3. Experimental
3.1. General
All reactions were carried out under a positive atmosphere of argon in dried glassware unless otherwise noted. Solvents and materials were obtained from commercial suppliers and used without further purification. Column chromatography was performed on Merck silica gel 60 (230-400 mesh). Reactions and chromatography fractions were analyzed employing pre-coated silica gel plate (Merck Silica Gel 60 F254). All melting points were measured on YANACO MP-500P micro melting point apparatus and are uncorrected. IR spectra were measured on JASCO FT/IR-410. The 1H- and 13C- NMR spectra were recorded on JEOL AL-400 or JEOL ECP-500 instruments with tetramethylsilane as internal standard. Low-resolution and high-resolution mass spectra were recorded on JEOL JMS-01SG-2 or JMS-HX/HX 110A mass spectrometer.
3.2. Representative Synthetic Procedure: Preparation of 6a
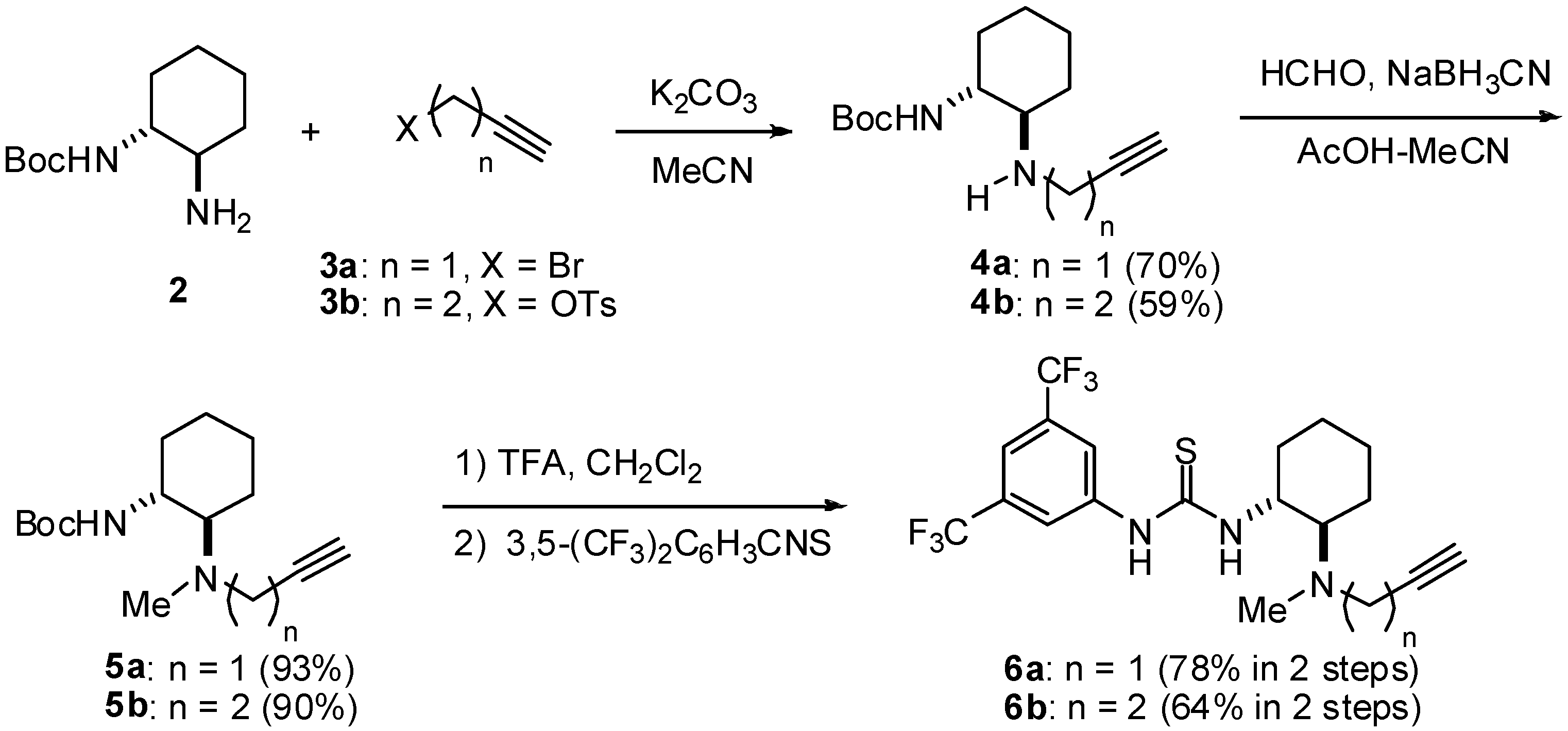
tert-Butyl (1R,2R)-2-(2-Propynylamino)cyclohexylcarbamate (4a): To a stirred mixture of 2 (1.50 g, 7.0 mmol) and K2CO3 (1.03 g, 8.4 mmol) in MeCN (30 mL) at room temperature, propagyl bromide (832 mg, 7.0 mmol) in MeCN (40 mL) was added. After being stirred at room temperature for 3 h, the mixture was quenched with water (20 mL) and extracted with CHCl3 (100 mL × 3). The extracts were dried over NaSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography with hexane/AcOEt (1:1) to afford 4a (1.24 g, 70%). Colorless crystals; [α]D24 -18.3 (c 0.94, CHCl3); Mp 109-110 °C; 1H-NMR (400 MHz, CDCl3) δ 4.46 (br, 1H), 3.52 (dd, J = 17.6, 2.4 Hz, 1H), 3.39 (dd, J = 17.6, 2.4 Hz, 1H), 3.32 (br, 1H), 2.45 (ddd, J = 10.4, 10.4, 6.0 Hz, 1H), 2.20 (dd, J = 2.4, 2.4 Hz, 1H), 2.04-2.06 (m, 1H), 2.05 (br, 1H), 1.66-1.73 (m, 1H), 1.45 (s, 9H), 1.04-1.42 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ 155.9, 82.6, 79.4, 71.0, 59.3, 54.4, 35.3, 32.9, 31.1, 28.4, 24.8, 24.3 ppm; IR (ATR) 3349, 3313, 3251, 2973, 2935, 2859, 1718, 1679, 1519 cm-1; MS (FAB) 253 (MH+, 100); Anal. Calcd. for C14H24N2O2: C, 66.63; H, 9.59; N, 11.10; Found; C, 66.66; H, 9.73; N, 10.94.
tert-Butyl (1R,2R)-2-{Methyl-(2-propynyl)amino}cyclohexylcarbamate (5a): To a stirred mixture of 2a (1.10 g, 4.4 mmol) in MeCN (30 mL) at room temperature, 37% aq HCHO (707 mg, 8.7 mmol) was added. After the mixture was stirred at room temperature for 15 min and 45 min, NaBH3CN (274 mg, 4.4 mmol) and AcOH (9 mL), respectively, were added. After being stirred at the same temperature for 4 h, the mixture was quenched with 1N aq NaOH (150 mL) and extracted with CHCl3 (150 mL × 3). The extracts were dried over NaSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography with hexane/AcOEt (8:1) to afford 5a (1.08 g, 93%). Colorless oil; [α]D24 -41.9 (c 1.1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 5.09 (br, 1H), 3.35 (t, J = 2.8 Hz, 2H), 3.21-3.30 (m, 1H), 2.40-2.46 (m, 2H), 2.28 (s, 3H), 2.20 (t, J = 2.8 Hz, 1H), 1.89-1.92 (m, 1H), 1.75-1.78 (m, 1H), 1.63-1.66 (m, 1H), 1.45 (s, 9H), 1.05-1.29 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ 156.2, 81.4, 78.9, 72.2, 65.1, 51.9, 42.7, 36.1, 33.2, 28.5, 25.3, 24.5, 23.31 ppm; IR (ATR) 3311, 1694, 1484 cm-1; MS (FAB) 267 (MH+, 84), 211 (100); Anal. Calcd. for C15H26N2O2: C, 67.63; H, 9.84; N, 10,52; Found; C, 67.40; H, 10.11; N, 10.44.
1-{3,5-Bis(trifluoromethyl)phenyl}-3-(1R,2R)-2-[{methyl(2-propynyl)amino}cyclohexyl]thiourea (6a): To a stirred mixture of 5a (100 mg, 0.38 mmol) in CH2Cl2 (1 mL) at room temperature, TFA (1 mL) was added. After being stirred at the same temperature for 3 h, the mixture was basified with 3 N NaOHaq (5 mL) and extracted with CHCl3 (5 mL × 3). The extracts were dried over NaSO4, filtered, and concentrated in vacuo. A mixture of the resulting crude product and 3,5-bis(trifluoromethyl)-phenylisothiocyanate (82 mg, 0.30 mmol) in THF (1.5 mL) was stirred at room temperature for 11 h. After concentration in vacuo, the mixture was purified by silica gel column chromatography with hexane/AcOEt/NEt3 (150:50:1) to afford 6a (112 mg, 78% in two steps). Pale yellow oil; [α]D24 -17.5 (c 1.2, CHCl3); 1H-NMR (400 MHz, acetone-d6) δ 9.47 (br, 1H), δ 8.28 (s, 2H), 7.67 (s, 1H), 7.51 (br, 1H), 4.25 (br, 1H), 3.44 (dd, J = 16.8, 2.4 Hz, 1H), 3.37 (dd, J = 16.8, 2.4 Hz, 1H), 2.77-2.84 (m, 1H), 2.64 (t, J = 2.4 Hz, 1H), 2.45-2.48 (m, 1H), 2.37 (s, 3H), 2.04-2.06 (m, 1H), 1.78-1.82 (m, 1H), 1.67-1.70 (m, 1H), 1.18-1.43 (m, 4H) ppm; 13C-NMR (126 MHz, acetone-d6): 181.1, 142.7, 132.1 (q, JC-F = 33.7 Hz), 124.3 (q, JC-F = 273 Hz), 123.0, 117.2, 82.0, 73.8, 66.0, 56.4, 43.0, 36.5, 32.9, 25.9, 25.4, 24.2; IR (ATR) 3309, 2937, 1531, 1467 cm-1; MS (FAB) 438 (MH+, 100); HRMS (FAB+) [C19H22F6N3S]+: 438.1439; Found. 438.1432
tert-Butyl (1R,2R)-2-(3-Butynylamino)cyclohexylcarbamate (4b): Colorless crystals; [α]D24 -30.5 (c 1.0, CHCl3); Mp 89-90 °C; 1H-NMR (400 MHz, CDCl3) δ 4.60 (br, 1H), 3.22 (br, 1H), 2.84-2.90 (m, 2H), 2.66-2.72 (m, 1H), 2.33-2.40 (m, 2H), 2.23-2.30 (m, 1H), 2.09 (d, J = 12.0 Hz, 1H), 1.98-2.03 (m, 1H), 1.98 (dd, J = 2.4, 2.4 Hz, 1H), 1.65-1.72 (m, 2H), 1.45 (s, 9H), 1.12-1.32 (s, 4H) ppm; 13C- NMR (126 MHz, CDCl3) δ 155.9, 82.6, 79.2, 69.5, 60.5, 54.4, 44.7, 32.8, 31.6, 28.4, 24.8, 24.6, 19.9 ppm; IR (ATR) 3280, 2933, 2857, 1708, 1525 cm-1; MS (FAB) 267 (MH+, 100); Anal. Calcd. for C15H26N2O2: C, 67.63; H, 9.84; N, 10,52; Found; C, 67.36; H, 9.74; N, 10.35.
tert-Butyl (1R,2R)-2-{3-Butynyl(methyl)amino}cyclohexylcarbamate (5b): Colorless crystals; Mp 60-61 °C; [α]D26 -37.7 (c 1.6, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 5.45 (br, 1H), 3.15-3.23 (m, 1H), 2.65-2.70 (m, 1H), 2.47-2.54 (m, 2H), 2.29-2.33 (m, 3H), 2.21 (s, 3H), 1.98 (t, J = 2.7 Hz, 1H), 1.59-1.78 (m, 3H), 1.44 (s, 9H), 1.03-1.26 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ 156.4, 83.0, 78.6, 69.1, 66.4, 52.0, 36.1, 33.1, 28.5, 25.5, 24.5, 23.0, 18.6 ppm; IR (ATR) 3383, 2974, 2929, 2857, 2361, 1707, 1483 cm-1: MS (FAB) 281 (MH+, 100); Anal. Calcd. for C16H28N2O2: C, 68.53; H, 10.06; N, 9.99; Found; C, 68.38; H, 10.34; N, 9.78.
1-{3,5-Bis(trifluoromethyl)phenyl}-(1R,2R)-3-[2-{2-butynyl(methyl)amino}cyclohexyl]thiourea (6b): White amorphous; [α]D27 -20.2 (c 1.0, CHCl3); 1H-NMR (400 MHz, acetone-d6) δ 9.34 (br, 1H), δ 8.28 (s, 2H), 7.66 (s 1H), 7.47 (br, 1H), 4.18 (br, 1H), 2.78 (m, 1H), 2.64 (m, 1H), 2.48-2.55 (m, 2H), 2.23-2.32 (m, 2H), 2.30 (s, 3H), 2.03 (t, J = 2.4 Hz, 1H), 1.90-1.94 (m, 1H), 1.77-1.80 (m, 1H), 1.63-1.67 (m, 1H), 1.13-1.32 (m, 4H) ppm; 13C-NMR (126 MHz, acetone-d6) δ 181.3, 142.8, 132.1 (q, JC-F = 27.7 Hz), 126.5 (q, JC-F = 270 Hz), 123.1, 117.2, 83.6, 70.4, 66.9, 56.4, 52.6, 37.5, 33.0, 25.4, 23.9, 19.2 ppm; IR (ATR) 3195, 3047, 2935, 1530, 1467 cm-1; MS (FAB) 452 (MH+, 100); Anal. Calcd. for C20H23F6N3S: C, 53.27; H, 5.13; N, 9.37; Found; C, 53.25; H, 5.15; N, 9.31.
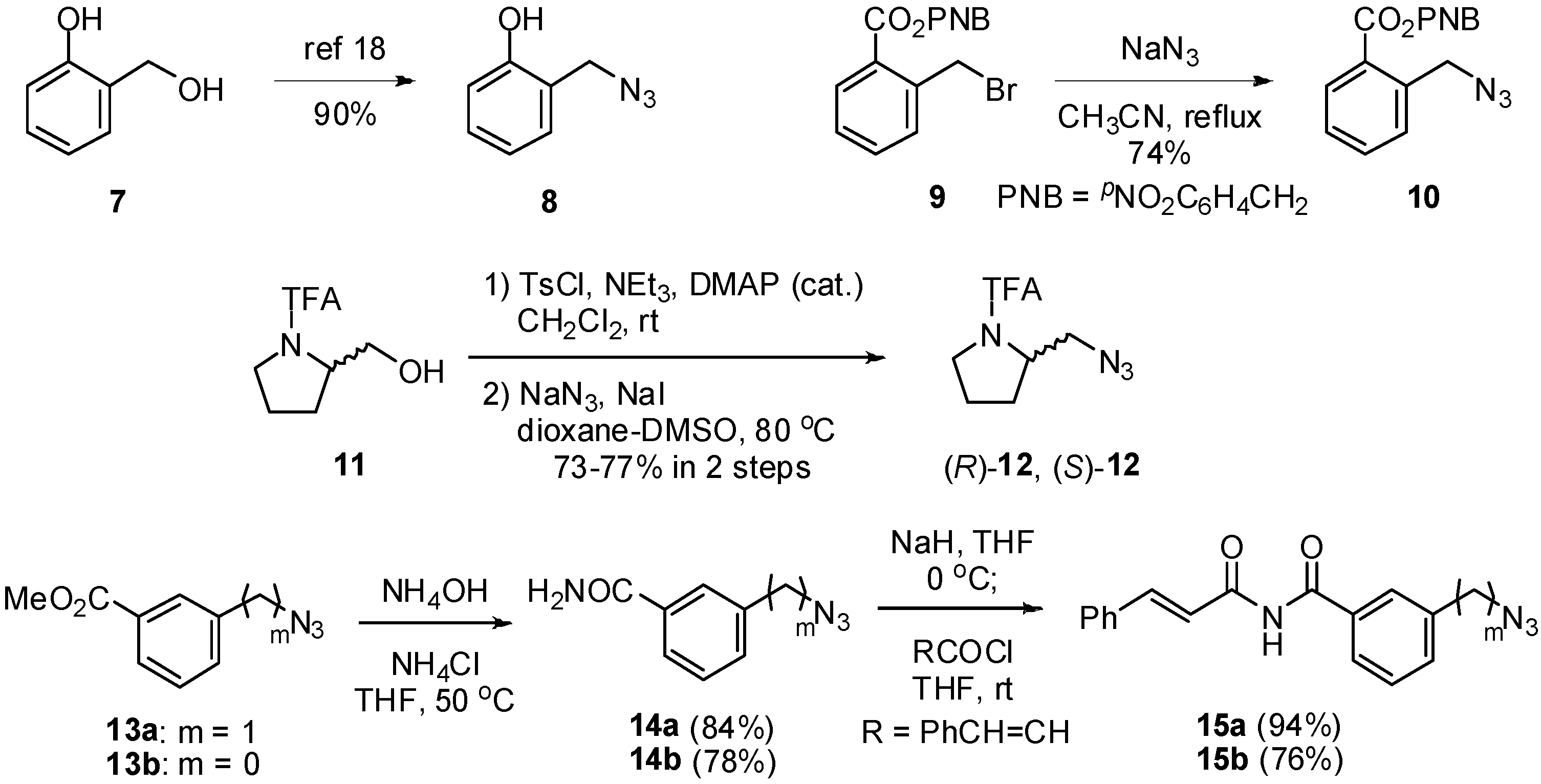
3.3. Synthesis of Azides
4-Nitrobenzyl 2-(Azidomethyl)benzoate (10): To a stirred solution of NaN3 (294 mg, 4.5 mmol) in MeCN (2 mL), 9 (660 mg, 1.9 mmol) in MeCN (3 mL) was added at room temperature. The mixture was refluxed for 20 h. After water (10 mL) was added, the organic layer was extracted with AcOEt (10 mL × 3). The extracts were dried over MgSO4, filtered, and concentrated in vacuo. The resulting residue was purified by silica gel column chromatography with hexane/AcOEt (7:1) to afford 10 (433 mg, 74%). Colorless crystals; Mp 33-34 °C; 1H-NMR (400 MHz, CDCl3) δ 8.26 (d, J = 6.8 Hz, 2H), 8.08 (dd, J = 7.8, 1.2 Hz, 1H), 7.58-7.63 (m, 3H), 7.51 (d, J = 7.1 Hz, 1H), 7.42-7.46 (m, 1H), 5.46 (s, 2H), 4.82 (s, 2H) ppm; 13C-NMR (126 MHz, CDCl3) δ 166.0, 147.8, 143.0, 137.7, 133,2, 131,2, 130.1, 129.0, 128.3, 128.0, 123.9, 65.4, 53.1 ppm; IR (ATR) 2941, 2094, 1715, 1603 cm-1; MS (FAB) 313 (MH+, 13),136 (100); Anal. Calcd. for C15H12N4O4: C, 57.69; H, 3.87; N, 17.94; Found: C, 57.72; H, 3.64; N, 18.19.
(S)-1-{2-(Azidomethyl)-1-pyrrolidinyl}-2,2,2-trifluoroethanone [(S)-12]: To a mixture of (S)-11 (1.25 g, 6.3 mmol), NEt3 (0.77 g, 7.6 mmol) and DMAP (77 mg, 0.63 mmol) in CH2Cl2 (20 mL), TsCl (1.45 g, 7.6 mmol) was added at 0 °C. The mixture was stirred at room temperature for 3 h. The mixture was diluted with AcOEt (100 mL), and then washed with sat. aq NaHCO3 (50 mL × 2) and brine (50 mL). The extracts were dried over MgSO4, filtered, and concentrated in vacuo to give the corresponding tosylate. The crude tosylate was added to a mixture of NaN3 (1.03 g, 15.9 mmol) and NaI (190 mg, 1.3 mmol) in DMSO-1,4-dioxane (1:3 v/v, 30 mL) at room temperature. The mixture was stirred at 80 °C for 24 h. After addition of water (50 mL), the mixture was extracted with Et2O (50 mL × 3). The organic layers were washed with H2O (50 mL × 2) and brine (50 mL). The extracts were dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography with hexane/AcOEt (7:1) to afford (S)-12 (1.02 g, 73%). Colorless oil; [α]D26 -97.7 (c 2.3, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 4.26-4.28 (m, 1H), 3.75 (dd, J = 12.4, 8.8 Hz, 1H), 3.63-3.72 (m, 2H), 3.48 (dd, 12.4, 2.8 Hz, 1H), 1.94-2.12 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ 156.0 (q, J = 37.8 Hz), 113.8 (q, J = 282 Hz), 58.3, 51.3, 47.5, 27.3, 24.5 ppm; IR (ATR) 2983, 2101, 1685 cm-1; MS (FAB) 223(MH+, 8), 154 (100); Anal. Calcd. for C7H9F3N4O: C, 37.84; H, 4.08; N, 10.52; Found: C, 37.68; H, 4.00; N, 25.44.
3-Azidobenzamide (14b): A mixture of 13b (2.62 g, 15 mmol), NH4Cl (395 mg, 7.4 mmol), 28% NH4OHaq (50 mL) and THF (5 mL) was stirred at 50 °C for 24 h. The mixture was extracted with AcOEt (75 mL × 3) and washed with brine (50 mL). The extracts were dried over MgSO4, filtered, and concentrated in vacuo to give 14b (1.86 g, 78%). Colorless crystals; Mp 142-143 °C; 1H-NMR (400 MHz, CDCl3) δ 7.52-7.54 (m, 2H), 7.43 (t, J = 8.1 Hz, 1H), 7.18 (ddd, J = 8.1, 2.4, 1.0 Hz, 1H) ppm; 13C-NMR (126 MHz, CDCl3) δ 168.2, 141.0, 135.1, 130.1, 123.4, 122.4, 118.2 ppm; IR (KBr) 3442, 3358, 2111, 1657, 1581, 1443 cm-1; MS (FAB) 436 (MH+) 163 (100); Anal. Calcd. for C7H6N4O: C, 51.85; H, 3.73; N, 34.55; Found; C, 52.13; H, 3.92; N, 34.53.
3-(Azidomethyl)benzamide (14a): A procedure similar to that of 14b afforded 14a (4.33g, 84%) from 13a. Colorless crystals; Mp 82-83 °C; 1H-NMR (400 MHz, CDCl3) δ 7.80 (s, 1H), 7.76 (dt, J = 6.4, 2.0 Hz, 1H), 7.46-7.51 (m, 2H), 4.42 (s, 2H) ppm; 13C-NMR (126 MHz, CDCl3) δ 169.3, 136.1, 134.0, 131.4, 129.1, 127.1, 127.0, 54.2 ppm; IR (KBr) 3369, 3181, 2112, 2086, 1621, 1582 cm-1; MS (FAB) 177 (MH+, 100); Anal. Calcd. for C8H8N4O: C, 54.45; H, 4.58; N, 31.80; Found; C, 54.43; H, 4.36; N, 32.02.
(E)-3-Azido-(N-cinnamoyl)benzamide (15b): A mixture of NaH (1.40 g, 59 mmol) and 14b (3.79 g, 23 mmol) in THF (200 mL) was stirred for 30 min at 0 °C. To a solution of cinnamoyl chloride (3.90 g, 23 mmol) THF (30 mL) was added the resulting mixture, and stirred for 2 h at room temperature. The reaction mixture was quenched with 1 aq N HCl (100 mL). The aqueous layer was extracted with AcOEt (150 mL x 3). The combined organic layers were washed with brine (100 mL), then dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by recrystallization (CHCl3/hexane). The collected mother liquid was purified again by silica gel column chromatography with CHCl3 . The desired product 7a (5.18 g, 76%) was combined. Pale brown crystals; Mp 148-149 °C; 1H-NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 7.96 (d, J = 15.9 Hz, 1H), 7.83 (d, J = 15.9 Hz, 1H), 7.60-7.68 (m, 4H), 7.52 (t, J = 7.8 Hz, 1H), 7.43-7.45 (m, 3H), 7.29 (dd, J = 2.2, 1.0 Hz, 1H) ppm; 13C-NMR (126 MHz, CDCl3) δ 167.7, 165.1, 147.1, 141.4, 134.8, 134.5, 130.8, 130.4, 128.9, 128.7, 123.8, 123.6, 119.1, 118.7 ppm; IR (KBr) 3261, 2099, 1703, 1667, 1608, 1350 cm-1; MS (FAB) 293 (MH+, 100); Anal. Calcd. for C16H12N4O2: C, 65.75; H, 4.14; N, 19.17; Found; C; 65.76; H, 4.39; N, 19.13.
(E)-3-(Azidomethyl)-(N-cinnamoyl)benzamide (15a): A procedure similar to that of 14b afforded 15a (6.99 g, 94%) from 14a. Colorless crystals; Mp 120-121 °C; 1H-NMR (400 MHz, CDCl3) δ 8.99 (s, 1H), 7.91-7.96 (d, J = 15.6 Hz, 1H), 7.85-7.92 (m, 2H), 7.84 (d, J = 15.6 Hz, 1H), 7.64-7.67 (m, 2H), 7.53-7.60 (m, 2H), 7.40-7.43 (m, 3H), 4.47 (s, 1H) ppm; 13C-NMR (126 MHz, CDCl3) δ 168.8, 165.6, 146.8, 136.6, 134.5, 133.6, 132.6, 130.7, 129.4, 128.9, 128.6, 127.7, 127.6, 119.4, 54.2 ppm; IR (CHCl3) 3020, 2102, 1681, 1618, 1339, 1216 cm-1; MS (FAB) 306 (MH+, 97) 131 (100); Anal. Calcd. for C17H14N4O2: C, 66.66; H, 4.61; N, 18.29; Found; C; 66.55; H, 4.91; N, 18.11.
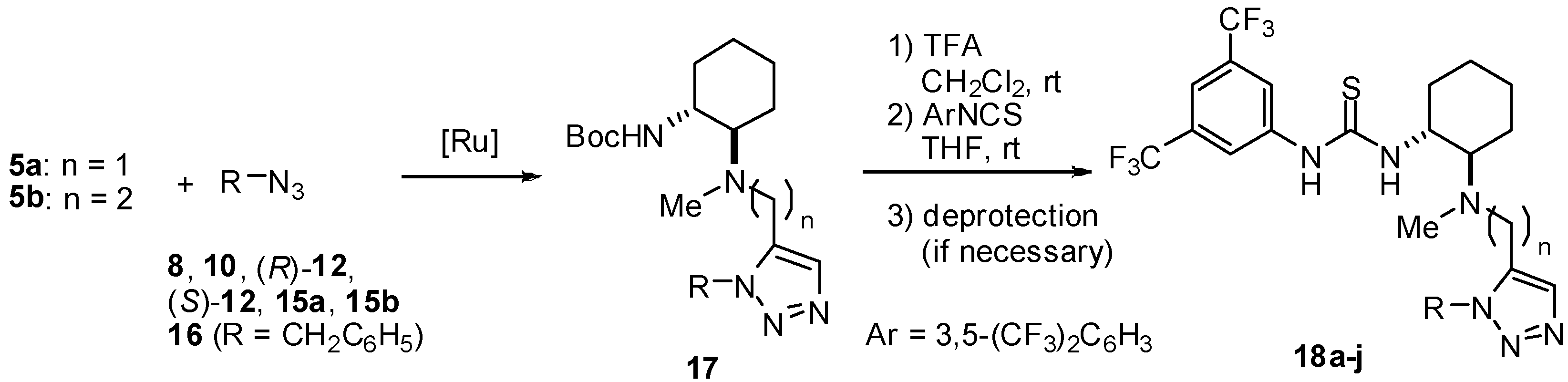
3.4. General Procedure for Ru-Catalyzed Huisgen Reactions
To a solution of [Cp*RuCl]4 (2.5 mol%) in DMF, 5 (1.0 eq) and azide (1.0 eq) were successively added at room temperature .The mixture was heated to 110 °C under microwave irradiation with stirring for 20 min. The resulting mixture was diluted with AcOEt and brine, and then extracted with AcOEt twice and washed with brine three times. The extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography.
tert-Butyl [(1R,2R)-2-[{(1-Benzyl-1H-1,2,3-triazol-5-yl)methyl}(methyl)amino]cyclohexyl] carbamate (17a): White amorphous solid; [α]D28 -3.1 (c 0.95, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.57 (s, 1H), 7.29-7.35 (m, 3H), 7.15-7.17 (m, 2H), 5.70 (d, J = 15.5 Hz, 2H), 4.60 (br, 1H), 3.52 (d, J = 16.0 Hz), 3.47 (d, J = 16.0 Hz), 2.20-2.23 (m, 1H), 2.14 (s, 3H), 2.06-2.09 (m, 1H), 1.75-1.79 (m, 2H), 1.65-1.67 (m, 1H), 1.44 (s, 9H), 1.03-1.24 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ 155.6, 135.1, 134.7, 134.4, 128.9, 128.2, 127.1, 79.2, 65.5, 51.9, 51.2, 36.2, 33.4, 28.5, 25.1, 24.8, 22.6 ppm; IR (ATR) 3315, 2930, 1701 cm-1; MS (FAB) 400 (MH+, 80), 344 (100); HRMS (FAB) [C22H34N5O2]+: 400.2713; Found. 400.2731.
tert-Butyl [2-[[{1-(2-Hydroxybenzyl)-1H-1,2,3-triazol-5-yl}methyl](methyl)amino]cyclohexyl] carb-amate (17b): Pale brown amorphous solid; [α]D27 +6.5 (c 1.1, CHCl3); 1H-NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), δ 8.31 (s, 1H), 7.12 (dd, J = 7.6 Hz, 7.6 Hz, 1H), 6.83-6.85 (m, 1H), 6.74 (dd, J = 7,6, 7.6 Hz, 2H), 6.40 (br, 1H), 5,59 (s, 2H), 3.75 (d, J = 14.4 Hz, 1H), 3.64 (d, J = 14.4 Hz, 1H), 3.37 (br, 1H), 2.28-2.33 (m, 1H), 2.08 (s, 3H), 1.59-1.79 (m, 4H), 1.03-1.25 (m, 4H) ppm; IR (ATR) 3387, 3160, 2935, 1661 cm-1; MS (FAB) 416 (MH+, 100); HRMS (FAB) [C22H34N5O3]+: 416.2662; Found. 416.2651.
4-Nitrobenzyl 2-[[5-[[{(1R,2R)-2-(tert-Butylcarbamoyl)cyclohexyl}(methyl)amino]methyl]-1H-1,2,3-triazol-1-yl]methyl]benzoate (17c): Pale brown amorphous solid; [α]D28 -9.8 (c 1.4, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 8.26 (dd, J = 8.8, 2.0 Hz, 2H), δ 8.10 (d, J = 7.8 Hz, 1H), 7.62 (s, 1H), 7.60 (dd, J = 8.8, 2.0 Hz, 2H), 7.50 (dd, J = 7.8, 7.6 Hz, 1H), 7.42 (dd, J = 7.8, 7.6 Hz, 1H), 6.78 (d, J = 7.8 Hz, 1H), 6.12 (d, J = 16.6 Hz, 1H), 5,93 (d, J = 16.6 Hz, 1H), 5.45 (s, 2H), 4.90 (s, 1H), 3.65 (d, J = 14.4 Hz, 1H), 3.47 (d, J = 14,4 Hz, 1H), 3.35-3.40 (m, 1H), 2.20-2.23 (m, 2H), 2.14 (s, 3H), 1.71-1.75 (m, 2H), 1.61-1.63 (m, 1H), 1.41 (s, 9H), 1.02-1.27 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ 166.1, 155.8, 147.8, 142.8, 137.6, 135.5, 134.1, 133.5, 131.1, 128.54 128.5, 128.2, 127.3, 123.9, 79.0, 66.2, 65.5, 51.5, 49.8, 46.6, 36.2, 33.8, 28.4, 25.1, 24.8, 23.2 ppm; IR (ATR) 2934, 1716, 1523 cm-1; MS (FAB) 580 (MH+, 100); HRMS (FAB) [C30H39N6O6]+: 579.2931; Found. 579.2930.
tert-Butyl [(1R,2R)-2-[Methyl[[1-[{(R)-1-(2,2,2-trifluoroacetyl)pyrrolidin-2-yl}methyl]-1H-1,2,3-triazol-5-yl]methyl]amino]cyclohexyl]carbamate (17d): Pale brown amorphous solid; [α]D26 -12.3 (c 0.92, CHCl3);1H-NMR (400 MHz, acetone-d6) δ 7.53 (s, 1H), 5.70 (br, 1H), 4.68 (br, 3H), 3.75 (d, J = 14.2 Hz, 1H), 3.73 (d, J = 14.2 Hz, 1H), 3.72 (br, 2H), 3.46 (br, 1H), 2.75-2.78 (m, 1H), 2.08 (s, 3H), 1.88-2.00 (m, 2H), 1.77-1.81 (m, 1H), 1.62-1.66 (m, 1H), 1.38 (s, 9H), 1.10-1.38 (m, 4H) ppm; 13C-NMR (126 MHz, acetone-d6) δ 156.7 (q, J = 37.2 Hz), 156.3, 136.1, 134.9, 117.3 (q, J = 289 Hz), 78.2, 67.6, 59.4, 51.9, 48.2, 48.2, 35.0, 34.7, 28.7, 28.6, 27.2, 26.0, 24.4, 23.6 ppm; IR (ATR) 3370, 2931, 2858, 1683, 1525 cm-1; MS (FAB) 489 (MH+, 62), 180 (100); HRMS (FAB) [C22H36F3N6O3]+: 489.2801; Found. 489.2802.
tert-Butyl [(1R,2R)-2-[Methyl[[1-[{(S)-1-(2,2,2-trifluoroacetyl)pyrrolidin-2-yl}methyl]-1H-1,2,3-triazol-5-yl]methyl]amino]cyclohexyl]carbamate (17e): Pale brown amorphous solid; [α]D26 -17.9 (c 1.7, CHCl3); 1H-NMR (400 MHz, acetone-d6) δ 7.53 (s, 1H), 5.57 (br, 1H), 5.03 (br, 1H), 4.52-4.57 (m, 1H), 4.43 (dd, J = 13.6, 9.3 Hz, 1H), 3.87 (s, 2 H), 3.75-3.80 (m, 2H), 3.47 (br, 1H), 2.61 (br, 1H), 2.05 (s, 3H), 1.86-2.05 (m, 2H), 1.75-1.80 (m, 1H), 1.65-1.70 (m, 1H), 1.38 (s, 9H), 1.10-1.38 (m, 4H) ppm; 13C-NMR (126 MHz, acetone-d6) δ 156.5 (q, JC-F = 36.0 Hz), 156.1, 136.4, 134.7, 117.3 (q, JC-F = 287 Hz), 78.3, 66.4, 59.7, 57.7, 52.0, 48.1, 47.9, 47.5, 35.1, 28.7, 28.7, 27.3, 25.9, 24.4, 23.7 ppm; IR (ATR) 3371, 2933, 2858, 1684, 1522 cm-1; MS (FAB) 489 (MH+, 100); HRMS (FAB) [C22H36F3N6O3]+: 489.2801; Found. 489.2798.
tert-Butyl [(1R,2R)-2-[Methyl[2-[1-[{(R)-1-(2,2,2-trifluoroacetyl)pyrrolidin-2-yl}methyl)-1H-1,2,3-triazol-5-yl]ethyl]amino]cyclohexyl]carbamate (17f): Pale brown oil; [α]D26 -5.5 (c 0.77, CHCl3); 1H- NMR (500 MHz, CDCl3) δ 7.54 (s, 1H), 4.95 (br, 1H), 4.59 (d, J = 10.9 Hz, 1H), 4.40 (d, J = 10.9 Hz, 1H), 4.38-4.40 (m, 1H), 3.66-3.68 (m, 2H), 3.27 (br, 1H), 2.87-2.89 (m, 2H), 2.79-2.81 (m, 1H), 2.62-2.65 (m, 1H), 2.20-2.35 (m, 3H), 2.26 (s, 3H), 1.90-2.02 (m, 2H), 1.62-1.82 (m, 4H), 1.44 (s, 9H), 1.02-1.25 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ 156.4 (q, JC-F = 37.2 Hz), 155.9, 136.4, 132.6, 116.0 (q, JC-F = 287 Hz), 78.8, 66.1, 58.8, 52.8, 51.6, 47.3 (q, JC-F = 3.6 Hz), 47.2, 36.1, 33.4, 28.4, 26.9, 25.3, 24.6, 23.9, 23.3, 22.2 ppm; IR (ATR) 3373, 2932, 1692 cm-1; MS (FAB) 503 (MH+, 83), 241 (100); HRMS (FAB) [C23H37F3N6O3]+ 503.2879; Found. 503.2882.
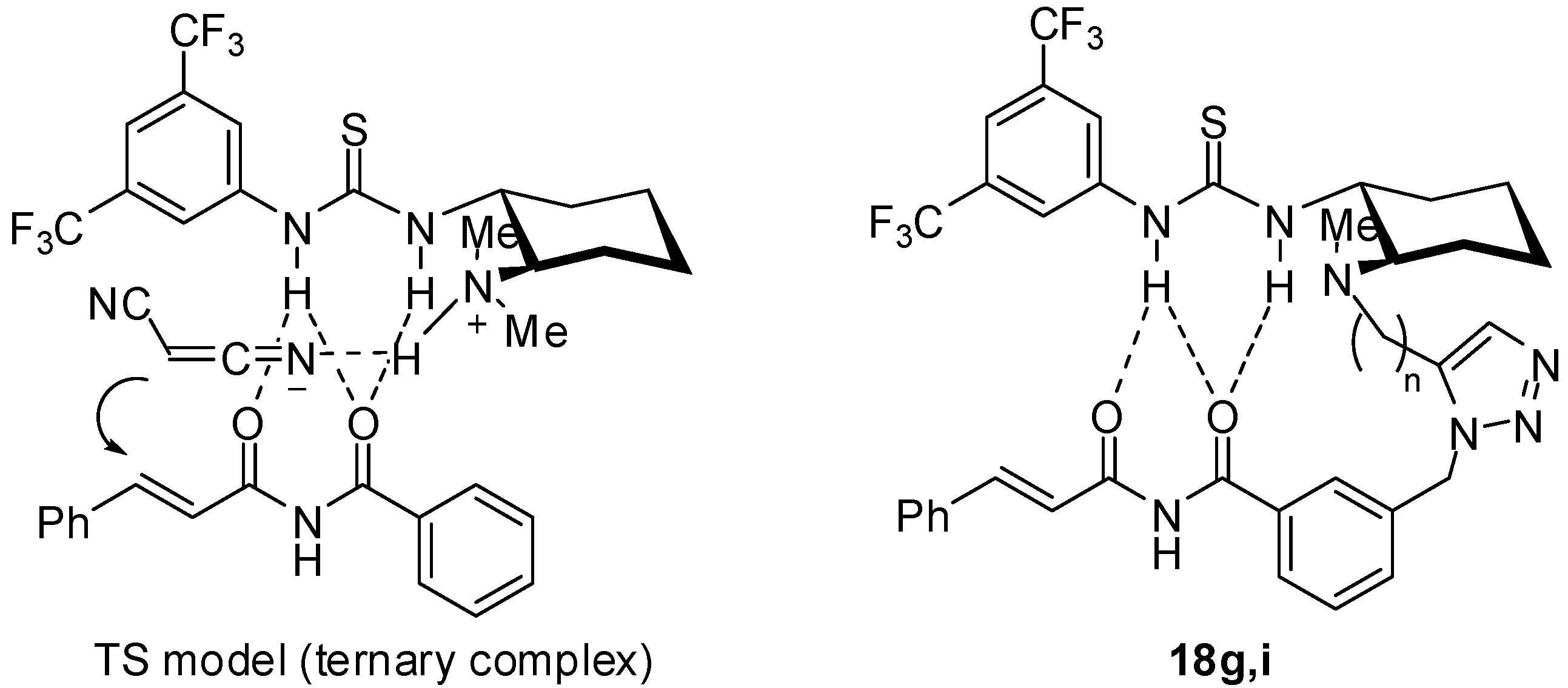
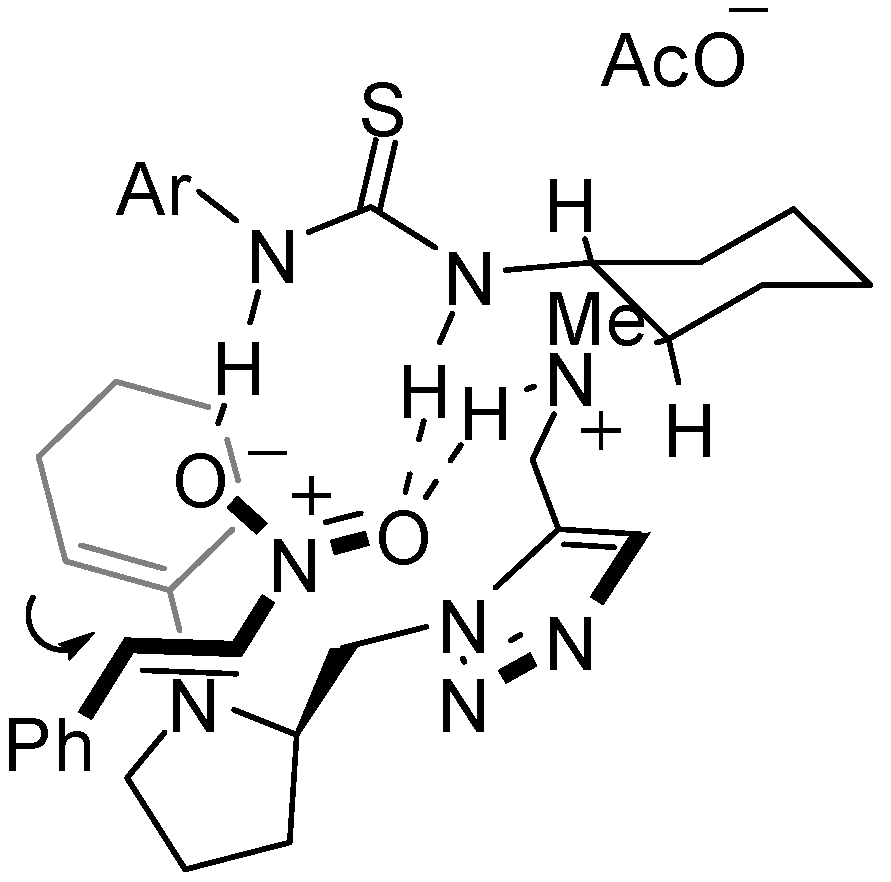
tert-Butyl (1R,2R)-2-[[[1-{3-(Cinnamoylcarbamoyl)benzyl}-1H-1,2,3-triazol-5-yl]methyl](methyl) amino]cyclohexylcarbamate (17g): Pale brown amorphous solid; [α]D26 +1.4 (c 2.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.85-7.92 (m, 3H), 7.62-7.72 (m 3H), 7.58-7.65 (m, 4H), 7.50-7.53 (m, 3H), 7.40-7.42 (m, 3H), 5.58 (s, 2H), 5.06 (br, 1H), 3.79 (d, J = 14.9 Hz, 1H), 3.63 (d, J = 14.9 Hz, 1H), 3.30 (br, 1H), 2.25 (br, 2H), 2.17 (s, 3H), 1.91 (m, 1H), 1.78 (m, 1H), 1.66 (m, 1H), 1.36 (s, 9H), 0.90-1.25 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ; 167.1 165.6, 155.9, 146.1, 135.9, 134.9, 134.5, 134.2, 133.6, 132.4, 130.5, 129.6, 128.8, 128.4, 128.3, 127.2, 119.6, 79.6, 66.4, 65.7, 51.2, 37.2, 33.7, 28.3, 25.0, 24.7, 22.6, 15.2 ppm; IR (ATR) 3293, 2931, 1679, 1623 cm-1; MS (FAB) 573 (MH+, 8) 149 (100); HRMS (FAB) [C33H43N6O4]+: 573.3189; Found. 573.3199
tert-Butyl (1R,2R)-2-[[2-[1-{3-(Cinnamoylcarbamoyl)benzyl}-1H-1,2,3-triazol-5-yl]ethyl(methyl) amino]cyclohexylcarbamate (17i): Pale brown amorphous solid; [α]D27 -2.1 (c 0.85, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 9.96 (br, 1H), 7.89-7.93 (m 3H), 7.61-7.69 (m, 3H), 7.55 (s, 1H), 7.48 (t, J = 7,8 Hz 1H), 7.40-7.42 (m, 4H), 5.69 (d, J = 15.6 Hz, 1H), 5.54 (d, J = 15.6 Hz, 1H), 4.92 (br, 1H), 2.99 (br, 1H), 2.76 (m, 1H), 2.63 (m, 1H), 2.50 (m, 1H), 2.22 (s, 3H), 2.04-2.22 (m, 3H), 1.60-1.76 (m, 3H), 0.88-1.28 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ; 167.1, 165.5, 156.2, 146.4, 135.5, 134.7, 134.0, 133.4, 131.3, 130.6, 129.8, 128.9, 128.6, 128.2, 126.6, 119.5, 67.5, 51.9, 51.8, 51.1, 50.9, 33.6, 28.5, 25.2, 24.7, 23.0 ppm; IR (ATR) 3343, 2927, 2856, 1674 cm-1; MS (FAB) 587 (MH+, 12) 149 (100); HRMS (FAB) [C33H43N6O4]+: 586.3346; Found. 586.3340.
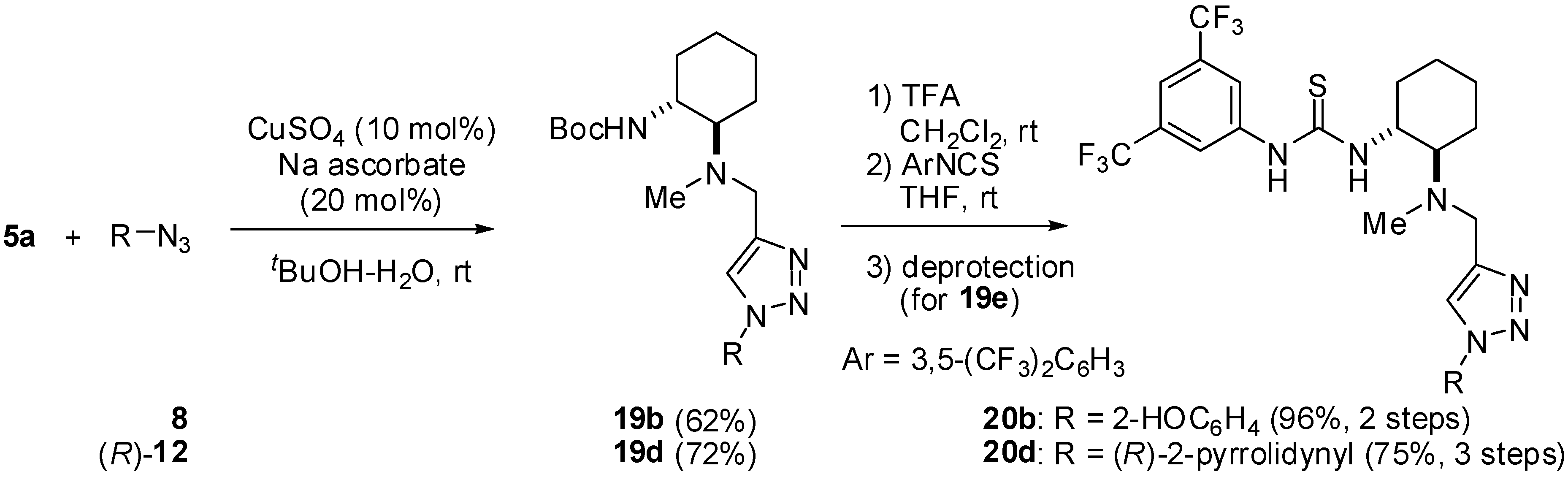
3.5. General Procedure for Cu-Catalyzed Huisgen Reaction
To a solution of CuSO4 (10 mol%) and sodium ascorbate (20 mol%) in t-BuOH-H2O (1 : 1 v/v), 5a (1.0 eq) and azide (1.0 eq) were successively added at room temperature. After being stirred for an appropriate time (4-6 h), the mixture was diluted with H2O. The residue was extracted with CHCl3 three times. The combined organic layers were washed with water twice and brine. The organic phase were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography.
tert-Butyl [(1R,2R)-2-[[{1-(2-Hydroxybenzyl)-1H-1,2,3-triazol-4-yl}methy](methyl)amino] cyclo- hexyl]carbamate (19b): White amorphous solid; [α]D27 -2.4 (c 1.0, CHCl3); 1H-NMR (400 MHz, DMSO-d6) δ 9.82 (s, 1H), 7.15 (dd, J = 8.0, 7.6 Hz, 1H), 6.99 (d, J = 6.8 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 6.76 (dd, J = 7.6, 6.8 Hz, 1H), 6.31 (br, 1H), 5,45 (s, 2H), 3.72 (d, J = 14.0 Hz, 1H), 3.52 (d, J = 14.0 Hz), 3.28 (br, 1H), 2.31-2.36 (m, 1H), 2.11 (s, 3H), 1.86-1.90 (m, 1H), 1.72-1.77 (m, 1H), 1.65-1.70 (m, 1H), 1.36 (s, 9H), 1.11-1.25 (m, 4H) ppm; IR (ATR) 2925, 1715 cm-1; MS (FAB) 416 (MH+, 100); HRMS (FAB) [C22H34N5O3]+: 416.2662; Found. 416.2661.
tert-Butyl [(1R,2R)-2-[Methyl[[1-[{(R)-1-(2,2,2-trifluoroacetyl)pyrrolidin-2-yl}methyl]-1H-1,2,3-triazol-4-yl]methyl]amino]cyclohexyl]carbamate (19d): Pale brown amorphous solid; [α]D24 -5.6 (c 3.3, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.43 (s, 1H), 5,11 (br, 1H), 4.70 (dd, J = 14.0, 6.3Hz, 1H), 4.59 (dd, J = 14.0, 2.9 Hz, 1H), 4.46 (br, 1H), 3.80 (d, J = 13.8 Hz, 1H), 3.63 (d, J = 13.8 Hz, 1H), 3.40-3.45 (m, 1H)¸ 3.29-3.33 (m, 1H), 2.37-2.39 (m, 1H), 2.28-2.32 (m, 1H), 2.21 (s, 3H), 2.00-2.15 (m, 2H), 1.85-1.90 (m, 2H), 1.78-1.81 (m, 1H), 1.57-1.66 (m, 2H), 1.44 (s, 9H), 1.02-1.31 (m, 4H) ppm; IR (ATR) 3372, 2931, 1692 cm-1; MS (FAB) 489 (MH+, 100); HRMS (FAB) [C22H36F3N6O3]+: 489.2801; Found. 489.2799.
3.6. General Procedure for the Synthesis of Thioureas 18 and 20
To a stirred mixture of appropriate substrates in CH2Cl2 at room temperature, TFA was added (CH2Cl2 : TFA = 1:1). After being stirred at room temperature for 1-3 h, the mixture was made basic with sat. aq NaHCO3 and extracted three times with CHCl3. The combined organic layers were dried over NaSO4, filtered, and concentrated in vacuo to give the corresponding amine. A solution of the crude amine and 3,5-bis(trifluoromethyl)phenylisothiocyanate (1.0 eq) in THF was stirred at room temperature for 2-10 h. The mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography to give the corresponding thiourea. If necessary, the following deprotonation reaction was carried out. To a mixture of the protected compound in THF, LiOH (10 eq) in H2O was added (THF/ H2O = 1:1). After being stirred at room temperature for 2-10 h, the mixture was quenched with sat. aq NaHCO3 or sat. aq NH4Cl. The mixture was extracted three times with AcOEt. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography.
1-[(1R,2R)-2-[{(1-Benzyl-1H-1,2,3-triazol-5-yl)methyl}(methyl)amino]cyclohexyl]-3-{3,5-bis-(tri-
fluoromethyl)phenyl}thiourea (18a): White amorphous solid; [α]D27 +27.1 (c 1.5, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 9.00 (br, 1H), 8.08 (s, 2H), 7.60 (br, 1H), 7.52 (s, 1H), 7.42 (s, 1H), 7.21-7.30 (m, 3H), 6.98-7.02 (m, 2H), 5.72 (d, J = 15.6 Hz, 1H), 5.59 (d, J = 15.6 Hz, 1H), 4.46 (br, 1H), 3.64 (d, J = 14.9 Hz, 1H), 3.34 (d, J = 14.9 Hz, 1H), 2.37 (s, 3H), 2.27-2.36 (m, 2H), 1.71-1.88 (m, 3H), 1.12-1.38 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ 180.4, 141.1, 135.7, 134.02, 133.5, 131.6 (q, JC-F = 34.8 Hz), 129.1, 128.5, 126.6, 123.1 (q, JC-F = 274 Hz), 122.3, 117.3, 63.8, 54.1, 52.2, 46.1, 37.8, 33.1, 25.3, 24.9, 22.0 ppm; IR (ATR) 3333, 2935, 1534 cm-1; MS (FAB) 571 (MH+, 100); HRMS (FAB) [C26H29F6N6S]+: 571.2079; Found. 571.2075.
1-{3,5-Bis(trifluoromethyl)phenyl}-3-[(1R,2R)-2-[[{1-(2-hydroxybenzyl)-1H-1,2,3-triazol-4-yl}-
methyl](methyl)amino]cyclohexyl]thiourea (18b): White amorphous solid; [α]D24 +56.0 (c 0.56, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 8.27 (br, 1H), 7.60 (s, 2H), 7.52 (s, 1H), 7.49 (s, 1H), 7.26 (s, 1H), 7.23 (br, 1H), 7.15 (dd, J = 8.0, 8.0 Hz, 1H), 7.08 (br, 1H), 6.84 (dd, J = 8.0, 8.0 Hz, 1H), 6.78 (d, J = 8.0 Hz, 1H), 5.58(d, J = 14.6 Hz, 1H), 5.28 (d, J = 14.6 Hz, 1H), 4.64 (br, 1H), 4.02 (d, J = 13.9 Hz, 1H), 3.60 (d, J = 13.9 Hz, 1H), 2.63 (m, 1H), 2.40 (m, 1H), 2.18 (s, 3H), 2.02 (m, 1H), 1.91 (m, 1H), 1.88 (m, 1H), 1.77 (m, 1H), 1.74 (m, 1H), 1.14-1.40 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ 180.1, 154.1, 139.9, 134.1, 131.9, 131.7, 131.4, 130.6, 123.0, 122.9 (q, JC-F = 274 Hz), 121.2, 121.0, 118.1, 117.2, 66.5, 54.9, 47.5, 44.3, 38.0, 33.1, 25.0, 24.7, 22.4 ppm; IR (KBr) 3316, 2938, 1540 cm-1; MS (FAB) 587 (MH+, 100); Anal. Calcd. for C26H28F6N6OS: C, 53.24; H, 4.81; N, 14.33; Found: C, 53.14; H, 4.84; N, 14.18.
2-[[5-[[[(1R,2R)-2-[3-{3,5-Bis(trifluoromethyl)phenyl}thioureido]cyclohexyl](methyl)amino]
methyl]-1H-1,2,3-triazol-1-yl]methyl]benzoic Acid (18c): Colorless crystals; [α]D27 +116 (c 0.68, CHCl3); Mp 53-54°C; 1H-NMR (400 MHz, CD3OD) δ 8.17 (s, 2H), 7.85 (d, J = 7.6 Hz, 1H), 7.70 (s, 1H), 7.46 (s, 1H), 7.29-7.37 (m, 2H), 7.16 (d, J = 7.6 Hz, 1H), 5.93 (d, J = 14.4, 1H), 5.74 (d, J = 14.4, 1H),4.57-4.61 (m, 1H), 4.24 (d, J = 14.2 Hz, 1H), 4.09 (d , J = 14.2 Hz, 1H), 1.65-1.72 (m, 2H), 3.16-3.20 (m, 1H), 2.47 (s, 3H), 2.26-2.28 (m, 1H), 2.05-2.09 (m, 1H), 1.83-1.87 (m, 1H), 1.66-1.70 (m, 1H), 1.18-1.46 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ 180.9, 173.7, 141.6, 135.4, 134.7, 134.0, 133.3, 132.4, 131.2 (q, JC-F = 34 Hz), 131.1, 130.9, 129.4, 128.8, 123.3 (q, JC-F = 277 Hz), 116.8, 68.2, 53.2, 52.1, 38.7, 32.3, 30.4, 29.7, 24.2, 23.0, 22.9 cm-1; IR (KBr) 3241, 2942, 1712 cm-1; MS (FAB) 615 (MH+, 100); HRMS (FAB) [C27H28F6N6O2S]+: 614.1899; Found. 614.1893.
1-{3,5-Bis(trifluoromethyl)phenyl}-3-[(1R,2R)-2-[methyl[[1-{(R)-pyrrolidin-2-ylmethyl}-1H-1,2,3-
triazol-5-yl]methyl]amino]cyclohexyl]thiourea (18d): White amorphous solid; [α]D24 -0.4 (c 0.86, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 8.06 (s, 2H), 7.59 (s, 1H), 7.55 (s, 1H), 4.54 (dd, J = 13.7, 6.3 Hz, 1H), 4.35 (dd, J = 13.7, 5.9 Hz, 1H), 4.26-4.30 (m, 1H), 3.84-3.89 (m, 1H), 3.71-3.77 (m, 2H), 2.88-3.02 (m, 2H), 2.60 (br, 1H), 2.42-2.47 (m, 1H), 2.23 (s, 3H), 1.71-1.99 (m, 7H), 1.00-1.40 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ 180.9, 141.4, 134.4, 131.8 (q, JC-F = 33.7 Hz), 123.2 (q, JC-F = 274 Hz), 122.7, 117.3, 64.5, 57.5, 57.4, 54.7, 51.9, 46.1, 36.0, 32.9, 28.8, 25.0, 24.6, 24.5, 21.8 ppm; IR (ATR) 3253, 2935, 2860, 1543 cm-1; MS (FAB) 564 (MH+, 41), 41 (100); HRMS (FAB) [C24H33F6N7S]+ 564.2344; Found. 564.2334.
1-{3,5-Bis(trifluoromethyl)phenyl}-3-[(1R,2R)-2-[methyl[[1-{(S)-pyrrolidin-2-ylmethyl}-1H-1,2,3-
triazol-5-yl]methyl]amino]cyclohexyl]thiourea (18e): Pale brown amorphous solid; [α]D27 +1.0 (c 2.6, CHCl3); 1H-NMR (400 MHz, DMSO-d6) δ 8.26 (s, 2H), 7.72 (s, 1H), 7.59 (s, 1H), 4.23-4.28 (m, 3H), 3.88 (d, J = 14.4 Hz, 1H), 3.66 (d, J = 14.4 Hz, 1H), 2.78-2.97 (m, 2H), 2.62-2.68 (m, 1H), 2.12 (s, 3H), 1.16-2.15 (m, 12H) ppm; IR (ATR) 2931, 1692 cm-1; MS (FAB) 564 (MH+, 100); HRMS (FAB) [C24H33F6N7S]+: 564.2344; Found. 564.2349.
1-{3,5-Bis(trifluoromethyl)phenyl}-3-[(1R,2R)-2-[methyl[2-[1-[{(R)-1-(2,2,2-trifluoroacetyl) pyrrol-idin-2-yl}methyl]-1H-1,2,3-triazol-5-yl]ethyl]amino]cyclohexyl]thiourea (18f): Pale brown amorphous solid; [α]D27 -18.0 (c 1.6, CHCl3); 1H-NMR (500 MHz, pyridine-d5) δ 11.82 (br, 1H), 8.62 (br, 1H), 8.48 (s, 2H), 7.87 (s, 1H), 7.66 (s, 1H), 4.86 (br, 1H), 4.50 (br, 1H), 4.48 (dd, J = 13.8, 4.3 Hz, 1H), 4.62 (dd, J = 13.8, 8.3 Hz, 1H), 3.72-3.77 (m, 1H), 2.95-3.02 (m, 2H), 2.44-2.89 (m, 5H), 2.36 (s, 3H), 1.40-1.85 (m, 7H), 0.99-1.35 (m, 4H) ppm; 13C-NMR (126 MHz, pyridine-d5) δ 182.7, 144.4, 138.4, 134.3, 133.2 (q, JC-F = 32.5 Hz), 125.7 (q, JC-F = 249 Hz), 118.5, 68.9, 60.6, 57.5, 54.4, 52.9, 48.3, 39.8, 34.6, 31.2, 27.3, 27.2, 26.8, 24.8 ppm (one peak for a nonaromatic carbon is missing); IR (ATR) 3329, 3019, 1735 cm-1; MS (FAB) 578 (MH+, 50), 369 (100); HRMS (FAB) [C25H34F6N7S]+ 578.2501; Found. 578.2498.
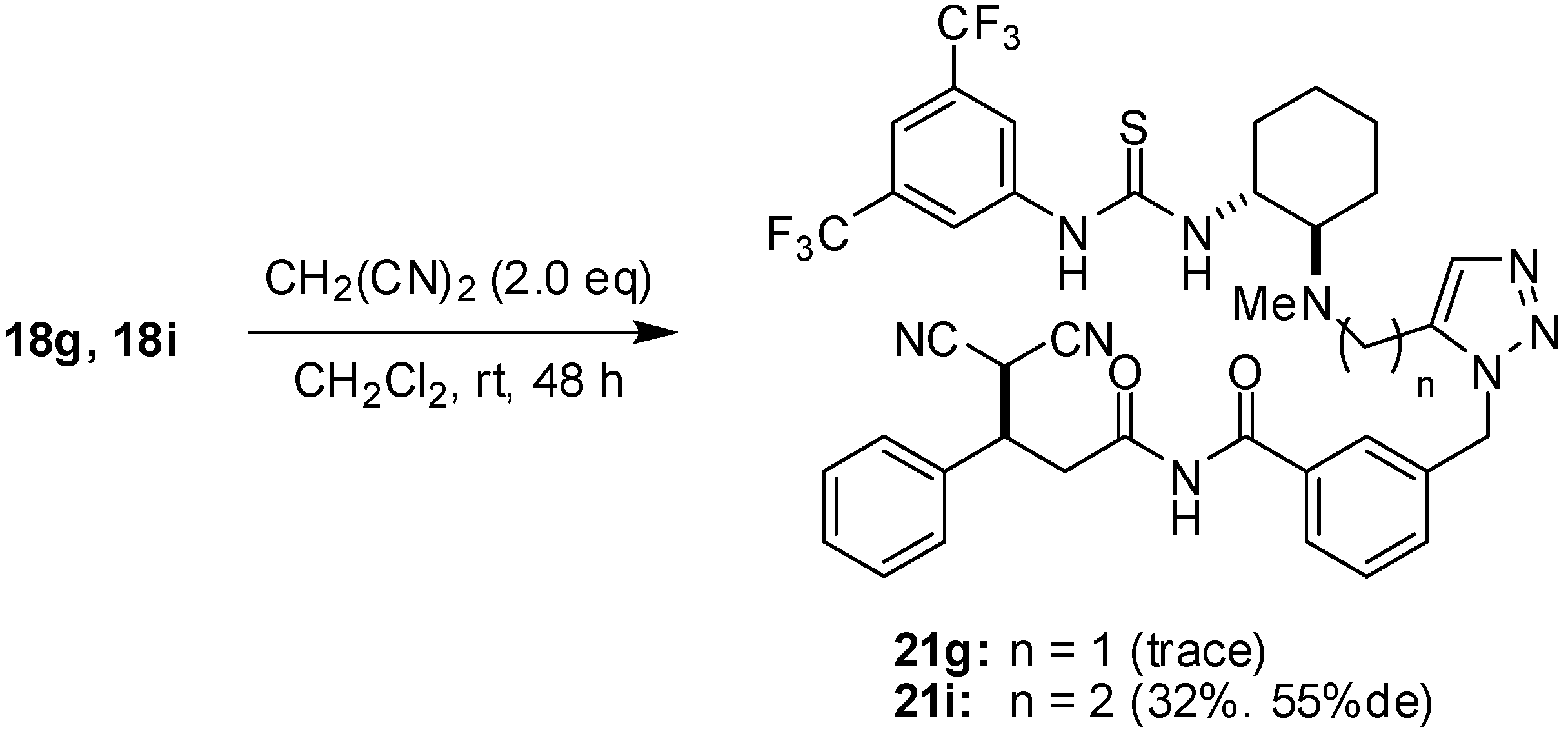
3-[[5-[[[(1R,2R)-2-[3-{3,5-Bis(trifluoromethyl)phenyl}thioureido]cyclohexyl](methyl)amino]
methyl]-1H-1,2,3-triazol-1-yl]methyl]-N-cinnamoylbenzamide (18g): Colorless crystals; Mp 134-137 °C; [α]D26 -32.0 (c 1.2, CHCl3); 1H-NMR (500 MHz, acetone-d6) δ 10.14 (s, 1H), 9.29 (s, 1H), 8.22 (s, 2H), 7.86-7.88 (m, 2H), 7.79 (d, J = 15.5 Hz, 1H), 7.58-7.68 (m, 5H), 7.39-7.50 (m, 6H), 5.70 (s, 2H), 4.53 (br, 1H), 3.88 (d, J = 14.4 Hz, 1H), 3.74 (d, J = 14.4 Hz, 1H), 2.73-2.75 (m, 1H), 2.32-2.35 (m, 1H), 2.25 (s, 3H), 1.96-2.01 (m, 1H), 1.77-1.81 (m, 1H), 1.65-1.69 (m, 1H), 1.18-1.46 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ; 180.9, 167.2, 167.0, 145.3, 142,8, 137.8, 135.9, 135.6, 134.9, 134.8, 133.8, 133.2, 131.9, 131.6, 131.3, 129.9, 129.8, 129.1, 128.7, 128.5, 125.4, 123.3, 123.1, 121.5, 117.2, 66.5, 55.6, 51.6, 46.2, 37.8, 33.4, 30.3, 30.1, 25.9, 25.6, 23.8 ppm; IR (ATR) 3127, 1738, 1635, 1528 cm-1; MS (FAB) 744 (MH+, 31) 369 (100); Anal. Calcd. for C36H35F6N7O2S: C, 58.13; H, 4.74; N, 13.18; Found: C, 57.73; H, 4.84; N, 12.87.
3-[[5-[2-[[(1R,2R)-2-[3-{3,5-Bis(trifluoromethyl)phenyl}thioureido]cyclohexyl](methyl)amino]
ethyl]-1H-1,2,3-triazol-1-yl]methyl]-N-cinnamoylbenzamide (18i): White amorphous solid; [α]D24 -161 (c 0.74, CHCl3); 1H-NMR (500 MHz, acetone-d6) δ 10.15 (s, 1H), 9.22 (s, 1H), 8.27 (s, 2H), 7.91 (d, J = 15.9 Hz, 1H), 7.60-7.64 (m, 3H), 7.55 (s, 1H), 7.51 (d, J = 15.9 Hz, 1H), 7.39-7.45 (m, 6H), 5.68 (s, 2H), 4.26 (br, 1H), 2.75-2.88 (m, 4H), 2.48-2.52 (m, 1H), 2.30-2.42 (m, 2H), 2.24 (s, 3H), 1.75-2.78 (m, 1H), 1.67-1.70 (m 1H), 1.58-1.61 (m, 1H), 1.18-1.28 (m, 4H) ppm; 13C-NMR (126 MHz, CDCl3) δ; 180.9, 167.2, 167.1, 145.3, 142.7, 137.9, 136.8, 135.7, 135.0, 133.7, 132.6, 132.1, 131.8, 131.6, 131.3, 130.0, 129.9, 129.1, 128.5, 128.3, 125.5, 123.4, 123.3, 121.5, 117.2, 67.6, 56.2, 52.2, 51.1, 37.6, 33.0, 25.9, 25.5, 23.9, 23.2 ppm; IR (ATR) 3298, 2934, 1726 cm-1; MS (FAB) 758 (MH+, 30), 369 (100); HRMS (FAB) [C37H38F6N7O2S]+: 758.2712; Found. 758.2717.
1-{3,5-Bis(trifluoromethyl)phenyl}-3-[(1R,2R)-2-[[{1-(2-hydroxybenzyl)-1H-1,2,3-triazol-4-yl}
methyl](methyl)amino]cyclohexyl]thiourea (20b): White amorphous solid; [α]D24 +39.5 (c 0.56, CHCl3); 1H-NMR (500 MHz, acetone-d6, 50 °C) δ 8.67 (br, 1H), 8.36 (s, 2H), 7.95 (s, 1H), 7.62 (s, 1H). 7.12 (m, 2H), 6.88 (d, J = 7.8 Hz, 1H), 6.76 (t, J = 7.5 Hz, 1H), 5.52 (s, 2H), 2.27-2.77 (br, 3H), 2.04 (s, 3H), 1.86-1.95 (m, 2H), 1.68-1.75 (m, 3H), 1.20-1.43 (m, 4H) ppm; IR (KBr) 3266, 3057, 1674 cm-1; MS (FAB) 587 (MH+, 100); HRMS (FAB) [C26H30F6N6OS]+ 587.2028; Found. 587.2031.
1-{3,5-Bis(trifluoromethyl)phenyl}-3-[(1R,2R)-2-[methyl[[1-{(R)-pyrrolidin-2-ylmethyl}-1H-1,2,3-
triazol-4-yl]methyl]amino]cyclohexyl]thiourea (20d): Colorless crystals, Mp 179-181 °C; [α]D24 -0.16 (c 3.5, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 8.10 (s, 2H), 7.79 (s, 1H), 7.49 (s, 1H), 4.26 (dd, J = 13.8, 5.8 Hz, 1H), 4.19 (dd, J = 13.8, 7.5 Hz, 1H), 4.14 (br, 1H), 3.77 (d, J = 13.8 Hz, 1H), 3.52 (d, J = 13.8 Hz, 1H), 3.36-3.40 (m, 2H), 2.68-2.80 (m, 2H), 2.50-2.57 (m, 1H), 2.32-2.36 (m, 1H), 2.17 (s, 3H), 1.88-1.92 (m, 1H), 1.55-1.77 (m, 5H), 1.05-1.37 (m, 4H) ppm; 13C-NMR (126 MHz, CD3OD) δ 181.6, 147.4, 143.3, 132.7 (q, JC-F = 33.6 Hz), 125.3, 124.8 (q, JC-F = 273 Hz), 123.1, 117.4, 67.2, 67.1, 66.9, 59.4, 56.7, 55.4, 47.1, 37.5, 33.5, 30.1, 26.4, 25.9, 24.2 ppm; IR (ATR) 3375, 2484, 1476 cm-1; MS (FAB) 564 (MH+, 38), 70 (100); Anal. Calcd. for C24H31F6N7S: C, 51.16; H, 5.54; N, 17.24; Found; C, 51.12; H, 5.43; N, 17.24.

















{kind=link}
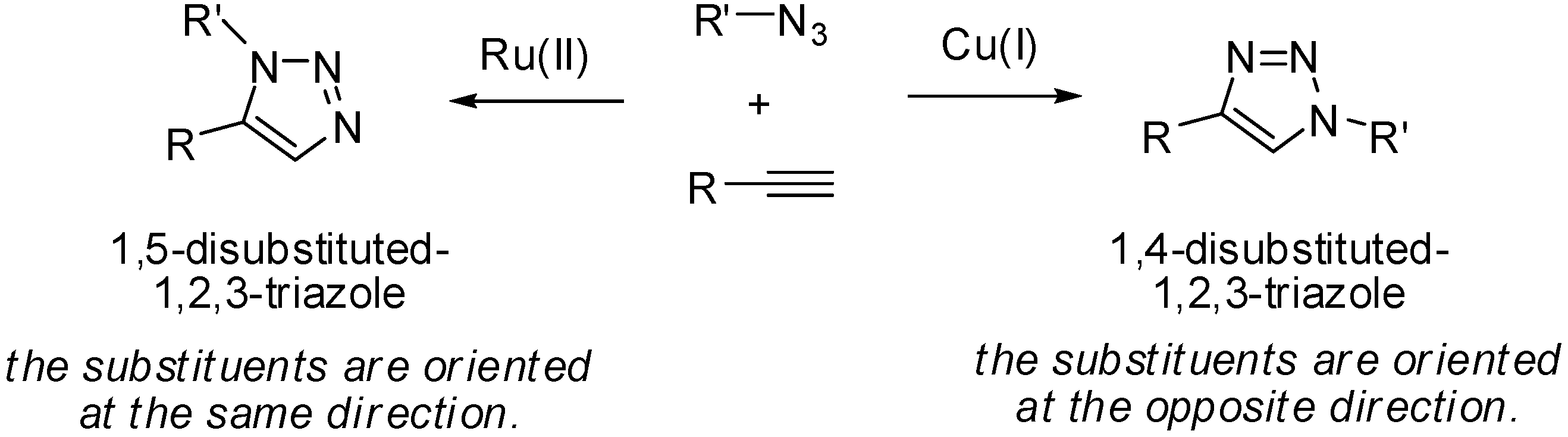{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}