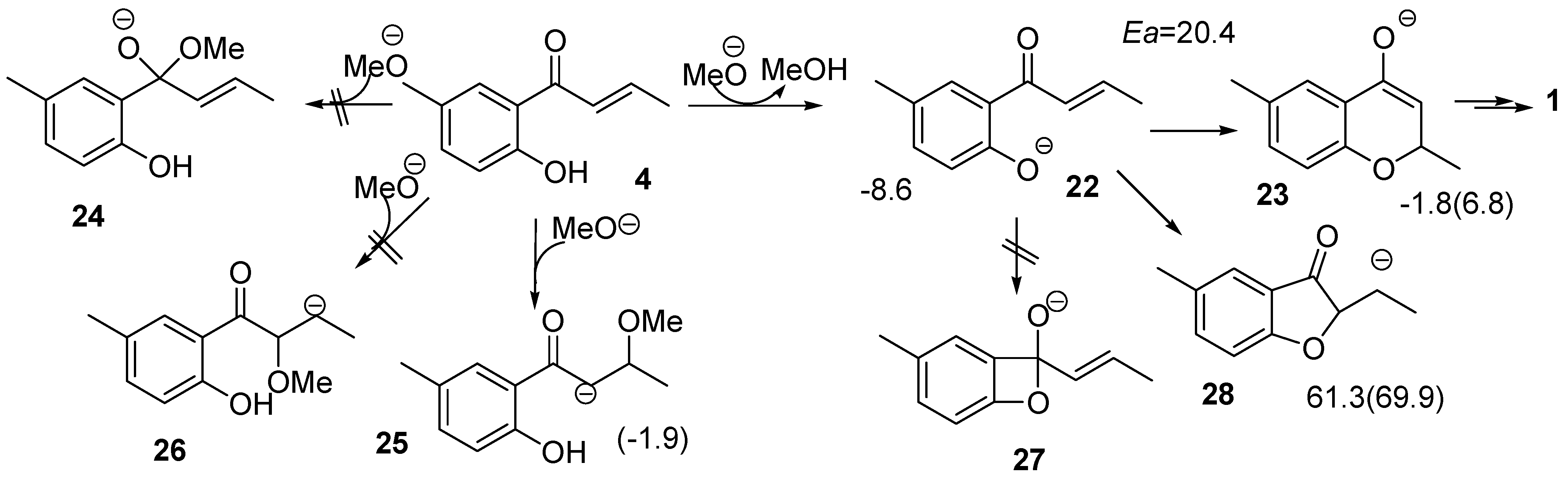
2.3. Route D Using the Michael Reaction



3. Experimental
3.1. Density Functional Theory (DFT) Calculations
3.2. Synthesis of 1-(2-fluoro-5-methylphenyl)-3-hydroxy-1-butanone (2a)
3.3. Synthesis of 1-(2-hydroxy-5-methylphenyl)but-2-en-1-one (4)
3.4. 2,6-dimethylchroman-4-one (1)
4. Conclusions

Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
Appendix



References and Notes
- Corey, J.E.; Cheng, X.-M. The Logic of Chemical Synthesis; John Wiley & Sons: New York, NY, USA, 1989. [Google Scholar]
- Gasteiger, J. Computer-assisted synthesis design. Present state and future perspectives. Chim. Ind. (Milan) 1982, 64, 714–721. [Google Scholar]
- Funatsu, K.; Sasaki, S. “AIPHOS”, Computer Chemistry Series 2; Kyoritsu Shuppan: Tokyo, Japan, 1994. [Google Scholar]
- Satoh, K.; Funatsu, K. A novel approach to retrosynthetic analysis using knowledge bases derived from reaction databases. J. Chem. Inf. Comput. Sci. 1999, 39, 316–325. [Google Scholar] [CrossRef]
- AIPHOS/KOSP V1.0 (2003) was commercially available from Fujitsu Limited (Tokyo, Japan). Available online: http://www.fujitsu.com/global/ accessed on 15 November 2010.
- Hori, K.; Yoshimura, K.; Ohno, H.; Onimura, K.; Ohishi, T. Theoretical study on the polymerization mechanism of substituted maleimides by using a chiral catalyst with Zn2+. Tetrahedron 2003, 59, 6301–6309. [Google Scholar] [CrossRef]
- Hori, K.; Sonoda, T.; Harada, M.; Yamazaki-Nishida, S. Theoretical study on the reactivity of phenyl cation with a propyl group at ortho-position. Tetrahedron 2000, 56, 1429–1436. [Google Scholar] [CrossRef]
- Musashi, Y.; Sakaki, S. Theoretical study of rhodium(III)-catalyzed hydrogenation of carbon dioxide into formic acid. Significant differences in reactivity among rhodium(III), rhodium(I), and ruthenium(II) complexes. J. Am. Chem. Soc. 2002, 124, 7588–7603. [Google Scholar] [CrossRef]
- Hori, K.; Sadatomi, H.; Okano, K.; Sumimoto, M.; Miyamoto, A.; Hayashi, S.; Yamamoto., H. An attempt method for developing new synthetic routes by fusing computational chemistry and chemoinformatics: Syntheses of ethyl and benzyl methacrylates. J. Comput. Aided Chem. 2007, 8, 12–18. [Google Scholar] [CrossRef]
- Hori, K.; Okano, K.; Yoshimura, K.; Nishida, A.; Yamamoto., H. An attempt judging possibility of synthesis routes from the TOSP program using DFT calculations. Application to synthesis routes for substituted fran-2,3-dione. J. Comput. Aided Chem. 2005, 6, 30–36. [Google Scholar] [CrossRef]
- Hori, K.; Yamaguchi, T.; Okano, K. A system fusing computational and information chemistry for developing new synthesis routes of compounds. J. Comput. Aided Chem. 2004, 5, 26–34. [Google Scholar]
- Comey, N.; Hook, I.; Sheridan, H.; Walsh, J. Isolation of (S)-(-)-2,3-dihydro-2,6-dimethyl-4H-benzopyran-4-one from roots of Leontopodium alpinum. J. Nat. Prod. 1997, 60, 148–149. [Google Scholar] [CrossRef]
- Asolkar, R.N.; Kamat, V.P.; Wagner-Dobler, I.; Laatsch, H. Limnazine, the first bacterial azine derivative from Bacillus sp. GW90a. J. Nat. Prod. 2002, 65, 1664–1666. [Google Scholar] [CrossRef]
- Hodgetts, K.J. Asymmetric synthesis of (S)-2,6-dimethylchroman-4-one. ARKIVOC 2001, xi, 74–79. [Google Scholar]
- Beswick, P.J.; Widdowson, D.A. The synthesis of 3-methylene-2,3-dihydrobenzofuran stabilized as its tricarbonylchromium complex. Synthesis 1985, 492–493. [Google Scholar]
- Benarab, A.; Guillaumet, G. Synthesis and Serotonergic Activity of 8-[4-(2,3-Dihydro-1,4-dioxino[2,3-b]pyridine-2-yl-methylamino)butyl]-8-azaspiro[4,5]decane-7,9-dione. Heterocycles 1993, 36, 2327–2333. [Google Scholar] [CrossRef]
- Sanchez, I.; Pujol, M.D. A convenient synthesis of pyrrolo[2,1-c][1,4]benzoxazines. Tetrahedron 1999, 55, 5593–5598. [Google Scholar] [CrossRef]
- Moukri, M.; Pujol, M.D.; Akss ira, M.; Leger, J.M.; Jarry, C.; Guillaumet, G. Synthesis of new compounds containing the 2,3-dihydro[1,4]dioxino[2,3-b]pyridine heterocyclic system as a substructure. Tetrahedron 2003, 59, 3665–3672. [Google Scholar] [CrossRef]
- Schenk, S.; Weston, J.; Anders, E. Density functional investigation of the Mitsunobu reaction. J. Am. Chem. Soc. 2005, 127, 12566–12576. [Google Scholar] [CrossRef]
- Mitsunobu, O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1981, 1–28. [Google Scholar] [CrossRef]
- Kato, K.; Mitsunobu, O. Oxidation of mercaptans with diehyl azodicarboxylate and trivalent phosphorus compounds. J. Org. Chem. 1970, 35, 4227–4229. [Google Scholar] [CrossRef]
- Mitsunobu, O.; Yamada, M. Preparation of esters of carboxylic and phosphoric acid via quaternary phosphonium salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. [Google Scholar] [CrossRef]
- Kürti, L.; Czakó, B. Stragtegic Application of Named Reactions in Organic Synthesis; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Baldwin, J.E.; Thomas, R.C.; Kruse, L.; Dilberman, L. Rules for ring closure: ring formation by conjugate addition of oxygen nucleophiles. J. Org. Chem. 1977, 42, 3846–3852. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03, Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr.; Hay, P.J. Modern Theoretical Chemistry; Schaefer III, H.F., Ed.; Plenum: NewYork, NY, USA, 1976; Volume 3. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- GaussView 3.0 was commercially available from Gaussian Inc. Available online: http://www.gaussian.com/ accessed online 2 October 2003.
- Fukui, K. The path of chemical reactions - the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hori, K.; Sadatomi, H.; Miyamoto, A.; Kuroda, T.; Sumimoto, M.; Yamamoto, H. Towards the Development of Synthetic Routes Using Theoretical Calculations: An Application of In Silico Screening to 2,6-Dimethylchroman-4-one. Molecules 2010, 15, 8289-8304. https://doi.org/10.3390/molecules15118289
Hori K, Sadatomi H, Miyamoto A, Kuroda T, Sumimoto M, Yamamoto H. Towards the Development of Synthetic Routes Using Theoretical Calculations: An Application of In Silico Screening to 2,6-Dimethylchroman-4-one. Molecules. 2010; 15(11):8289-8304. https://doi.org/10.3390/molecules15118289
Chicago/Turabian StyleHori, Kenji, Hirotaka Sadatomi, Atsuo Miyamoto, Takaaki Kuroda, Michinori Sumimoto, and Hidetoshi Yamamoto. 2010. "Towards the Development of Synthetic Routes Using Theoretical Calculations: An Application of In Silico Screening to 2,6-Dimethylchroman-4-one" Molecules 15, no. 11: 8289-8304. https://doi.org/10.3390/molecules15118289