4.2. Synthesis and characterization
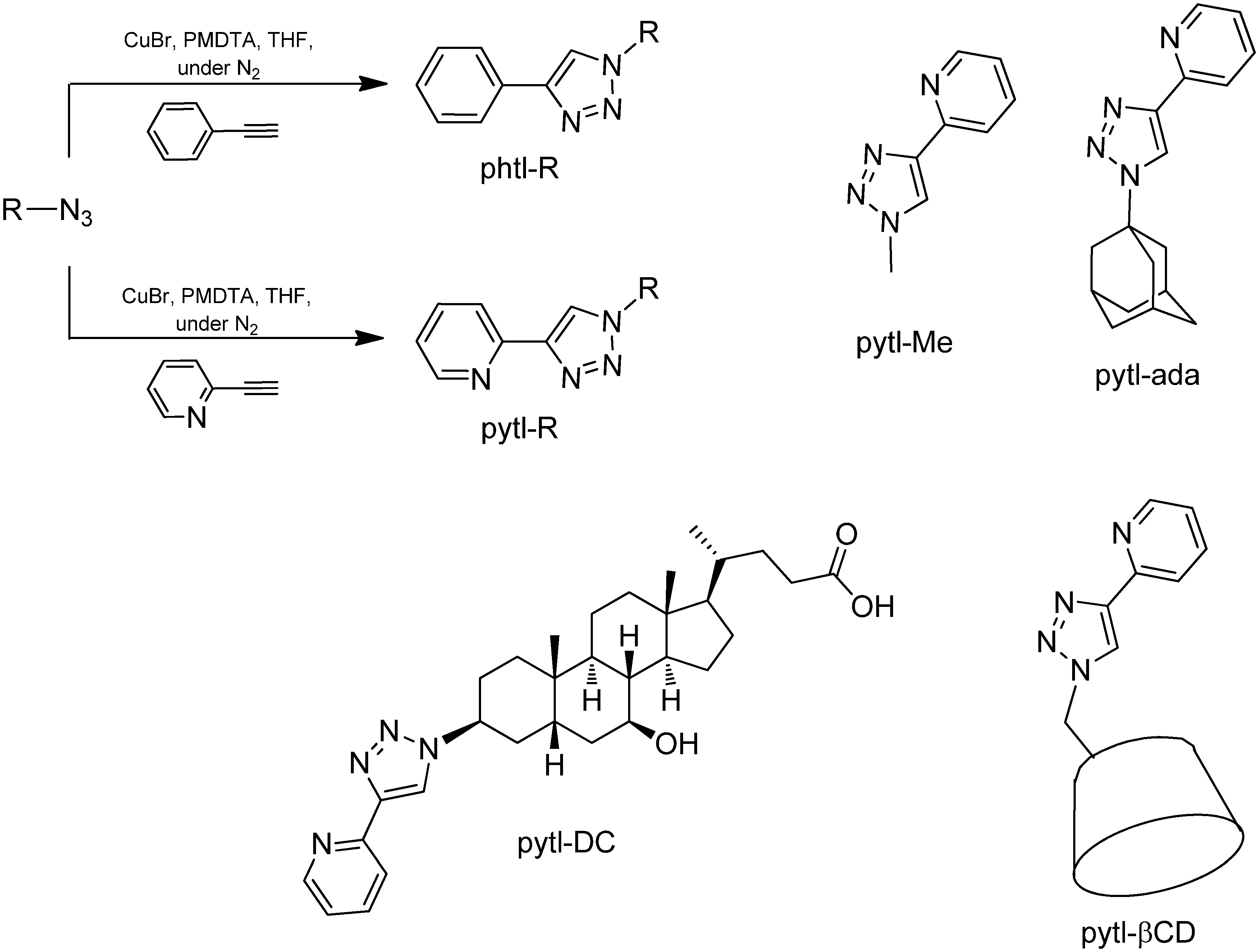
1-Methyl-4-phenyl-1H-1,2,3-triazole (
1): Dry DMF was deoxygenated prior to use by performing three freeze-pump-thaw cycles. A two-necked round bottom flask was carefully dried with flame, under N
2 flow. In the cooled flask, NaN
3 (198 mg, 3 mmol) was added together with deoxygenated dry DMF (40 mL). CH
3I (0.3 mL, 4.8 mmol) was added dropwise and the solution was stirred in the dark overnight. The methyl-azide formed is a highly explosive intermediate and therefore it was not isolated [
40]. The ‘clicking’ reagents were subsequently added to the reaction mixture and a large excess of ethynylbenzene (658.9 μL, 6 mmol) and of catalyst were used to ensure the complete consumption of the methyl-azide. A solution of CuBr (430 mg, 3 mmol) and PMDTA (0.67 mL, 3.2 mmol) was prepared by dissolving both reagents in oxygen-free dry DMF (10 mL), bubbling nitrogen through the solution for 20 min to prevent oxidation of Cu(I). When the CuPMDTA complex dissolved, an aliquot of the solution (5 mL), together with compound ethynylbenzene (0.45 mL, 4.5 mmol) were added to the flask. The resulting mixture was stirred in the dark at room temperature under a nitrogen atmosphere for 24 hours. The reaction was followed by TLC (eluent: Et
2O). After removal of the solvent
in vacuo (CAUTION: this solution may still contain methyl azide in case the conversion with phenylacetylene was not 100 %) the solid obtained was purified by column chromatography (Et
2O/Heptane 50/50 followed by Et
2O/ethyl acetate 90/10). Product
1 was obtained as a white solid (208 mg, overall yield 43%). Crystals were grown by slow evaporation of a solution of
1 in CHCl
3/heptane. The structure was further confirmed by X-ray single crystal diffraction.
1H-NMR (300 MHz, CDCl
3): δ ppm 7.86–7.78 (m, 2H), 7.73 (s, 1H), 7.47–7.38 (m, 2H), 7.37–7.29 (m, 1H), 4.14 (s, 3H);
13C-NMR (75 MHz, CDCl
3): δ ppm 148.0, 130.6, 128.8, 128.1, 125.7, 120.5, 36.7; HRMS (ES+, CHCl
3/CH
3OH): m/z calcd for C
9H
10N
3: 160.08747; found: 160.08768 [M+H]
+.
1-Adamantyl-4-phenyl-1H-1,2,3-triazole (2): Distilled THF was bubbled with N2 for 1h prior to use in order to remove the oxygen. Ethynylbenzene (185.6 μL, 1.69 mmol) and 1-adamantylazide (300 mg, 1.69 mmol) were added under nitrogen atmosphere in a Schlenk tube and dissolved in deoxygenated THF (15 mL). A solution of CuBr (242.4 mg, 1.69 mmol) and PMDTA (365.8 μL, 1.75 mmol) was prepared by dissolving both reagents in deoxygenated THF (15 mL) and bubbling nitrogen through the solution for 20 min to prevent oxidation of Cu(I). When the CuPMDTA complex dissolved, the solution became slightly green, and an aliquot of it (3 mL) was added to the Schlenk tube. The resulting mixture was stirred in the dark, at room temperature under a nitrogen atmosphere for 2.5 hours. The reaction was followed by TLC (eluent: Et2O/heptane 80/20). No workup was done and after removal of the solvent in vacuo, the blue-green solid obtained was directly purified by column chromatography (Et2O/heptane 30/70). Product 2 was obtained as a white solid (358.8 mg, 76%). Crystals were grown by slow evaporation of a solution of 2 in CHCl3/heptane. The structure was further confirmed by X-ray single crystal diffraction. 1H-NMR (300 MHz, CDCl3): δ ppm 7.86–7.81 (m, 2H), 7.82 (s, 1H), 7.45–7.38 (m, 2H), 7.34–7.28 (m, 1H), 2.32–2.26 (m, 9H), 1.85–1.79 (m, 6H); 13C-NMR (75 MHz, CDCl3): δ ppm 137.8, 131.1, 128.8, 127.8, 125.6, 118.2, 59.6, 43.0, 35.9, 29.5; HRMS (ES+, CHCl3/CH3OH): m/z calcd for C18H22N3: 280.18137; found: 280.18165 [M+H]+.
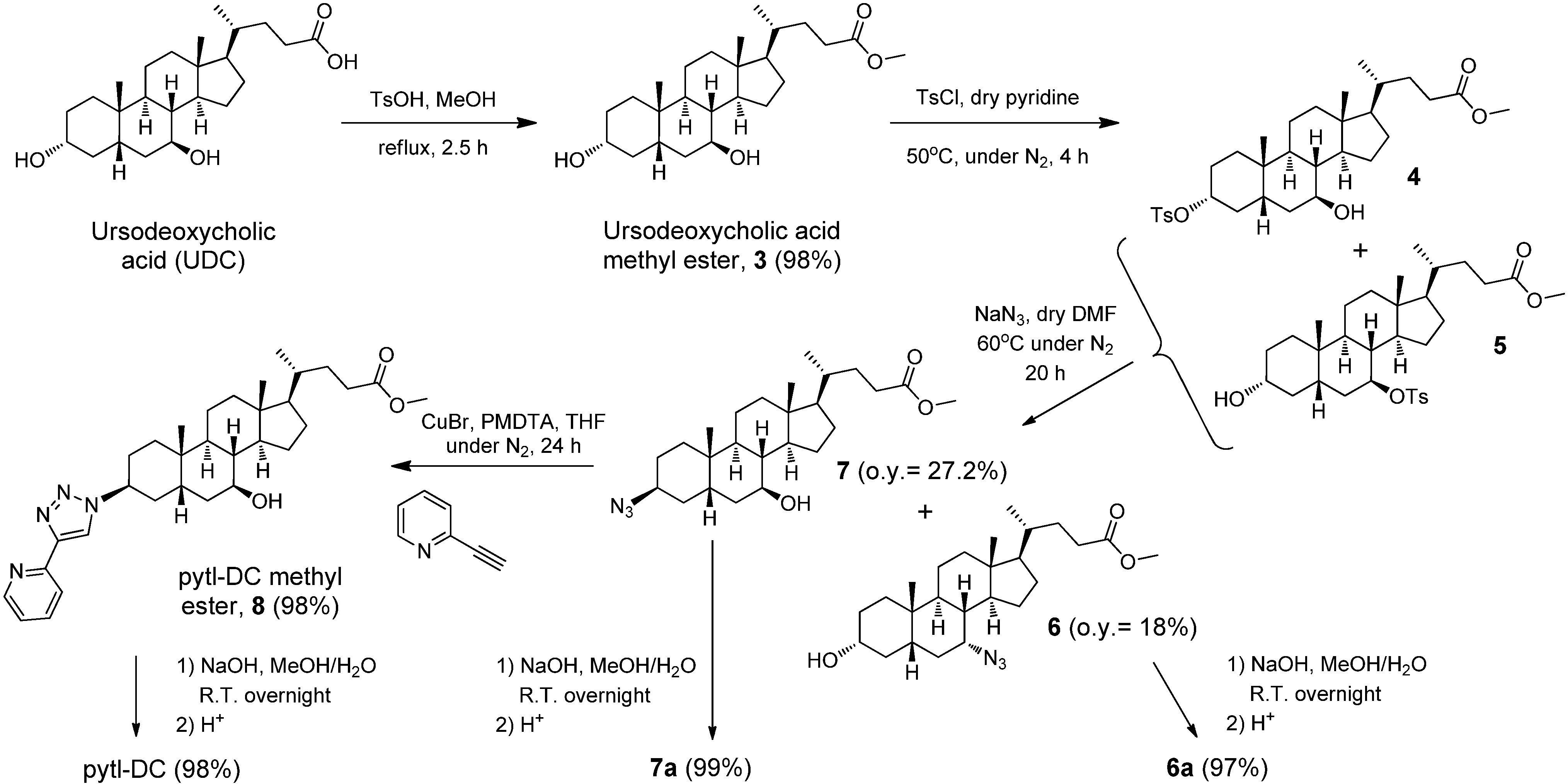
3α,7β-Dihydroxy-5β-cholan-24-oic acid methyl ester (3): Ursodeoxycholic acid (UDC, 277.1 mg, 0.71 mmol) was dissolved in methanol (10 mL). A catalytic amount of p-toluensolfonic acid was added and the solution was stirred under reflux for 2.5 hours. The reaction was followed by TLC (eluent: ethyl acetate/heptane 80/20). After removal of the solvent in vacuo, the crude product was dissolved in CHCl3 and washed with aqueous solution of K2CO3 (1M, 1 × 100 mL). The organic phase was dried over Na2SO4 anhydrous and the solvent removed under reduced pressure to yield 3 as a white solid (282.3 mg, 98%). 1H-NMR (300 MHz, CDCl3): δ ppm 3.64 (s, 3H), 3.61–3.48 (m, 2H), 2.41–2.10 (m, 2H), 2.01–0.96 (m, 24H), 0.92 (s, 3H), 0.90 (d, J = 6.4 Hz, 3H), 0.65 (s, 3H); 13C-NMR (75 MHz, CDCl3): δ ppm 174.7, 71.3, 71.2, 55.7, 54.9, 51.4, 43.69, 43.66, 42.4, 40.1, 39.2, 37.3, 36.9, 35.2, 34.9, 34.0, 31.01, 30.96, 30.3, 28.5, 26.8, 23.3, 21.1, 18.3, 12.1.
Mixture of 3α-(4-Methylphenyl)sulfonyloxy-7β-hydroxy-5β-cholan-24-oic acid methyl ester (4) and 3α-hydroxy-7β-(4-Methylphenyl)sulfonyloxy-5β-cholan-24-oic acid methyl ester (5): Compound 3 (254.5 mg, 0.62 mmol) was added under nitrogen atmosphere in a Schlenk tube and dissolved in dry pyridine (5 mL). Tosyl chloride (178.4 mg, 0.94 mmol) was added and the solution was heated to 50 ºC under nitrogen for 4 hours. The reaction was followed by TLC (eluent: ethyl acetate/heptane 50/50). After removal of the solvent in vacuo, the crude product was dissolved in ethyl acetate, washed with HCl (1N, 3 × 80 mL) and water (1 × 80 mL). The organic phase was dried over Na2SO4 anhydrous and the solvent removed under reduced pressure. The crude product (348.4 mg) was further reacted without purification.
3α-Hydroxy-7α-azido-5β-cholan-24-oic acid methyl ester (6) and 3β-azido-7β-hydroxy-5β-cholan-24-oic acid methyl ester (7): The crude material containing compounds 4+5 (348.4 mg) was added under nitrogen atmosphere in a Schlenk tube and dissolved in dry DMF (15 mL). Sodium azide (201.5 mg, 3.1 mmol) was added and the solution was heated to 60 ºC under nitrogen for 20 hours. The reaction was followed by TLC (eluent: ethyl acetate/heptane 50/50). After removal of most of the solvent in vacuo, the crude material was dissolved in ethyl acetate and washed with water (2 × 80 mL). The organic phase was dried over Na2SO4 anhydrous and the solvent removed under reduced pressure. The crude material was purified by column chromatography (eluent: ethyl acetate/heptane 15/85). Product 6 was obtained as a slightly yellow viscous oil (48.2 mg, overall yield = 18%). Product 7 was obtained as a slightly yellow viscous oil (72.8 mg, overall yield = 27.2%).
6: 1H-NMR (300 MHz, CDCl3): δ ppm 3.73–3.68 (m, 1H), 3.66 (s, 3H), 3.55–3.43 (m, 1H), 2.43–0.95 (m, 26H), 0.92 (d, J = 6.1 Hz, 3H), 0.91 (s, 3H), 0.63 (s, 3H); 13C-NMR (75 MHz, CDCl3): δ ppm 174.7, 71.8, 60.5, 55.6, 51.5, 51.1, 42.6, 41.0, 39.3, 38.3, 38.2, 35.3, 35.2, 34.9, 33.7, 31.0, 30.9, 30.6, 30.4, 28.0, 23.3, 22.9, 20.4, 18.3, 11.7.
7: 1H-NMR (300 MHz, CDCl3): δ ppm 3.93–3.88 (m, 1H), 3.66 (s, 3H), 3.59–3.46 (m, 1H), 2.41–2.15 (m, 2H), 2.04–0.99 (m, 24H), 0.97 (s, 3H), 0.92 (d, J = 6.4 Hz, 3H), 0.67 (s, 3H); 13C-NMR (75 MHz, CDCl3): δ ppm 174.6, 71.3, 58.3, 55.8, 54.9, 51.5, 43.7, 43.6, 40.1, 38.9, 37.8, 36.3, 35.2, 34.4, 31.2, 31.01, 30.98, 30.3, 28.5, 26.8, 24.5, 23.7, 21.3, 18.3, 12.1.
3α-Hydroxy-7α-azido-5β-cholan-24-oic acid (6a) and 3β-azido-7β-hydroxy-5β-cholan-24-oic acid (7a): A solution of NaOH was prepared by adding an aqueous solution of NaOH (2 N, 2 mL) into CH3OH (7 mL). 6a (20 mg, 0.046 mmol) or 7a (20 mg, 0.046 mmol) were dissolved in this solution and the resulting mixture was stirred at room temperature overnight. The reaction was followed by TLC (eluent: ethyl acetate). While cooling the reaction in ice bath, HCl (4 N) was added until pH = 7. After removal of the solvent in vacuo, the crude material obtained was purified by column chromatography (eluent: ethyl acetate). Product 6a was obtained as a white solid (18.6 mg, 97%). Product 7a was obtained as a white solid (19 mg, 99%). Single crystals of 6a and 7a were grown by slow evaporation of their slightly acidic solution in H2O/acetonitrile. The structures were further confirmed by X-ray single crystal diffraction. Alternatively, compounds 6a and 7a could be isolated from the crude mixture by semipreparative HPLC equipped with a reversed-phase column. The mobile phase was a gradient of H2O and acetonitrile (30% to 0% v/v of H2O in 35 min) both containing 0.1% v/v TFA, with a flow rate of 4 mL min-1.
6a: 1H-NMR (300 MHz, CDCl3): δ ppm 3.73–3.68 (m, 1H), 3.56–3.44 (m, 1H), 2.46–0.95 (m, 26H), 0.95–0.90 (m, 6H), 0.64 (s, 3H); 13C-NMR (75 MHz, CDCl3): δ ppm 179.0, 71.8, 60.4, 55.6, 51.1, 42.6, 41.0, 39.3, 38.3, 38.1, 35.3, 35.2, 34.9, 33.7, 30.8, 30.7, 30.6, 30.4, 28.0, 23.6, 22.9, 20.5, 18.2, 11.8.
7a: 1H-NMR (300 MHz, CDCl3): δ ppm 3.93–3.89 (m, 1H), 3.57–3.45 (m, 1H), 2.45–2.22 (m, 2H), 2.04–1.0 (m, 24H), 0.98 (s, 3H), 0.94 (d, J = 6.4 Hz, 3H), 0.69 (s, 3H); 13C-NMR (75 MHz, CDCl3): δ ppm 178.8, 71.4, 58.3, 55.8, 54.9, 43.8, 43.6, 40.1, 38.9, 37.9, 36.3, 35.2, 34.5, 31.3, 30.8, 30.3, 29.7, 28.6, 26.8, 24.6, 23.8, 21.4, 18.4, 12.1.
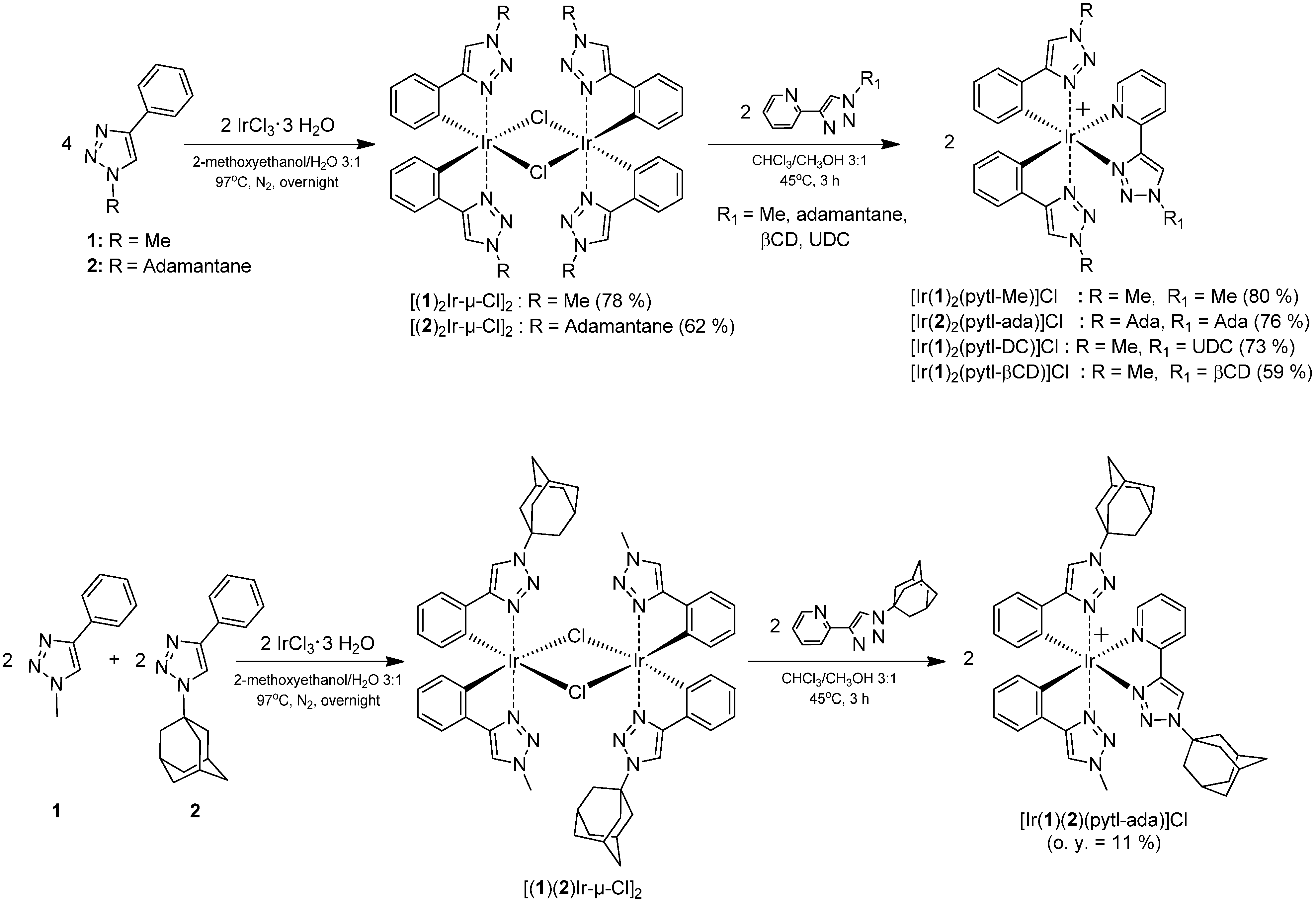
3β-(4'-(Pyridin-6'-yl)-1'H-1',2',3'-triazol-1'-yl)-7β-hydroxy-5β-cholan-24-oic acid methyl ester (
8) (
Scheme 5): Distilled THF was bubbled with N
2 for 1 hour prior to use in order to remove the oxygen. 2-Ethynylpyridine (58.6 μL, 0.58 mmol) and
7 (250 mg, 0.58 mmol) were added under nitrogen atmosphere in a Schlenk tube and dissolved in deoxygenated THF (10 mL). A solution of CuBr (83.2 mg, 0.58 mmol) and PMDTA (125.4 μL, 0.6 mmol) was prepared by dissolving both reagents in deoxygenated THF (10 mL) and bubbling nitrogen through the solution for 20 min to prevent oxidation of Cu(I). When the CuPMDTA complex dissolved, the solution became slightly green, and an aliquot of it (4 mL) was added to the Schlenk tube. The resulting mixture was stirred in the dark, at room temperature under a nitrogen atmosphere for 24 hours. The reaction was followed by TLC (eluent: ethyl acetate). After removal of the solvent
in vacuo, the blue-green solid obtained was directly purified by column chromatography (eluent: ethyl acetate/heptane 50/50 followed by ethyl acetate/heptane 60/40). Product
8 was obtained as a white solid (303.9 mg, 98%).
1H-NMR (400 MHz, CDCl
3): δ ppm 8.57 (ddd,
J = 4.9, 1.7, 0.9 Hz, 1H), 8.24 (s, 1H), 8.20 (ddd,
J = 8.0, 1.0, 1.0, Hz, 1H), 7.77 (ddd,
J = 7.8, 7.8, 1.8 Hz, 1H), 7.22 (ddd,
J = 7.6, 4.9, 1.1 Hz, 1H), 4.72–4.68 (m, 1H), 3.66 (s, 3H), 3.67–3.57 (m, 1H), 2.40–1.02 (m, 26H), 0.94–0.91 (m, 6H), 0.68 (s, 3H);
13C-NMR (75 MHz, CDCl
3): δ ppm 174.5, 150.3, 149.1, 147.6, 136.8, 122.6, 121.0, 120.1, 70.9, 56.5, 55.8, 54.8, 51.3, 43.5, 43.2, 39.9, 39.4, 37.6, 36.2, 35.1, 34.2, 30.94–30.77 (m), 30.4, 28.4, 26.7, 24.8, 23.4, 21.3, 18.2, 12.0; HRMS (ES+, CH
3OH): m/z calcd for C
32H
46N
4NaO
3: 557.34676; found: 557.34578 [M+Na]
+.
Scheme 5.
Structure and atom labeling of 8.
Scheme 5.
Structure and atom labeling of 8.
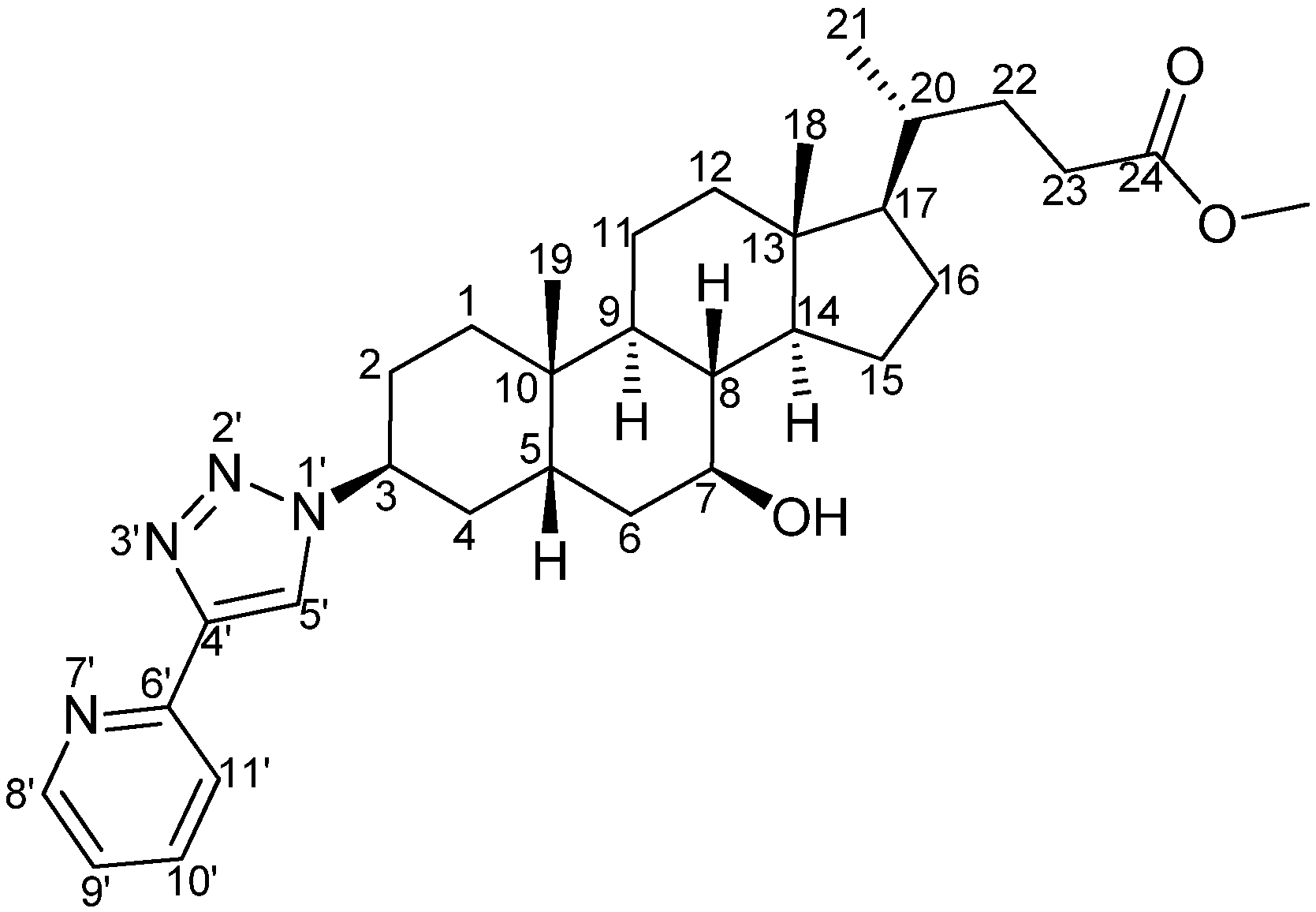
3β-(4'-(Pyridin-6'-yl)-1'H-1',2',3'-triazol-1'-yl)-7β-hydroxy-5β-cholan-24-oic acid (pytl-DC) (
Scheme 6): A solution of NaOH was prepared by adding an aqueous solution of NaOH (2 N, 2 mL) into CH
3OH (7 mL).
8 (55.9 mg, 0.10 mmol) was dissolved in this solution and the resulting mixture was stirred at room temperature overnight. The reaction was followed by TLC (eluent: CH
3OH/ethyl acetate 10/90). While cooling the reaction in ice bath, HCl (4 N) was added until pH=7–8. After removal of the solvent
in vacuo, the crude material obtained was purified by column chromatography (CH
3OH/ethyl acetate 10/90). Pytl-DC was obtained as a white solid (55.6 mg, 98%). Crystals were grown by slow evaporation of a solution of pytl-DC in CH
3OH. The structure was further confirmed by X-ray single crystal diffraction.
1H-NMR (300 MHz, DMSO-d
6): δ ppm 8.67 (s, 1H), 8.59 (ddd,
J = 4.9, 1.8, 1.0 Hz, 1H), 8.05 (ddd,
J = 8.0, 1.1, 1.1 Hz, 1H), 7.89 (ddd,
J = 8.0, 7.8, 1.8 Hz, 1H), 7.33 (ddd,
J = 7.7, 4.9, 1.1 Hz, 1H), 4.74–4.68 (m, 1H), 3.44–3.32 (m, 1H), 2.29–0.93 (m, 26H), 0.89 (d,
J = 6.4 Hz, 3H), 0.85 (s, 3H), 0.63 (s, 3H);
13C-NMR (75 MHz, DMSO-d
6): δ ppm 175.8, 150.7, 149.9, 147.4, 137.6, 123.3, 122.9, 119.9, 69.8, 56.6, 56.2, 55.3, 43.5, 43.2, 40.2, 39.3, 38.1, 37.2, 35.3, 34.4, 31.6, 31.4, 31.0, 30.7, 28.6, 27.1, 24.8, 24.0, 21.6, 18.8, 12.5; HRMS (ES+, CH
3OH): m/z calcd for C
31H
44N
4NaO
3: 543.33111; found: 543.33058 [M+Na]
+.
Scheme 6.
Structure and atom labeling of pytl-DC.
Scheme 6.
Structure and atom labeling of pytl-DC.
[(1)2Ir-µ-Cl]2: A mixture 3/1 of 2-methoxyethanol/water (9 mL) was deoxygenated by bubbling nitrogen through it for 10-15 min. The ligand 1 (91.5 mg, 0.57 mmol) and IrCl3·3H2O (70 mg, 0.23 mmol) were added and the solution was heated to 97 ºC in the dark for 24 hours, under nitrogen atmosphere (without bubbling). After cooling to room temperature, the yellow solid was filtered off, washed with water (3 × 100 mL), diethyl ether (3 × 100 mL) and dried. After purification by column chromatography (eluent: from MeOH/CHCl3 5/95 to MeOH/CHCl3 10/90), the chloride-bridged complex [(1)2Ir-µ-Cl]2 was obtained as a yellow solid (97.4 mg, 78%). 1H-NMR (400 MHz, DMSO-d6): δ ppm 8.61 (s, 2H), 8.59 (s, 2H), 7.42 (ddd, J = 7.4, 1.4, 0.5 Hz, 2H), 7.41 (ddd, J = 7.5, 1.4, 0.5 Hz, 2H), 6.81 (ddd, J = 7.4, 7.4, 1.2 Hz, 2H), 6.80 (ddd, J = 7.4, 7.4, 1.2 Hz, 2H), 6.64 (ddd, J = 7.4, 7.4, 1.4 Hz, 2H), 6.63 (ddd, J = 7.4, 7.4, 1.5 Hz, 2H), 6.05 (ddd, J = 7.6, 1.2, 0.5 Hz, 2H), 6.00 (ddd, J = 7.6, 1.2, 0.5 Hz, 2H), 4.30–4.26 (m, 12H). ESI-MS (ES+, CHCl3/CH3OH): m/z = 1053.1 [M-Cl]+.
[(2)2Ir-µ-Cl]2: A mixture 3/1 of 2-methoxyethanol/water (9 mL) was deoxygenated by bubbling nitrogen through it for 10-15 min. The ligand 2 (50 mg, 0.18 mmol) and IrCl3⊕3H2O (21.4 mg, 0.072 mmol) were added and the solution was heated to 97 ºC in the dark for 24 hours, under nitrogen atmosphere (without bubbling). After cooling to room temperature, the yellow solid was filtered off, washed with water (3 × 80 mL) and dried. After purification by column chromatography (eluent: ethyl acetate), the chloride-bridged complex [(2)2Ir-µ-Cl]2 was obtained as a yellow solid (35 mg, 62%). 1H- NMR (400 MHz, CDCl3): δ ppm 7.83 (s, 2H), 7.77 (s, 2H), 7.27–7.22 (m, 4H), 6.75 (ddd, J = 7.4, 7.4, 1.1 Hz, 2H), 6.70 (ddd, J = 7.3, 7.3, 1.2 Hz, 2H), 6.64 (ddd, J = 7.2, 7.2, 1.1 Hz, 2H), 6.62 (ddd, J = 7.3, 7.3, 1.2 Hz, 2H), 6.15 (dd, J = 7.4, 0.8 Hz, 2H), 6.04 (dd, J = 7.6, 0.5 Hz, 2H), 2.48–2.28 (m, 36H), 1.90–1.79 (m, 24H); 13C-NMR (75 MHz, CDCl3): δ ppm 157.0, 156.8, 145.9, 143.9, 136.3, 135.8, 132.9, 132.3, 127.24, 127.22, 121.08, 121.06, 120.8, 120.6, 113.9, 113.7, 61.9, 61.4, 42.9, 42.8, 35.79, 35.75, 29.49, 29.46; ESI-MS (ES+, CHCl3/CH3OH): m/z = 807.0 [(M/2)+Na]+
[(1)(2)Ir-µ-Cl]2: A mixture 3/1 of 2-methoxyethanol/water (12 mL) was deoxygenated by bubbling nitrogen through it for 10-15 min. The ligands 1 (40 mg, 0.25 mmol) and 2 (70.2 mg, 0.25 mmol), and IrCl3⊕3H2O (62.5 mg, 0.21 mmol) were added and the solution was heated to 97 ºC in the dark for 24 hours, under nitrogen atmosphere (without bubbling). After cooling to room temperature, the yellow solid was filtered off, washed with water (3 × 80 mL) and dried. This reaction is expected to yield a mixture of chloride-bridged complexes [(X)(Y)Ir-µ-Cl]2 (X=Y=1,2). The reaction was followed by TLC (eluent: CH3OH/CHCl3 20/80) where several spots, corresponding to different dimeric species, could be observed. The crude material was further reacted without purification.
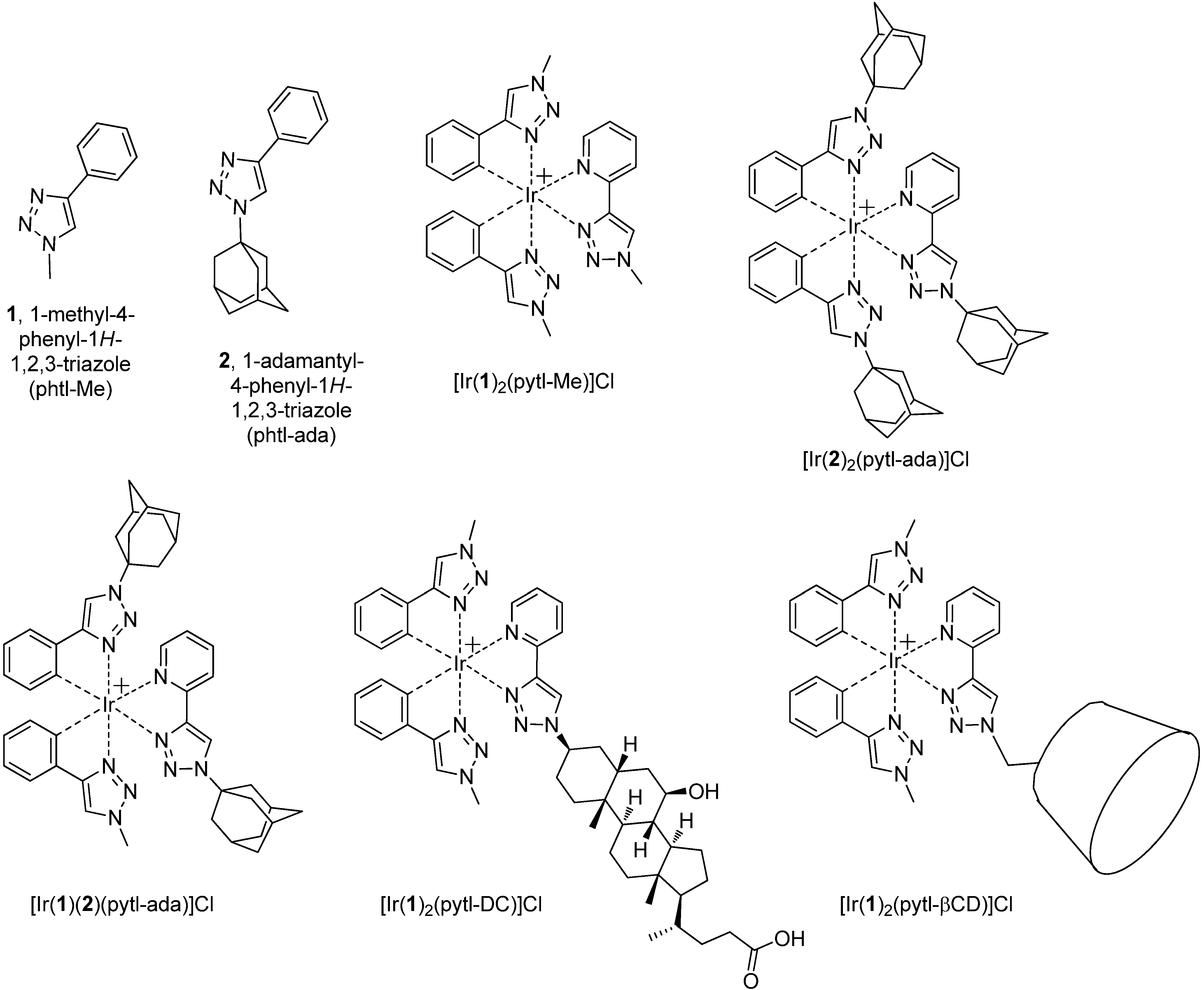
[Ir(1)2(pytl-Me)]Cl: To a suspension of the precursor [(1)2Ir-µ-Cl]2 (47.2 mg, 0.043 mmol) in MeOH/CHCl3 1/3 v/v (4 mL) was added pytl-Me as a solid (14.6 mg, 0.086 mmol). The suspension was heated to 45 ºC and stirred in the dark for 3 hours, after which time a clear and yellow solution was obtained. The reaction was followed by TLC (eluent: MeOH/CHCl3 30/70) where, under UV light at 366 nm, the compound appeared as a bright green-blue luminescent spot. After removal of the solvent in vacuo, the solid obtained was purified by PLC (eluent: MeOH/CHCl3 25/75). The product was obtained as a slightly yellow solid (47.4 mg, 80%). 1H-NMR (400 MHz, CDCl3): δ ppm 10.48 (s, 1H), 8.88 (d, J = 7.9 Hz, 1H), 7.93-7.87 (m, 2H), 7.77 (s, 1H), 7.73 (s, 1H), 7.43 (dd, J = 7.5, 1.1 Hz, 1H), 7.39 (dd, J = 7.4, 1.1 Hz, 1H), 7.12 (ddd, J = 7.5, 5.6, 1.3 Hz, 1H), 6.96 (ddd, J = 7.5, 7.5, 1.2 Hz, 1H), 6.92 (ddd, J = 7.4, 7.4, 1.2 Hz, 1H), 6.85 (ddd, J = 7.5, 7.5, 1.4 Hz, 1H), 6.81 (ddd, J = 7.5, 7.5, 1.4 Hz, 1H), 6.32–6.28 (m, 2H), 4.16 (s, 3H), 4.08 (s, 3H), 4.05 (s, 3H); 13C-NMR (75 MHz, CDCl3): δ ppm 157.9, 157.5, 151.1, 150.3, 149.3, 146.1, 142.3, 139.0, 135.3, 135.0, 132.75, 132.73, 128.9, 128.6, 128.1, 124.8, 123.7, 122.6, 122.3, 122.2, 122.1, 118.9, 118.8, 38.60, 38.56, 38.5; HRMS (ES+, CH3OH): m/z calcd for C26H24IrN10: 669.18146; found: 669.18464 M+.
[Ir(2)2(pytl-ada)]Cl: To a suspension of the precursor [(2)2Ir-µ-Cl]2 (20.0 mg, 0.013 mmol) in MeOH/CHCl3 1/3 v/v (4 mL) was added pytl-ada as a solid (7.29 mg, 0.026 mmol). The suspension was heated to 45 ºC and stirred in the dark for 3 hours, after which time a clear and yellow solution was obtained. The reaction was followed by TLC (eluent: MeOH/CHCl3 20/80) where, under UV light at 366 nm, the compound appeared as a bright green-blue luminescent spot. After removal of the solvent in vacuo, the solid obtained was purified by column chromatography (eluent: from CHCl3 to MeOH/CHCl3 5/95). The product was obtained as a slightly yellow solid (20.3 mg, 76%). 1H-NMR (400 MHz, CDCl3): δ ppm 10.56 (s, 1H), 9.16 (d, J = 7.8 Hz, 1H), 7.96 (bd, J = 5.4 Hz, 1H), 7.92 (ddd, J = 7.8, 7.8, 1.4 Hz, 1H), 7.76 (s, 1H), 7.71 (s, 1H), 7.34 (dd, J = 5.7, 1.3 Hz, 1H), 7.32 (dd, J = 5.6, 1.2 Hz, 1H), 7.07 (ddd, J = 7.2, 5.8, 1.2 Hz, 1H), 6.91 (ddd, J = 6.7, 6.7, 1.2 Hz, 1H), 6.88 (ddd, J = 6.6, 6.6, 1.2 Hz, 1H), 6.82 (ddd, J = 7.4, 7.4, 1.4 Hz, 1H), 6.77 (ddd, J = 7.5, 7.5, 1.4 Hz, 1H), 6.16 (dd, J = 7.6, 0.6 Hz, 1H), 6.13 (dd, J = 7.5, 0.6 Hz, 1H), 2.30–2.19 (m, 15H), 2.13–2.10 (m, 12H), 1.81–1.68 (m, 18H); 13C-NMR (75 MHz, CDCl3): δ ppm 156.7, 156.1, 151.7, 150.0, 148.9, 146.8, 143.3, 138.9, 136.1, 135.7, 133.0, 132.3, 128.2, 127.4, 125.2, 124.01, 123.95, 122.1, 121.7, 121.5, 121.2, 114.1, 114.0, 62.2, 61.9, 61.8, 42.70, 42.67, 42.5, 35.67, 35.63, 29.7, 29.5, 29.37, 29.36; HRMS (ES+, CH3OH): m/z calcd for C53H60IrN10: 1029.46316; found: 1029.46242 M+.
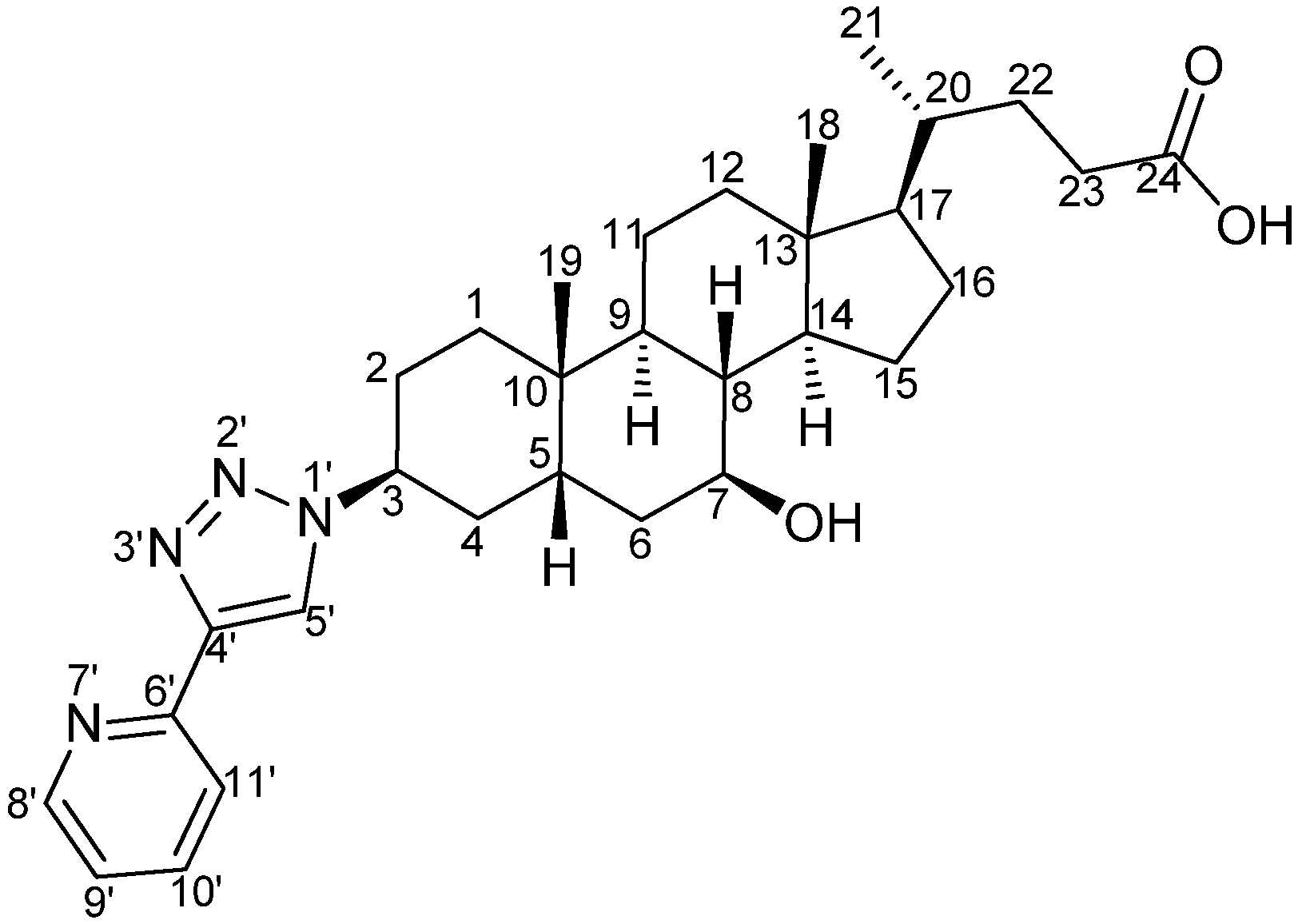
[Ir(1)(2)(pytl-ada)]Cl: To a suspension of the crude mixture of the precursors [(
X)(
Y)Ir-µ-Cl]
2 (
X=
1,
2;
Y=1,
2) (76.2 mg) in MeOH/CHCl
3 1/3 v/v (8 mL) was added pytl-ada as a solid (44.8 mg, 0.16 mmol). The suspension was heated to 45 ºC and stirred in the dark for 3 hours, after which time a yellow solution was obtained. The reaction was followed by TLC (eluent: MeOH/CHCl
3 20/80). The mixture of products obtained could not be separated by column chromatography. After removal of the solvent
in vacuo, the crude material was purified by using a semipreparative HPLC equipped with a reversed-phase column. The mobile phase was a gradient of H
2O and acetonitrile (50% for 20 min + gradient 50% to 100% v/v of acetonitrile in 40 min) both containing 0.1% v/v HCl. The complex [Ir(
1)(
2)(pytl-ada)]Cl exists in four stereoisomeric forms: the two diastereoisomers
A and
B (
Scheme 7), and their corresponding enantiomers (Λ,Δ). The diastereoisomeric species
A(Λ+Δ) and
B(Λ+Δ) could be separated by HPLC in the conditions described above. However, we did not perform their isolation and collected them as a mixture. The HPLC fractions containing the desired iridium complex were identified by ESI-MS. The retention time of the two peaks of [Ir(
1)(
2)(pytl-ada)]Cl were t = 41.9 min and t = 43.3 min. After collecting the fractions, the solvent was removed
in vacuo at 40 ºC. The product was obtained as a slightly yellow solid (21.1 mg, overall yield = 11%) containing the two diastereoisomers in a ratio 1:0.8 as calculated from the NMR spectrum.
1H-NMR (400 MHz, CDCl
3): δ ppm 10.49 (s, 0.8H), 10.45 (s, 1H), 9.12–9.05 (m, 1H+0.8H), 7.93–7.83 (m, 2H+1.6H), 7.77 (bs, 1.6H), 7.76 (s, 1H), 7.73 (s, 1H), 7.33 (dd,
J = 7.4,
J = 1.1 Hz, 1.6H), 7.32 (dd,
J = 7.2,
J = 1.1 Hz, 1H), 7.29 (dd,
J = 7.4,
J = 1.2 Hz, 1H), 7.08–7.02 (m, 1H+0.8H), 6.92–6.76 (m, 3H+2.4H), 6.75 (ddd,
J = 7.4,
J = 7.4,
J = 1.3 Hz, 0.8H) 6.74 (ddd,
J = 7.4,
J = 7.4,
J = 1.3 Hz, 1H), 6.31 (d,
J = 7.5 Hz, 0.8H), 6.24 (d,
J = 7.5 Hz, 1H), 6.12 (d,
J = 7.5 Hz, 1H), 6.08 (d,
J = 7.5 Hz, 0.8H), 4.02 (s, 2.4H), 4.00 (s, 3H), 2.28–2.22 (m, 9H+7.2H), 2.22–2.17 (m, 3H+2.4H), 2.14–2.09 (m, 6H+4.8H), 1.82–1.66 (m, 12H+9.6H);
13C-NMR (75 MHz, CDCl
3): δ ppm 157.8, 157.2, 156.6, 156.0, 151.48, 151.47, 150.1, 149.9, 148.84, 148.79, 146.5, 146.2, 143.2, 142.9, 138.9–138.8 (m), 135.9, 135.63, 135.60, 135.4, 133.1, 132.9, 132.4, 132.2, 128.2, 128.1, 127.5, 127.3, 125.1, 124.9, 124.3, 124.2, 123.7, 123.6, 122.23, 122.19, 122.1, 121.8, 121.7, 121.56, 121.55, 121.3, 119.2, 119.0, 114.3, 114.1, 62.4, 62.3, 61.9, 61.8, 42.62, 42.57, 42.4, 42.3, 38.5, 38.4, 35.60, 35.58, 35.55, 29.41–29.37 (m), 29.32, 29.30; HRMS (ES+, CH
3OH):
m/z calcd for C
44H
48IrN
10: 909.36926; found: 909.37012 M
+.
Scheme 7.
Structure of the diastereoisomers A and B of [Ir(1)(2)(pytl-ada)]Cl.
Scheme 7.
Structure of the diastereoisomers A and B of [Ir(1)(2)(pytl-ada)]Cl.
[Ir(1)2(pytl-DC)]Cl: To a suspension of the precursor [(1)2Ir-µ-Cl]2 (28.3 mg, 0.026 mmol) in MeOH/CHCl3 1/3 v/v (4 mL) was added pytl-DC as a solid (26.8 mg, 0.052 mmol). The suspension was heated to 45 ºC and stirred in the dark for 17 hours, after which time a clear and yellow solution was obtained. The reaction was followed by TLC (eluent: MeOH/CHCl3 20/80) where, under UV light at 366 nm, the compound appeared as a bright green-blue luminescent spot. After removal of the solvent in vacuo, the solid obtained was purified by PLC (eluent: MeOH/CHCl3 20/80). The product was obtained as a slightly yellow solid (39.1 mg, 73%) containing a mixture 1:1 of two diastereoisomers as also confirmed by the NMR spectrum. 1H-NMR (400 MHz, CD3OD): δ ppm 9.041 (s, 1H), 9.038 (s, 1H), 8.24–8.23 (m, 1H), 8.23 –8.22 (m, 3H), 8.20–8.16 (m, 2H), 8.08–7.99 (m, 4H), 7.50–7.46 (m, 2H), 7.44–7.40 (m, 2H), 7.34–7.30 (m, 2H), 6.97–6.92 (m, 2H), 6.88–6.79 (m, 4H), 6.71 (ddd, J = 7.4, J = 1.4, J = 1.4 Hz, 2H), 6.29 (ddd, J = 7.5, J = 1.1, J = 0.5 Hz, 1H), 6.28 (ddd, J = 7.6, J = 1.1, J = 0.5 Hz, 1H), 6.25–6.22 (m, 2H), 4.81–4.76 (m, 2H), 4.07–4.06 (m, 6H), 4.05 (bs, 6H), 3.51–3.42 (m, 2H), 2.37–0.89 (m, 64H), 0.72 (s, 6H); HRMS (ES+, CH3OH): m/z calcd for C49H60IrN10O3: 1029.44791; found: 1029.44633 M+.
[Ir(1)2(pytl-βCD)]Cl: To a suspension of the precursor [(1)2Ir-µ-Cl]2 (7.04 mg, 0.006 mmol) in MeOH/CHCl3 1/3 v/v (4 mL) was added pytl-βCD as a solid (20.1 mg, 0.013 mmol). The suspension was heated to 45 ºC and stirred in the dark for 3 hours, after which time a clear and yellow solution was obtained. The reaction was followed by TLC (eluent: MeOH/CHCl3 15/85) where, under UV light at 366 nm, the compound appeared as a bright green-blue luminescent spot. After removal of the solvent in vacuo, the solid obtained was purified by column chromatography (eluent: MeOH/CHCl3 5/95). The product was obtained as a slightly yellow solid (16.0 mg, 59%) containing a mixture 1:1 of two diastereoisomers as also confirmed by the NMR spectrum. 1H-NMR (300 MHz, CDCl3): δ ppm 9.73 (s, 1H), 9.63 (s, 1H), 8.87–8.68 (m, 2H), 7.95–7.87 (m, 2H), 7.81 (s, 1H), 7.78 (s, 1H), 7.744 (s, 1H), 7.736 (s, 1H), 7.40–7.21 (m, 6H), 7.17–7.11 (m, 2H), 6.97–6.65 (m, 8H), 6.30–6.21 (m, 2H), 6.17–6.06 (m, 2H), 5.57–5.46 (m, 1H), 5.39–5.03 (m, 13H), 4.95–4.83 (m, 1H), 4.78–4.69 (m, 1H), 4.17 (s, 3H), 4.12 (s, 3H), 4.11 (s, 3H), 4.05 (s, 3H), 4.15–2.98 (m, 200H), 2.96–2.89 (m, 1H), 2.81–2.74 (m, 1H); HRMS (ES+, CH3OH): m/z calcd for C87H130IrN10O34: 2051.83801; found: 2051.83965 M+.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


