2. Results and Discussion
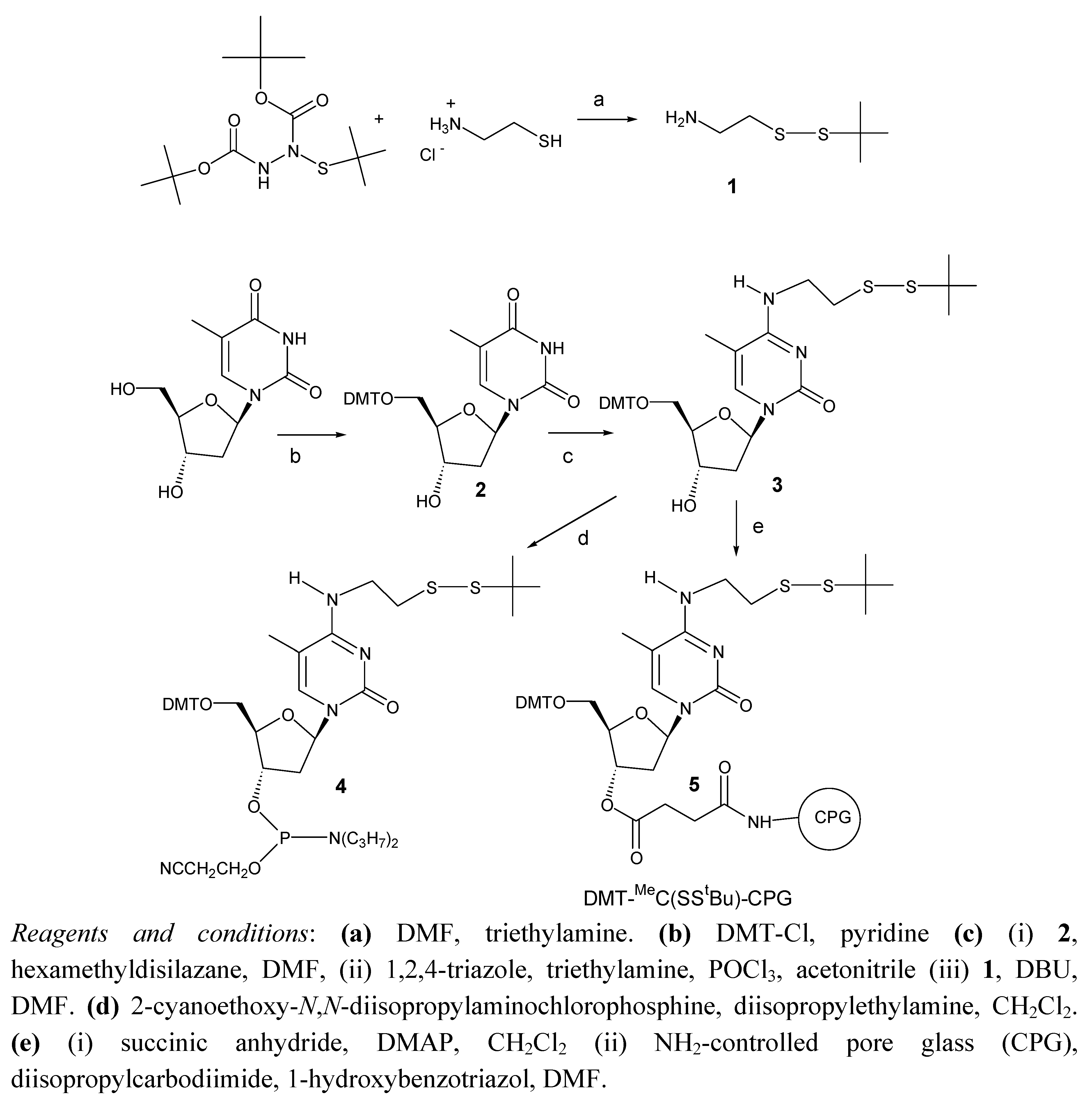
The synthesis of the protected phosphoramidite derivative of 2’-deoxy-[2-(t-butyldisulfanyl)ethyl]-5-methylcytidine is illustrated in
Scheme 1. First, 2-aminoethyl-
tert-butyl disulfide (
1) was prepared by reaction of cysteamine with di-
tert-butyl-1-(
tert-buylthio)-1,2-hydrazinedicarboxylate as described previously [
15]. After, dimethoxytritylthymidine (DMT-T,
2) was prepared by standard protocols. Then, the 3’-OH of DMT-T (
2) was protected with the transient trimethylsilyl group [
18]. Activation of position 4 of 5’-DMT-3’-trimethylsilyl-T was done with a solution of phosphoryl
tris(1,2,4-triazolide), previously prepared by mixing phosphoryl chloride and 1,2,4-triazole in the presence of a large excess of triethylamine [
18,
19,
20]. The resulting triazolyl derivative was reacted with 2-aminoethyl-
tert-butyl disulfide (
1) yielding the desired 5’-DMT-2’-deoxy-
N4-[2-(t-butyldisulfanyl)ethyl]-5-methylcytidine derivative (
3). Compound
3 was reacted with 2-cyanoethoxy-
N,
N-diisopropylaminochlorophosphine yielding the desired phosphoramidite
4. This phosphoramidite was incorporated into oligonucleotide sequence (5’-d (TTCCAXATTACCG)-3’ being X the position of the
N4- [2-(t-butyldisulfanyl)ethyl]-5-methylcytidine derivative. The addition of the new phosphoramidite proceeded with 95-99% coupling yields, similar to standard phosphoramidites. After the assembly of the sequence, ammonia deprotection yielded the desired oligonucleotide together with a more polar side compound that had 40 mass units less than the desired oligonucleotide (see below)
In order to determine the structure of the side compound a solid support functionalized with the protected 5’-DMT-2’-deoxy-N4-[2-(t-butyldisulfanyl)ethyl]-5-methylcytidine derivative was prepared. Compound 3 was then reacted with succinic anhydride to yield the 3’-hemisuccinate derivative that was reacted with amino-controlled pore glass (long chain amino alkyl-controlled pore glass, LCAA-CPG) yielding CPG solid support functionalized with 5’-DMT-2’-deoxy-N4-[2-(t-butyldisulfanyl)ethyl]-5-methylcytidine (5).
Scheme 1.
Synthesis of the protected phosphoramidite derivative of 2’-deoxy-[2-(t-butyldisulfanyl)ethyl]-5-methylcytidine.
Scheme 1.
Synthesis of the protected phosphoramidite derivative of 2’-deoxy-[2-(t-butyldisulfanyl)ethyl]-5-methylcytidine.
At this point we tested the stability of the disulfide bond of DMT-
MeC(SS
tBu)-CPG (
5) in the presence of the iodine solution (0.02 M iodine in water/ pyridine/ tetrahydrofuran) used during oligonucleotide synthesis. Alternatively the stability of the disulfide bond to a 10% solution of
tert-butylhydroperoxide in acetonitrile was studied. For comparison purposes we also studied the stability of the disulfide bond of the 2’-deoxycytidine derivative DMT-C(SS
tBu)-CPG (
6). This solid support was prepared as described [
15].
Both DMT-
MeC(SS
tBu)-CPG (
5) and DMT-C(SS
tBu)-CPG (
6) were treated with iodine solution for 5 min and 1 hr and
tert-butylhydroperoxide for 3 h. During oligonucleotide synthesis, solid supports are exposed to iodine solution for approx. 1 min per cycle and to
tert-butylhydroperoxide for 15 min per cycle. After the treatment with the oxidation solution, the DMT group was removed with trichloroacetic acid and the support treated with ammonia. The resulting nucleosides were analyzed by HPLC. Results are shown in
Scheme 2 and
Table 1.
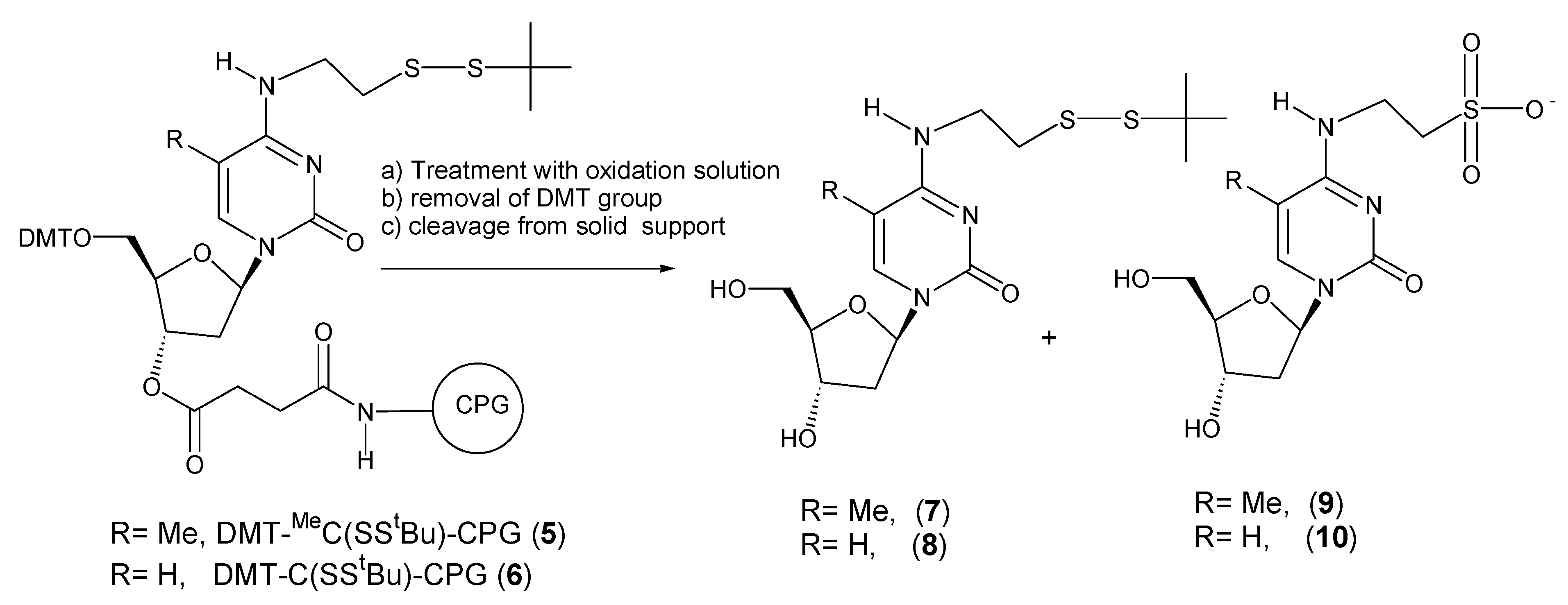
Scheme 2.
Products obtained after treatment of DMT-MeC(SStBu)-CPG (5) and DMT-C(SStBu)-CPG (6) with iodine or tert-butylhydroperoxide solutions. Reagents and conditions: (a) iodine or tert-butylhydroperoxide solution. (b) 3% trichloroacetic acid in dichloromethane. (c) concentrated ammonia, room temperature, 30 min.
Scheme 2.
Products obtained after treatment of DMT-MeC(SStBu)-CPG (5) and DMT-C(SStBu)-CPG (6) with iodine or tert-butylhydroperoxide solutions. Reagents and conditions: (a) iodine or tert-butylhydroperoxide solution. (b) 3% trichloroacetic acid in dichloromethane. (c) concentrated ammonia, room temperature, 30 min.
Table 1.
Products resulting from the treatment of DMT-MeC(SStBu)-CPG (5) and DMT-C(SStBu)-CPG (6) with iodine or tert-butylhydroperoxide solutions.
Table 1.
Products resulting from the treatment of DMT-MeC(SStBu)-CPG (5) and DMT-C(SStBu)-CPG (6) with iodine or tert-butylhydroperoxide solutions.
| Solid Support | Treatment | tBuSS- protected / oxidized (-SO3) |
|---|
| 5 | Iodine solution, 5 min | 95 : 5 |
| 5 | Iodine solution, 1 h | 3 : 97 |
| 5 | tert-Butylhydroperoxide solution, 3 h | 100 : 0 |
| 6 | Iodine solution, 5 min | 100 : 0 |
| 6 | Iodine solution, 1 h | 96 : 4 |
| 6 | tert-Butylhydroperoxide solution, 3 h | 100: 0 |
Treatment of solid support carrying 5’-DMT-2’-deoxy-
N4-[2-(t-butyldisulfanyl)ethyl]-5-methylcytidine derivative (
5) with iodine resulted on the formation of two nucleoside derivatives: the expected 2’-deoxy-
N4-[2-(t-butyldisulfanyl)ethyl]-5-methylcytidine (
7) (retention time 13.6 min, M = 388) and a new nucleoside derivative (retention time 6.2 min, M = 348). HPLC profiles are shown as
supplementary material. The new compound was identified as the sulfonic acid derivative
9 resulting from the removal of the
t-butylthio group, followed by oxidation of the resulting thiol group to the corresponding sulfonic acid. A 5-min iodine treatment (corresponding to five synthesis cycles) resulted on the formation of 5% of this side compound while 1 h treatment (60 synthesis cycles) resulted on the total conversion of the protected nucleoside to the oxidation product. The use of
tert-butylhydroperoxide solution prevented the oxidation reaction (3 h treatment equivalent to 12 synthesis cycles).
On the other hand treatment of solid support carrying 5’-DMT-2’-deoxy-
N4-[2-(t-butyldisulfanyl)ethyl]cytidine derivative
6, either with iodine or
tert-butylhydroperoxide yielded the expected
tBuS protected nucleoside as the major compound (> 96%). It has been also described that
tBuS protected cysteine is stable to iodine and
tert-butylhydroperoxide solutions [
21,
22]. We hypothesize that the electron donor properties of the methyl group at position 5 is responsible of a slightly higher electron density on the thiol group that may facilitate the oxidation of the sulfur with iodine with subsequent loss of the
tBuS group. Although some steric or other effects should be also present as the methyl group at C5 and the sulfur atom are separated by five bonds.
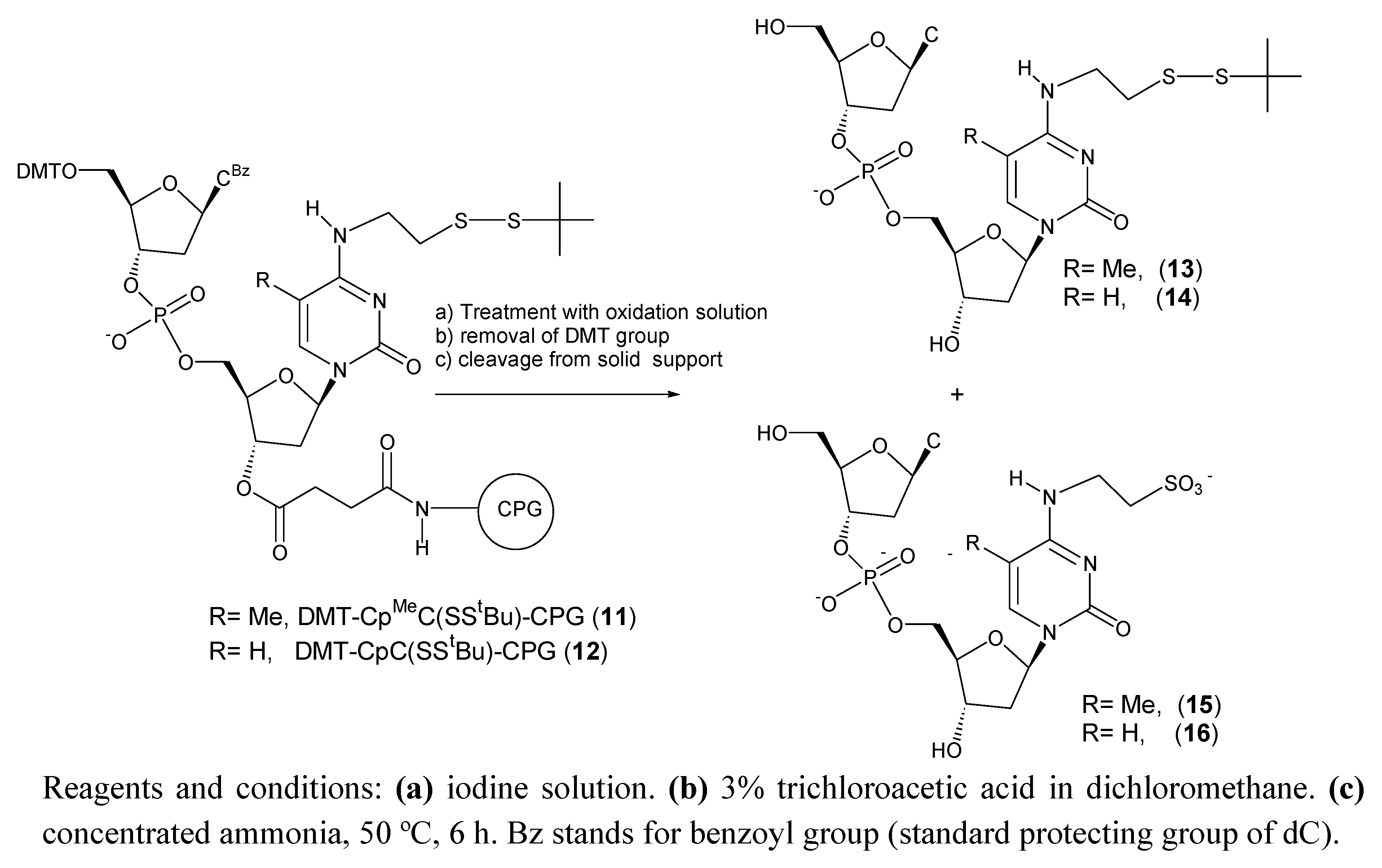
Next we studied the stability of the tBuS protected nucleosides on a dinucleotide sequence (DMT-CBzpMeC(SStBu)-CPG (11) and DMT-CBzpC(SStBu)-CPG (12)) to determine the effect a phosphate group in an environment similar to the one found inside of an oligonucleotide sequence. We used a similar protocol to the one described for the monomer except for the ammonia treatment that was performed at 50 ºC for 6 h (standard conditions for removal of nucleobase protecting groups).
Scheme 3.
Products obtained after treatment of DMT-CBzpMeC(SStBu)-CPG (11) and DMT-CBzpC(SStBu)-CPG (12) with iodine solution.
Scheme 3.
Products obtained after treatment of DMT-CBzpMeC(SStBu)-CPG (11) and DMT-CBzpC(SStBu)-CPG (12) with iodine solution.
Table 2 shows the results obtained in this study. The formation of the oxidation products were also observed but in a much lower efficiency. For example 1hr treatment of DMT-Cp
MeC(SS
tBu)-CPG (
11) with iodine solution yielded 19% of side compound (
Table 2). The same treatment on DMT-
MeC(SS
tBu)-CPG (
5) gave 97% of side product (
Table 1). A treatment of 3 h equivalent to 36 synthesis cycles generated a 38% of side compound (
Table 2). As observed in the monomer the side reaction is less efficient with the cytosine derivative than the 5-methylcytosine derivative. A treatment of 3 h of DMT-CpC(SS
tBu)-CPG (
12) with iodine solution generated a 12% of side compound instead of 38% that was observed for the support
11 (
Table 2). In summary the presence of a nucleotide attached at the 5’ position slows the efficiency of the oxidation of
tBuS protected nucleoside to sulfonic acid.
Table 2.
Products resulting from the treatment of DMT-CpMeC(SStBu)-CPG (11) and DMT-CpC(SStBu)-CPG (12) with iodine solution.
Table 2.
Products resulting from the treatment of DMT-CpMeC(SStBu)-CPG (11) and DMT-CpC(SStBu)-CPG (12) with iodine solution.
| Solid Support | Treatment | tBuSS- protected / oxidized (-SO3) |
|---|
| 11 | Iodine solution, 5 min | 96 : 4 |
| 11 | Iodine solution, 1 h | 81 : 19 |
| 11 | Iodine solution, 3 h | 62 : 38 |
| 12 | Iodine solution, 5 min | 100 : 0 |
| 12 | Iodine solution, 1 h | 93 : 7 |
| 12 | Iodine solution, 3 h | 88: 12 |
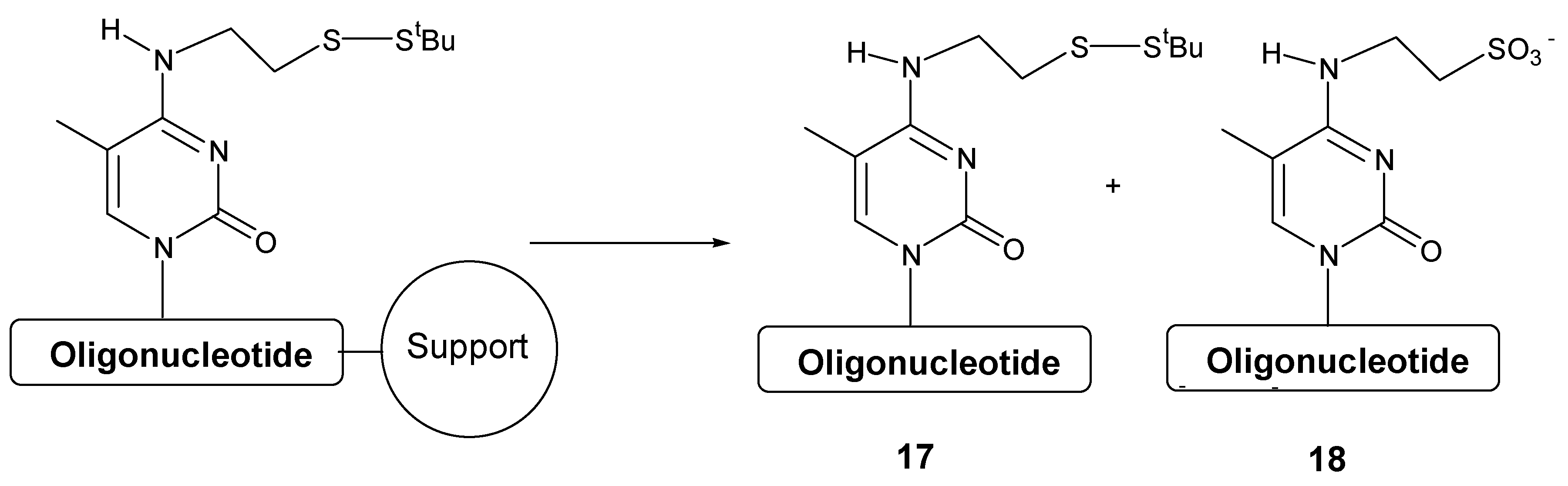
Next we studied the effect of the iodine or tert-butylhydroperoxide solutions in the purity of a short oligonucleotide containing one modified nucleoside in the middle of the sequence. Oligonucleotide sequence 5’-d(TTCCAXATTACCG)-3’ (being X the position of the N4- [2-(t-butyldisulfanyl)ethyl]-5-methylcytidine derivative) was prepared on 200 nmol scale using three different methods: a) LV200® polystyrene support using iodine solution; b) LV200® polystyrene support using tert-butylhydroperoxide solution and c) CPG support using iodine solution.
Scheme 4.
Potential products produced during the synthesis of oligonucleotides carrying N4- [2-(t-butyldisulfanyl)ethyl]-5-methylcytosine.
Scheme 4.
Potential products produced during the synthesis of oligonucleotides carrying N4- [2-(t-butyldisulfanyl)ethyl]-5-methylcytosine.
Table 3.
Products resulting from the synthesis of 5’-d(TTCCAXATTACCG)-3’.
Table 3.
Products resulting from the synthesis of 5’-d(TTCCAXATTACCG)-3’.
| Solid Support | Oxidation solution | tBuSS- protected 17 / oxidized 18 |
|---|
| polystyrene | Iodine | 41 : 59 |
| polystyrene | tert-Butylhydroperoxide | 1 : 99 |
| CPG | Iodine solution | 6 : 94 |
As it can be seen in
Table 3, the oxidation reaction is more pronounced on polystyrene support when iodine solution is used obtaining a 59% of the oligonucleotide carrying the sulfonic acid group
18. When using polystyrene and
tert-butylhydroperoxide solution as well as CPG and iodine solution only 1% and 6% of side compound
18 was observed. The formation of the side compound when using polystyrene support and iodine solution may be due to an increase of the local concentration of iodine by absorption of iodine on the polystyrene surface.
In order to analyze the stability of the disulfide bond in the presence of
tert-butylhydroperoxide , the oligonucleotide attached to polystyrene and oxidized with
tert-butylhydroperoxide was treated with the
tert-butylhydroperoxide solution for 6 and 12 h more. The resulting crudes were analyzed by HPLC.
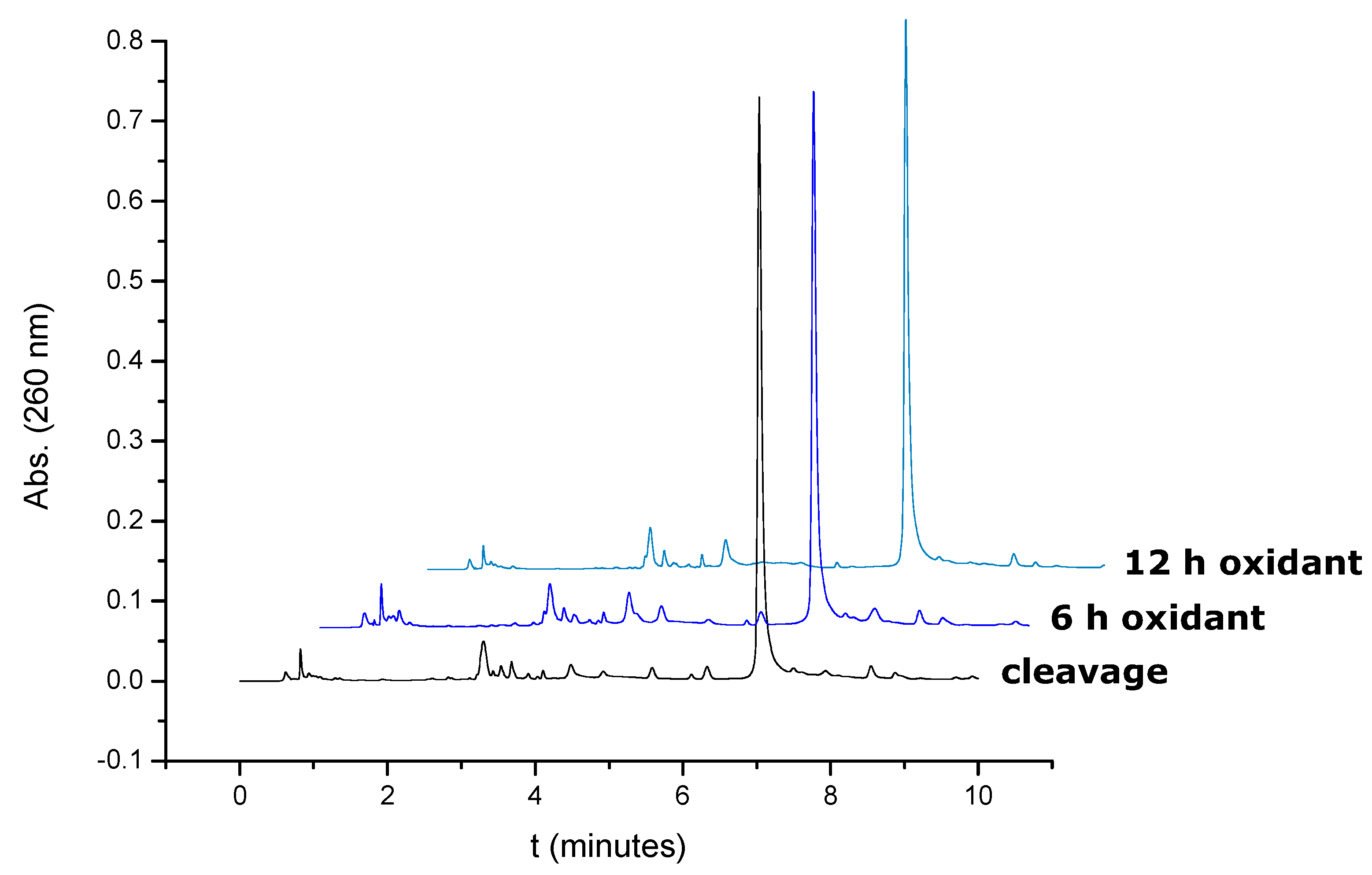
Figure 1 shows that the disulfide bond is stable to
tert-butylhydroperoxide solution at prolonged periods of time as no increase of the peaks around the elution of the oxidation product was observed.
Figure 1.
HPLC profiles of oligonucleotide 5’-d(TTCCAXATTACCG)-3’ after synthesis and after prolonged treatment of the support with tert-butylhydroperoxide solution for 6 and 12 h.
Figure 1.
HPLC profiles of oligonucleotide 5’-d(TTCCAXATTACCG)-3’ after synthesis and after prolonged treatment of the support with tert-butylhydroperoxide solution for 6 and 12 h.
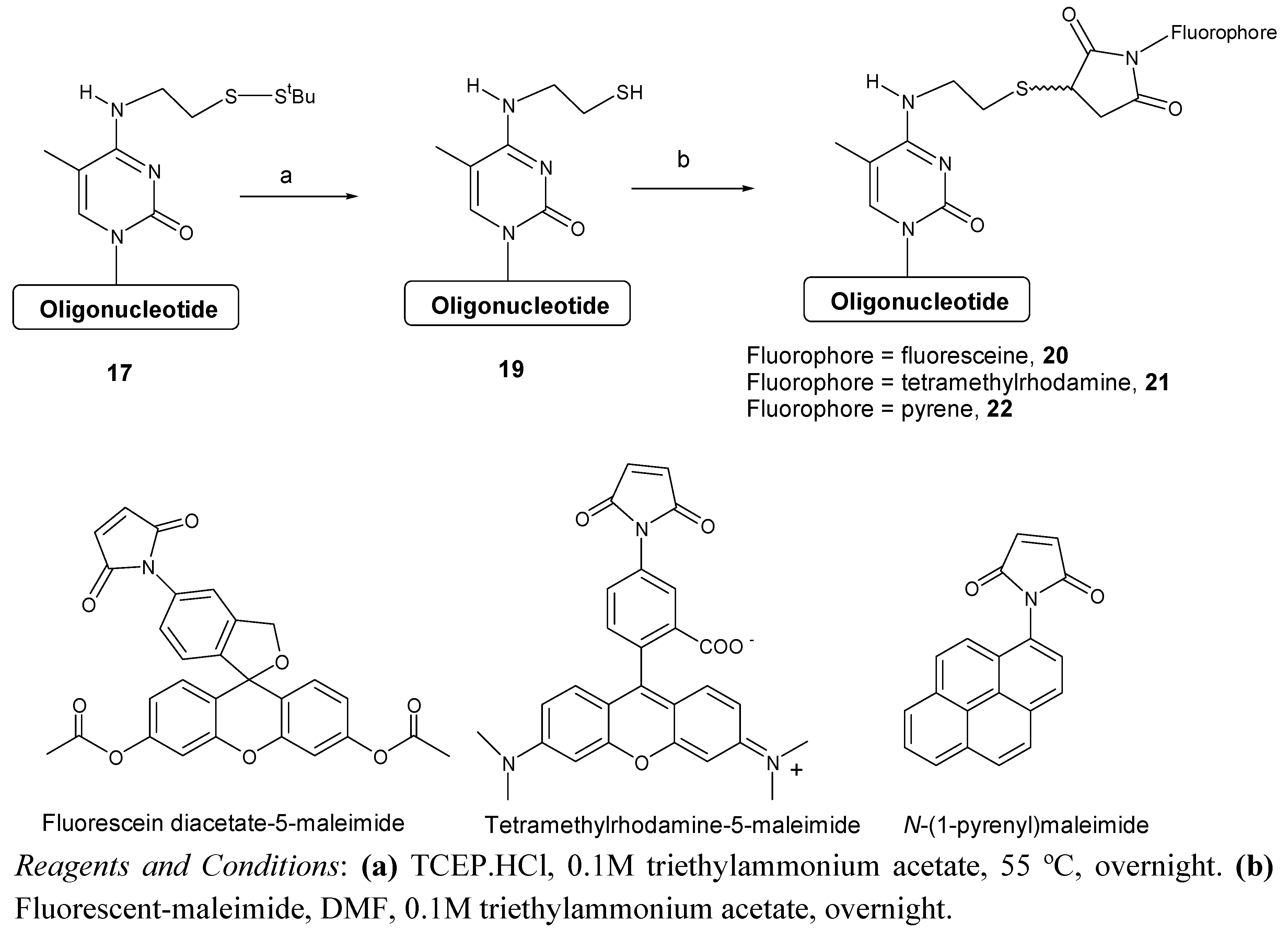
Finally, the use of the modified oligonucleotide for the preparation of oligonucleotides carrying fluorescent compounds was demonstrated. To this end oligonucleotide
17 was treated with tris(2-carboxyethyl)phosphine to remove the
tBuS group and the resulting free thiol oligonucleotide
19 was reacted with three different fluorescent maleimides (
Scheme 5). The desired fluorescent oligonucleotides were obtained as a mixture of isomers that were characterized by UV and mass spectrometry analysis. In the case of the fluorescein diacetate 5-maleimide we observed that the acetate groups are partially hydrolyzed in the reaction conditions yielding a complex HPLC profile (see
supplementary material). A short treatment with a bicarbonate solution resulted on the total hydrolysis of the acetate groups obtaining a more simple HPLC profile. Coupling efficiency based on the integration of the HPLC peaks was between 80-84% (
Table 4).
Table 4.
Oligonucleotide carrying fluorescent compounds prepared in this study.
Table 4.
Oligonucleotide carrying fluorescent compounds prepared in this study.
| Compound | Fluorophore | Yield (%)* | MS (expected) | MS (found) |
|---|
| 20 | Fluoresceine | 82 | 4372.1 | 4378.6 / 4369.4# |
| 21 | Tetramethylrhodamine | 80 | 4424.2 | 4422.6 |
| 22 | Pyrene | 84 | 4240.0 | 4240.5# |
Scheme 5.
Synthesis of oligonucleotides carrying fluorescent molecules.
Scheme 5.
Synthesis of oligonucleotides carrying fluorescent molecules.
3. Experimental
3.1. General
All reagents were purchased from Aldrich, Sigma or Fluka (Sigma-Aldrich Química S.A., Spain) and were used without further purification. Phosphoramidites and ancillary reagents used during oligonucleotide synthesis were from Applied Biosystems (PE Biosystems Hispania S.A., Spain). Flash column chromatography was carried out on silica gel SDS 0.063-0.2 mm/70-230 mesh. NMR spectra were recorded on a Varian Mercury 400 spectrometer operating at 400 MHz (
1H) and 100 MHz (
13C). Chemical shifts are reported in ppm relative to the singlet at
δ = 7.24 ppm of CHCl
3 for
1H-NMR and to the centre line of the triplet at
δ = 77.0 ppm of CDCl
3 for
13C-NMR. J values are given in Hz.
31P-NMR spectra were recorded on a Varian Inova 300 spectrometer. HPLC separations were performed using a Waters HPLC system. MALDI mass spectrometry was recorded on a Fisons VG Tofspec spectrometer and ESI mass spectra on a Fisons VG Platform II spectrometer. Oligonucleotide sequences were prepared using solid phase methodology. The syntheses of oligonucleotides were carried out on an Applied Biosystems Model 3400 DNA synthesizer. Details of the synthesis of 2-aminoethyl-tert-butyl disulfide (
1) [
15] and DMT-T (
2) are described as
supplementary material. HPLC profiles are shown as
supplementary material.
3.2. Synthesis of 2’-deoxy-5’-O-(4,4’-dimethoxytriphenylmethyl)-N4-(tert-butyldithio-ethyl)-5-methyl-cytidine (3)
DMT-T (2, 1.25 g, 2.30 mmol) was dried by evaporation of anhydrous ACN under reduced pressure, and the residue was dissolved in anhydrous DMF (30 mL) under argon. Hexamethyldisilazane (0.96 mL 4.6 mmol) was added with a syringe to the solution with exclusion of moisture. After 30 min of magnetic stirring at room temperature, the reaction was complete as judged by TLC (ethyl acetate, Rf = 0.61). Then, the solution was evaporated under reduced pressure. The residue was dissolved in toluene (4 × 10 mL) and concentrated to dryness. 1,2,4-Triazole (1.67 g, 24.15 mmol) was dried by evaporation of anhydrous ACN under reduced pressure, and the residue was dissolved in anhydrous ACN (40 mL) under argon. Triethylamine (3.7 mL, 26.45 mmol) was added and the solution was cooled on ice. Then, phosphorus oxychloride (0.53 mL, 5.75 mmol) was added with a syringe to the solution with exclusion of moisture. After 30 min of magnetic stirring at T = 0 ºC, the protected nucleoside dissolved in anhydrous ACN (40 mL) was added dropwise to the solution under argon. The reaction mixture was stirred at room temperature. After 6 hours, the reaction was complete as judged by TLC (ethyl acetate, Rf = 0.39). The solvent was removed under reduced pressure and the residue dissolved in DCM (50 mL). The solution was washed with saturated aqueous NaCl (50 mL). After drying the organic phase with Na2SO4, the solvent was evaporated under reduced pressure. The residue was dissolved in dry DMF (20 mL) under argon. Compound 1 (0.57 g, 3.45 mmol) and DBU (0.52 mL, 3.45 mmol) were added to the solution. The reaction mixture was stirred overnight at room temperature. DBU was added (0.34 mL, 2.30 mmol) and the reaction mixture was stirred at room temperature. After 6 hours, the solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate (100 mL). The solution was washed with saturated aqueous sodium chloride (100 mL). After drying the organic phase with sodium sulphate, the solvent was evaporated under reduced pressure. The residue was dissolved in a small amount of ethyl acetate and purified by chromatography on silica gel. The column was packed with silica gel using a 1% triethylamine solution in ethyl acetate. The product was eluted with a gradient of methanol from 0 to 5% in ethyl acetate. The pure compound was obtained as pale yellow foam (0.85 g, 53%). TLC (3% methanol in ethyl acetate) Rf = 0.25. 1H-NMR, δH (CDCl3): 7.61 (s, 1H), 7.45-6.70 (m, 13H), 6.38 (t, 1H, J = 6.6 Hz), 5.28 (t, 1H, J = 5.6 Hz), 4.50-4.47 (m, 1H), 4.04-4.01 (m, 1H), 3.79 (t, 2H, J = 6.0 Hz), 3.72 (s, 6H), 3.40 (dd, 1H, J = 10.4 and 3.2 Hz), 3.28 (dd, 1H, J = 10.6 and 3.0 Hz), 2.84 (t, 2H, J = 6.0 Hz), 2.55-2.50 (m, 1H), 2.20-2.13 (m, 1H), 1.43 (s, 3H), 1.26 (s, 9H). 13C-NMR, δC (CDCl3): 163.27 (C), 158.82 (CH), 156.54 (C), 144.76 (C), 137.77 (CH), 135.85 (C), 130.33 (CH), 128.39 (CH), 128.15 (CH), 127.21 (C), 113.45 (CH), 102.29 (C), 86.91 (C), 86.17 (CH), 86.16 (CH), 72.42 (CH), 63.83 (CH2), 55.48 (CH3), 48.53 (C), 42.30 (CH2), 39.87 (CH2), 39.27 (CH2), 30.11 (CH3), 12.65 (CH3); ESI-MS m/z (+ve mode [M+H]+) calc. for C37H45N3O6S2 691.90, found 692.28.
3.3. Synthesis of 2’-deoxy-5’-O-(4,4’-dimethoxytriphenylmethyl)-N4-(2-aminoethyldi-thioethyl)-5-methylcytidine-3-O-(2-cyanoethyl-N,N’-diisopropylphosphoramidite) (5)
Compound 3 (200 mg, 0.22 mmol) was dried by evaporation of anhydrous ACN in vacuo, and the residue was dissolved in anhydrous DCM (10 mL) under argon. N,N’-diisopropylethylamine (154 µL, 0.88 mmol) was added with exclusion of moisture. The solution was cooled on ice and 2-cyanoethoxy-N,N’-diisopropylaminochlorophosphine (73 µL, 0.33 mmol) was added dropwise with a syringe. Afterward, the solution was stirred at room temperature for 1 h. After this time, TLC (1% cyclohexane in ethyl acetate) showed the presence of the starting compound. As a consequence more 2-cyanoethoxy-N,N-diisopropylaminochlorophosphine (26 μL, 0.11 mmol) once the mixture was cooled on ice. After the addition the reaction mixture was allowed to stir at room temperature for 1 h. The solvent was removed in vacuo and the residue was dissolved in dichloromethane (20 mL). The solution was washed with 5% aqueous sodium hydrogen carbonate (20 mL) and with saturated aqueous sodium chloride (20 mL). After drying the organic phase with sodium sulphate, the solvent was evaporated under reduced pressure. The residue was dissolved in a small amount of ethyl acetate/cyclohexane 2:1 and purified by chromatography on silica gel. The column was packed with silica gel using a 5% triethylamine solution in ethyl acetate/cyclohexane 2:1. The product was eluted with ethyl acetate/cyclohexane 2:1. The pure compound was obtained as pale yellow foam (220 mg, 86%). TLC (1% cyclohexane in ethyl acetate) Rf = 0.39 and 0.26. 1H-NMR, δH (CDCl3): Most of signals are duplicated due to the presence of diastereoisomers 7.72 and 7.65 (2s, 1H), 7.43-6.81 (m, 13H), 6.47 and 6.43 (2t, 1H, J = 6.4 and 6.4 Hz respectively), 4.66-4.57 (m, 1H), 4.23-4.01 (m, 1H), 3.87-3.91 (m, 2H), 3.80 and 3.79 (2s, 6H), 3.61-3.46 (m, 4H), 2.93 (t, 2H, J = 5.8 Hz), 2.76 (td, 2H, J = 6.2 and 2.0 Hz), 2.66-2.56 (m, 1H), 2.61 and 2.38 (2t, 1H, J = 6.4 and 6.4 Hz respectively), 2.30-2.20 (m, 1H), 1.46 and 1.43 (2s, 3H), 1.34 (s, 9H), 1.29-1.14 (m, 12H). 13C-NMR, δC (CDCl3): Most of signals are duplicated due to the presence of diastereoisomers 163.24 and 163.22 (C), 158.89 and 158.87 (CH), 156.37 and 156.33 (C), 144. 63 (C), 137.65 and 137.60 (CH), 135.75 and 135.73 (C), 130.46 and 130.37 (CH), 128.56 and 128.46 (CH), 128.13 (CH), 127.29 and 127.25 (C), 117.81 (C), 113.49 (C), 113.42 (CH), 102.18 and 102.06 (C), 86.90 and 86.88 (C), 85.79 (CH), 85.40 and 85.28 (2d, J = 4.7 and 5.8 Hz), (CH), 73.78 and 72.76 (2d, J = 8.2 and 7.2 Hz) (CH), 63.34 and 62.84 (CH2), 58.58 and 58.37 (2d, J = 10.9 and 11.3 Hz) (CH2), 55.49 and 55.46 (CH3), 48.52 (C), 45.57 and 45.51 (CH2), 43.49 and 43.37 (2d, J = 9.2 and 9.7 Hz) (CH), 43.41 and 43.32 (CH), 39.81 (CH2), 39.32 (CH2), 30.11 (CH3), 24.76 and 24.75 (2d, J = 8.2 Hz and 7.2 Hz) (CH3), 23.17 and 23.14 (2d, J = 8.2 and 8.7 Hz) (CH3), 20.35 and 20.33 (2d, J = 7.1 and 6.9 Hz) (CH2), 12.57 and 12.55 (CH3). 31P NMR, δP (CDCl3, 81 MHz): 150.03 and 149.61, two diastereoisomers. ESI-MS m/z (+ve mode) [M+H]+ = 892.40, (M = 892.12 g/mol calculated for C46H62N5O7PS2).
3.4. Preparation of the solid support functionalized with 2’-deoxy-5’-O-(4,4’-dimethoxytriphenyl-methyl)-N4-(2-aminoethyldithioethyl)-5-methylcytidine (DMT-MeC(SStBu)-succinyl-CPG) (5)
5’-DMT-N4-(2-aminoethyldithioethyl)-5-methylcytidine was incorporated on a long-chain alkylamine-controlled pore glass support (LCAA-CPG), following the standard methodology via monosuccinate derivative 4. Compound 3 (50 mg, 0.07mmol) was dried by evaporation of anhydrous ACN under reduced pressure, and the residue was dissolved in anhydrous pyridine (5 mL) under argon. Succinic anhydride (18 mg, 0.18 mmol) and 4-dimethylaminopyridine (DMAP) (5 mg, 0.04 mmol) were added to the solution. After 4 hours of magnetic stirring at room temperature, the reaction was complete as judged by TLC (5% methanol in ethyl acetate) Rf = 0.11. The solvent was removed under reduced pressure and the residue was dissolved in DCM (20 mL). The solution was washed with saturated aqueous sodium chloride (15 mL). After drying the organic phase with sodium sulphate, the solvent was evaporated under reduced pressure. The monosuccinate derivative 4, which was used in the next step without further purification, was obtained as a pale yellow foam. 250 mg of commercial LCAA-CPG (CPG New Jersey, 73 µmol amino/g) were placed into a polypropylene syringe fitted with a polypropylene disc and washed sequentially with DMF, methanol, THF, DCM, and ACN. Then, a solution of 5’-DMT-N4-(2-aminoethyldithioethyl)-5-methylcytidine-3’-mono-succinate (4, 22 mg, 27 µmol), hydroxybenzotriazole (HOBt) (4 mg, 27 µmol) and diisopropyl-carbodiimide (DIP) (23 µL, 146 µmol) in 700 µL of DMF was prepared. The solution was added to the resin and left to react for 2 h. The resin was washed with DMF, methanol, DCM and ACN. After washings, the resin was treated with 500 µL of Ac2O/DMF 1:1 to cap free amino groups. The incorporation of nucleoside was determined by DMT quantification (34 µmol/g).
3.5. Study of the stability of DMT-MeC(SStBu)-CPG under oxidative conditions and comparison with DMT-C(SStBu)-CPG
The solid support functionalized with the cytidine derivative, 5’-DMT-
N4-(2-aminoethyldithioethyl)cytidine (DMT-C(SS
tBu)-CPG) (
6) was synthesized as described in [
15]. Five mg of each solid support (DMT-C(SS
tBu)-CPG) (
6) and (DMT-
MeC(SS
tBu)-CPG) (
5) were treated with oxidation solutions (iodine or
t-butylhydroperoxide). Iodine solution was the same solution used in synthesis of oligonucleotides (0.02 M iodine in water/ pyridine/ tetrahydrofuran). The 10%
t-butylhydroperoxide solution was prepared freshly by mixing 86.4 mL of acetonitrile and 14.3 mL of commercially available 70%
t-butylhydroperoxide solution in water. At different time intervals the solution were filtered and the resulting solid support washed with ACN. The resulting supports were treated with a solution of trichloroacetic acid 3% in DCM for 5 min to remove the DMT group and with aqueous concentrated ammonia for 30 min to cleave the product from the resin. The mixture was filtered and the ammonia solution was concentrated to dryness. The resulting products were analyzed by HPLC (Column: X-Bridge
TMOST C
18 (2.5 μm; 4.6 × 50 mm); solvent A: 100 mM triethylammonium acetate (pH = 7) and solvent B: 70% ACN in 100 mM triethylammonium acetate (pH = 7). Flow rate: 1 mL/min. Conditions: 2.5 min 100% A, then 5.5 min linear gradient from 0-12.5% B, then 12 min linear gradient from 12-100% B, and mass spectrometry. HPLC profiles are shown as
supplementary material. Compound
7: t
R = 13.6 min, ESI-MS m/z (negative mode [M-H]
-) calc for C
16H
27N
3O
4S
2 389.53, found 388.14. Compound
8: t
R = 13.2 min, ESI-MS m/z (negative mode [M-H]
-) calc for C
15H
25N
3O
4S
2 375.51, found 373.17. Compound
9: t
R = 6.2 min, ESI-MS m/z (negative mode [M-H]
-) calc for C
12H
19N
3O
7S 349.35, found 348.08. Compound
10: t
R = 4.7 min, ESI-MS m/z (negative mode [M-H]
-) calc for C
11H
17N
3O
7S 335.33, found 334.08.
3.6. Analysis of the stability of DMT-Cp(MeC(SStBu))-CPG and DMT-Cp(C(SStBu))-CPG solid supports to oxidative conditions
DMT-dCBz phosphoramidite was incorporated in the solid supports 5 and 6 previously synthesized in a DNA synthesizer (Applied Biosystems 3400) on 1 μmol scale using commercially available chemicals. The synthesis was carried out using the standard phosphite triester methodology. For the oxidation step was used a solution of tert-butyl hydroperoxide 10% in ACN instead of the commercially available solution of iodine 0.02 M. In both cases the synthesis was carried out without the removal of the 5’-DMT group.
The analysis of the stability of the thiolated nucleosides under oxidation conditions were studied as described for supports
5 and
6. Five mg of each solid support were treated with iodine solution. At different time intervals the solution were filtered and the resulting solid support washed with acetonitrile. The resulting supports were treated with a solution of trichloroacetic acid 3% in DCM for 5 min to remove the DMT group and with aqueous concentrated ammonia at 55 ºC for 6 h to cleave the products from the support and to remove the benzoyl group. The mixtures were filtered and the ammonia solutions were concentrated to dryness. The resulting products were characterized by HPLC (Column: X-Bridge
TMOST C
18 (2.5 μm, 4.6 × 50 mm); solvent A: 100 mM triethylammonium acetate (pH = 7) and solvent B: 70% ACN in 100 mM triethylammonium acetate (pH = 7). Flow rate: 1 mL/min. Conditions: 2.5 min 100% A, then 5.5 min linear gradient from 0-12.5% B, then 12 min linear gradient from 12-100% B, and mass spectrometry. HPLC profiles are shown as
supplementary material. Compound
13: t
R = 12.4 min, ESI-MS m/z (negative mode [M-H]
-) calc for C
25H
39N
6O
10PS
2 678.71, found 677.17. Compound
14: t
R = 12.2 min, ESI-MS m/z (negative mode [M-H]
-) calc for C
24H
37N
6O
10PS
2 664.68, found 663.18. Compound
15: t
R = 7.6 min, ESI-MS m/z (negative mode [M-H]
-) calc for C
21H
31N
6O
13PS 638.53, found 637.13. Compound
16: t
R = 7.0 min, ESI-MS m/z (negative mode [M-H]
-) calc for C
20H
29N
6O
13PS 624.50, found 623.13.
3.7. Synthesis of oligonucleotides containing a residue of N4-mercaptoethyl-5-methylcytosine
The oligonucleotide with the sequence
5’TTCCAXATTACCG
3’, where X stands for the modified nucleoside, was synthesized on 0.2 µmol scale employing three methodologies:
- a)
LV200® polystyrene, iodine solution, 1 min.
- b)
CPG; iodine solution, 1 min.
- c)
LV200® polystyrene, 10 % of tert-butylhydroperoxide in ACN, 15 min.
The protecting group for dC and dA was the benzoyl (Bz) group. The isobutyryl (
ibu) group was used for protection of dG. In all cases, at the end of the synthesis the DMT group was removed. The coupling efficiency for the modified phosphoramidite was between 95-99% (as measured by the absorbance of the DMT group). After the assembly of the sequence the solid supports were treated with concentrated aqueous ammonia for 6 h at 55 ºC. Under these conditions, all base and phosphate groups were also removed. The mixtures were filtered and the ammonia solutions were concentrated to dryness. The resulting crudes were analyzed by HPLC (Column: X-Bridge
TMOST C
18 (2.5 μm; 4.6 × 50 mm); solvent A: 5% ACN in 100 mM triethylammonium acetate (pH = 7) and solvent B: 70% ACN in 100 mM triethylammonium acetate (pH = 7). Flow rate: 1 mL/min. Conditions: 10 min linear gradient from 0-30%B. The different peaks observed in the HPLC profiles were analyzed by mass spectrometry. HPLC profiles are shown as
supplementary material. Compound
17: t
R = 7.5 min, MALDI-TOF m/z (negative mode [M-H]
-) calc for C
132H
175N
43O
77P
12S
2 4031.90, found 4032.52. Compound
18: t
R = 4.9 min, ESI-MS m/z (negative mode [M-H]
-) calc for C
128H
166N
6O
80P
12S 3991.80, found 3996.15.
3.8. Synthesis of oligonucleotide-fluorophore conjugates
The oligonucleotide obtained before was used to perform the reactions with fluorescein diacetate 5-maleimide, tetramethylrhodamine-5-maleimide and
N-(1-pyrenyl)maleimide For the preparation of each conjugate, 1.4 OD
260 units of oligonucleotide
17 were used. To cleave the disulfide bond, compound
17 was dissolved in 0.3 mL of 0.1 M triethylammonium acetate solution (pH = 7). Afterward, 34 μL of a 0.1 M tris(2-carboxyethyl)phosphine hydrochloride (TCEP. HCl) solution were added to the solution and allow to react at 55 ºC overnight. Under these conditions, the
tert-butylthiol group was completely removed. The resulting product was purified with Sephadex G-25 (NAP-5 column). The oligonucleotide
19 was eluted with 1 mL of sterile water. The solution was analyzed by HPLC (Column: X-Bridge
TMOST C
18 (2.5 μm, 4.6 × 50 mm); solvent A: 5% ACN in 100 mM triethylammonium acetate (pH = 7) and solvent B: 70% ACN in 100 mM triethylammonium acetate (pH = 7). Flow rate: 1 mL/min. Conditions: 10 min linear gradient from 5-40% B. Under these conditions the protected oligonucleotide eluted at t
R = 4.9 min and the deprotected oligonucleotide at t
R = 3.0 min. The solution was concentrated to 500 μL and 50 μL of a 1M triethylammonium acetate solution were added. Then, 100 μL of a solution 0.56 mM fluorescein diacetate 5-maleimide, tetramethylrhodamine-5-maleimide or
N-(1-pyrenyl)maleimide in DMF were added and allowed to react at room temperature overnight. The crude oligonucleotide was concentrated to dryness and the excess of reagents and salts were removed by using a NAP-5 column. The oligonucleotide was eluted with 1 mL of sterile water. All crudes were analyzed by HPLC using the conditions described above. HPLC profiles are shown as
supplementary material.
3.8.1. Conjugation with fluorescein diacetate 5-maleimide
The HPLC analysis showed the presence of four products related with the oligonucleotide-fluoresecein conjugate with tR = 6.6 min, (λmax = 262 and 451 nm), tR = 4.1 min, (λmax = 262 and 493 nm), tR = 3.6 min, (λmax = 262 and 490 nm) and tR = 3.4 min, (λmax = 262 and 486 nm). The analysis of all four bands by EM (MALDI-TOF) indicated that the high tR band corresponded to the monodeacetylated form of the oligonucleotide-fluorescein conjugate (20d). The band collected at tR = 4.1 min corresponded to the diacetylated form of the conjugate (20c). The other two bands (20a and 20b) shared the same mass found in 20c. To the resulting crude of synthesis were added 100 μL of a solution of 2 M sodium bicarbonate and left to react for 1 h at 55 ºC. The crude oligonucleotide was concentrated to dryness and the excess of salts were removed by using a NAP-5 column. The oligonucleotide was eluted with 1 mL of sterile water. The HPLC analysis of the crude revealed the presence of only two products related with the oligonucleotide-fluorescein conjugate, 20a and 20b. The coupling efficiency was determined from the HPLC profiles (~82%). Compound 20a: tR = 3.4 min, MALDI-TOF m/z (negative mode [M-H]-) calc for C152H180N44O84P12S 4372.08, found 4378.56. Compound 20b: tR = 3.6 min, MALDI-TOF m/z (negative mode [M-H]-) calc for C152H180N44O84P12S 4372.08, found 4369.44. Compound 20c: tR = 4.1 min, ESI-MS m/z (negative mode [M-H]-) calc for C152H180N44O84P12S 4372.08, found 4370.02. Compound 20d: tR = 6.6 min, ESI-MS m/z (negative mode [M-H]-) calc for C154H192N44O85P12S 4414.12, found 4413.63.
3.8.2. Conjugation with tetramethylrhodamine-5-maleimide
The HPLC analysis of the crude revealed the presence of four bands between tR = 7.6 min and tR = 8.3 min. Only the band with tR = 7.9 min showed an UV spectra consistent with an oligonucleotide (λmax = 262 nm) with rhodamine (λmax = 554 nm). The other three peaks showed λmax around 556 nm without the maxima at 260 nm, indicating that they are tetramethylrhodamine-5-maleimide derivatives that could not be removed with a NAP-5 column. This is due to the positive charge of tetramethylrhodamine that interacts with the negative charge of the phosphate bonds. The conjugate was characterized as well by EM (MALDI-TOF). The coupling efficiency was determined from the HPLC profile (~80%). Compound 21: tR = 7.9 min, ESI-MS m/z (negative mode [M-H]-) calc for C156H189N46O82P12S 4424.20, found 4422.58.
3.8.3. Conjugation with N-(1-pyrenyl)maleimide
The crude was analyzed by HPLC showing at least four bands (tR = 6.9, 7.3, 7.7 and 7.8 min) sharing the same UV spectra (λmax = 263 and 341 nm) and the same mass by mass spectrometry. All four bands corresponded to the oligonucleotide-pyrene conjugate (22), being different isomers. The coupling efficiency was determined from the HPLC profile (~84%). Compound 22: tR = 6.9, 7.3, 7.7 and 7.8 min, ESI-MS m/z (negative mode [M-H]-) calc for C148H177N44O79P12S 4240.01, found 4240.52.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



