Design and Synthesis of New N-(5-Trifluoromethyl)-1H-1,2,4-triazol-3-yl Benzenesulfonamides as Possible Antimalarial Prototypes

Abstract
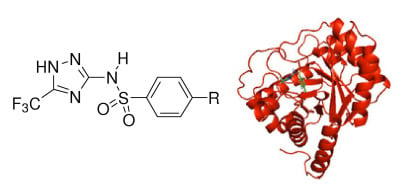
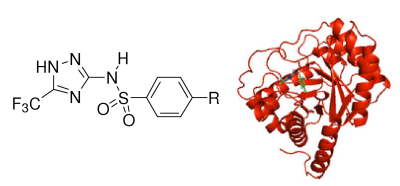
:
1. Introduction

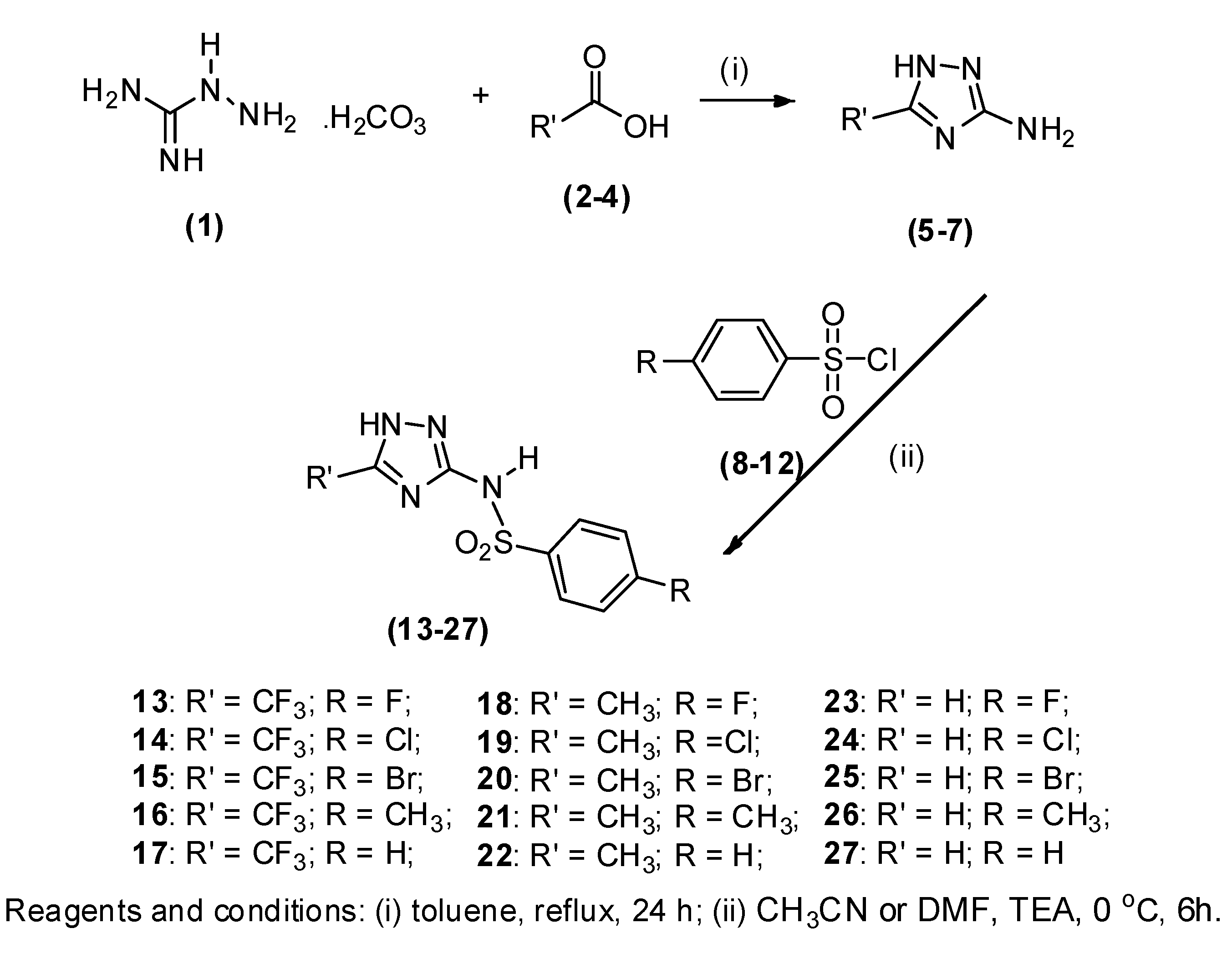
1.1. Chemistry
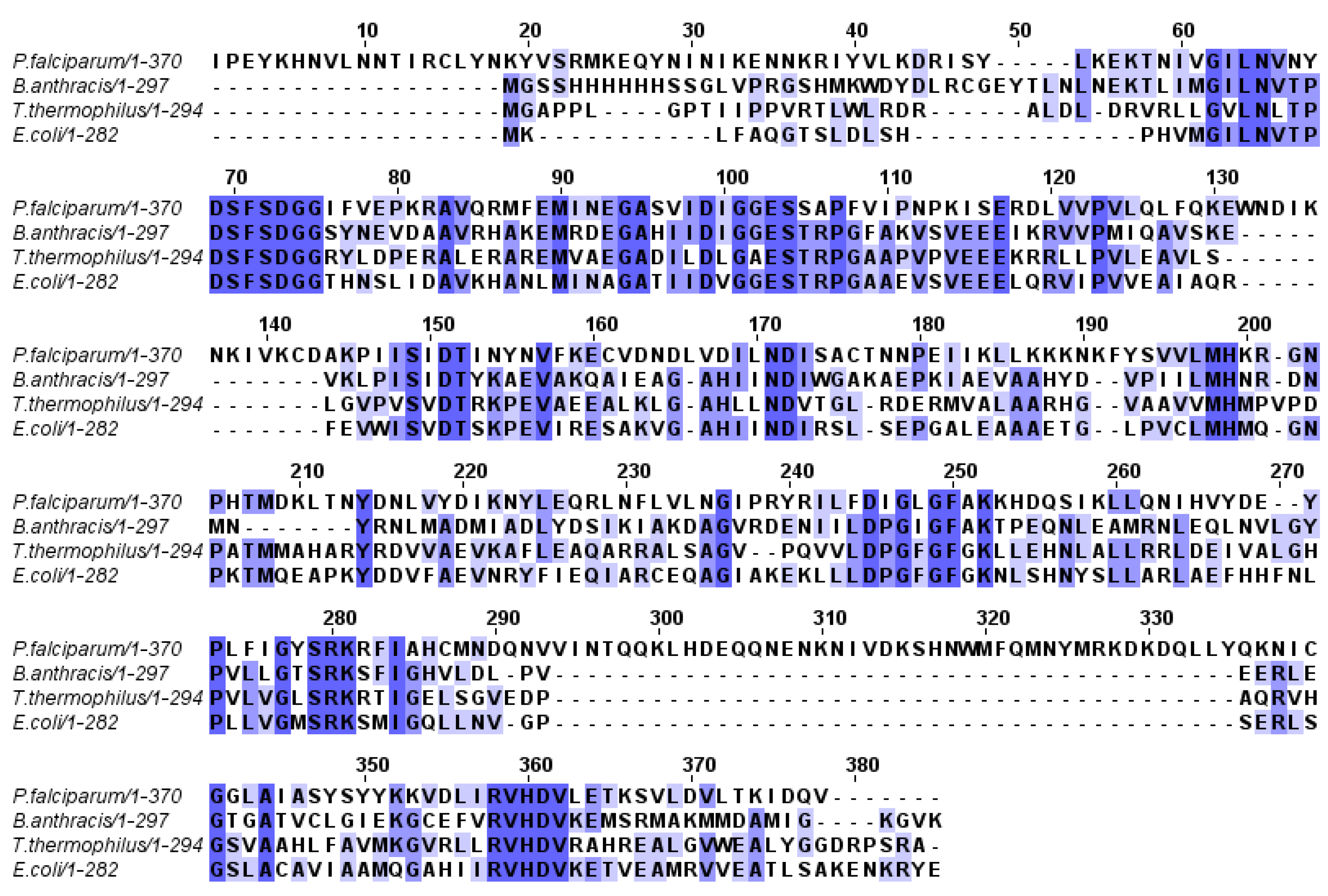
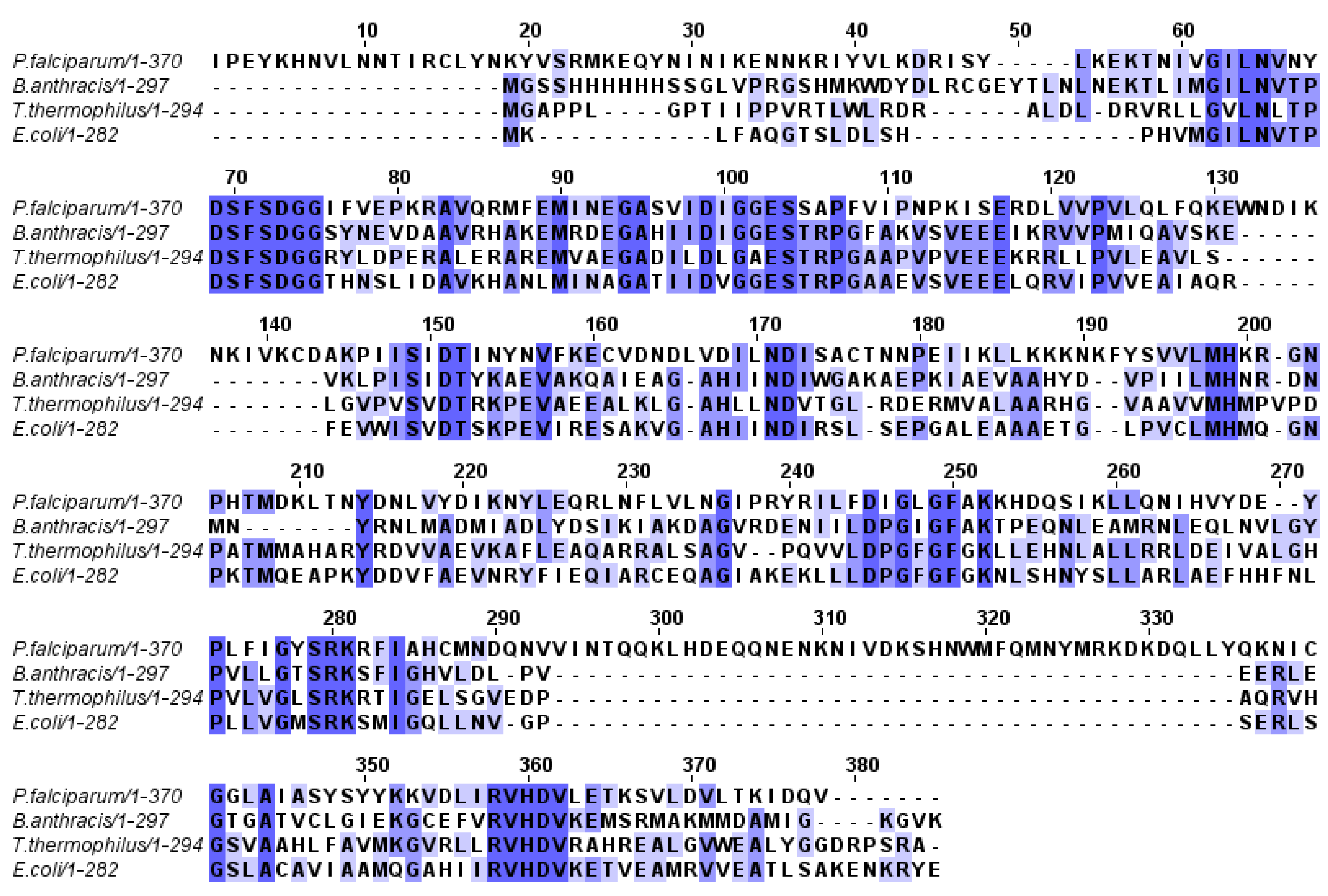
1.2. Molecular Modeling and Docking Simulation

2. Results and Discussion
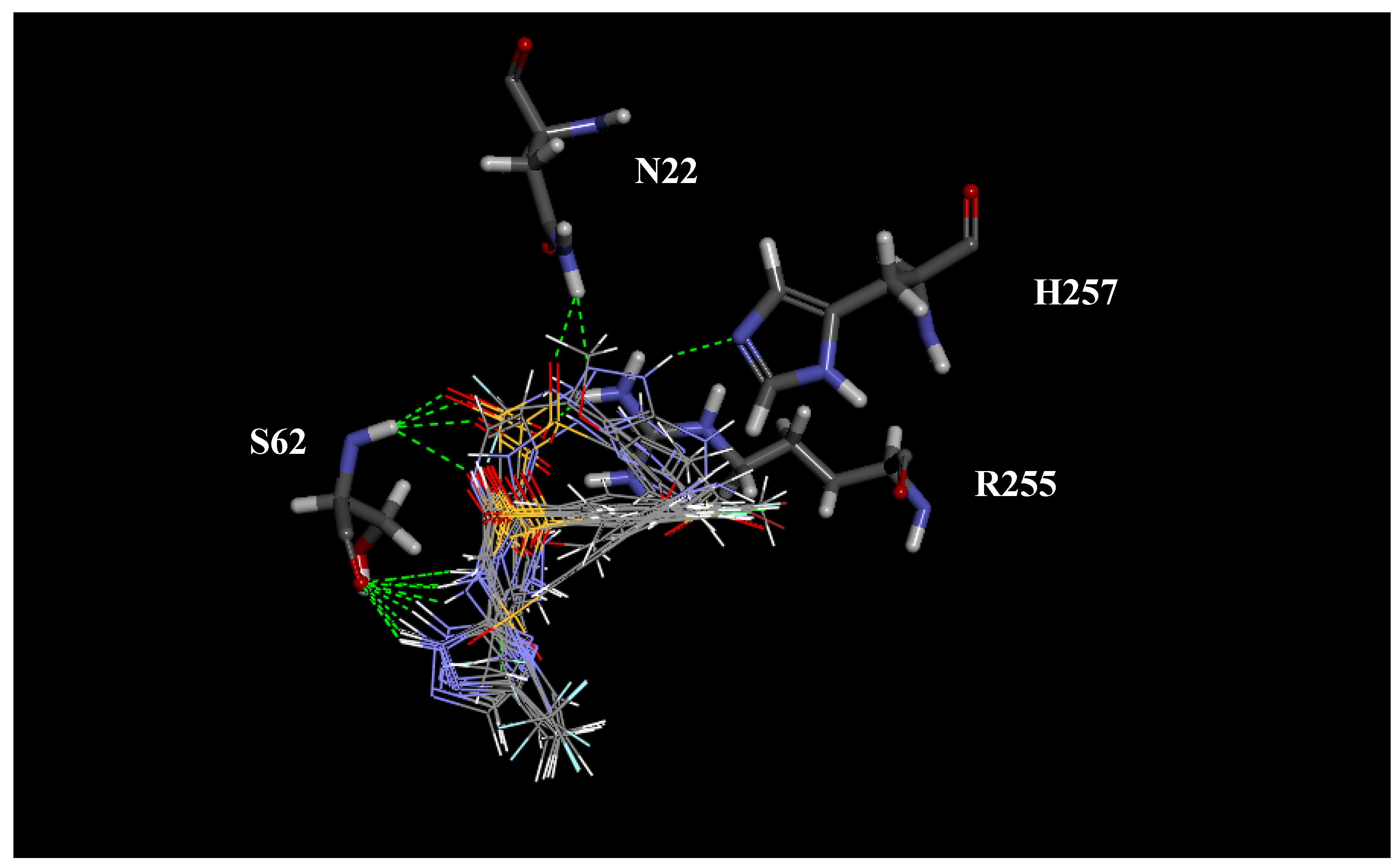
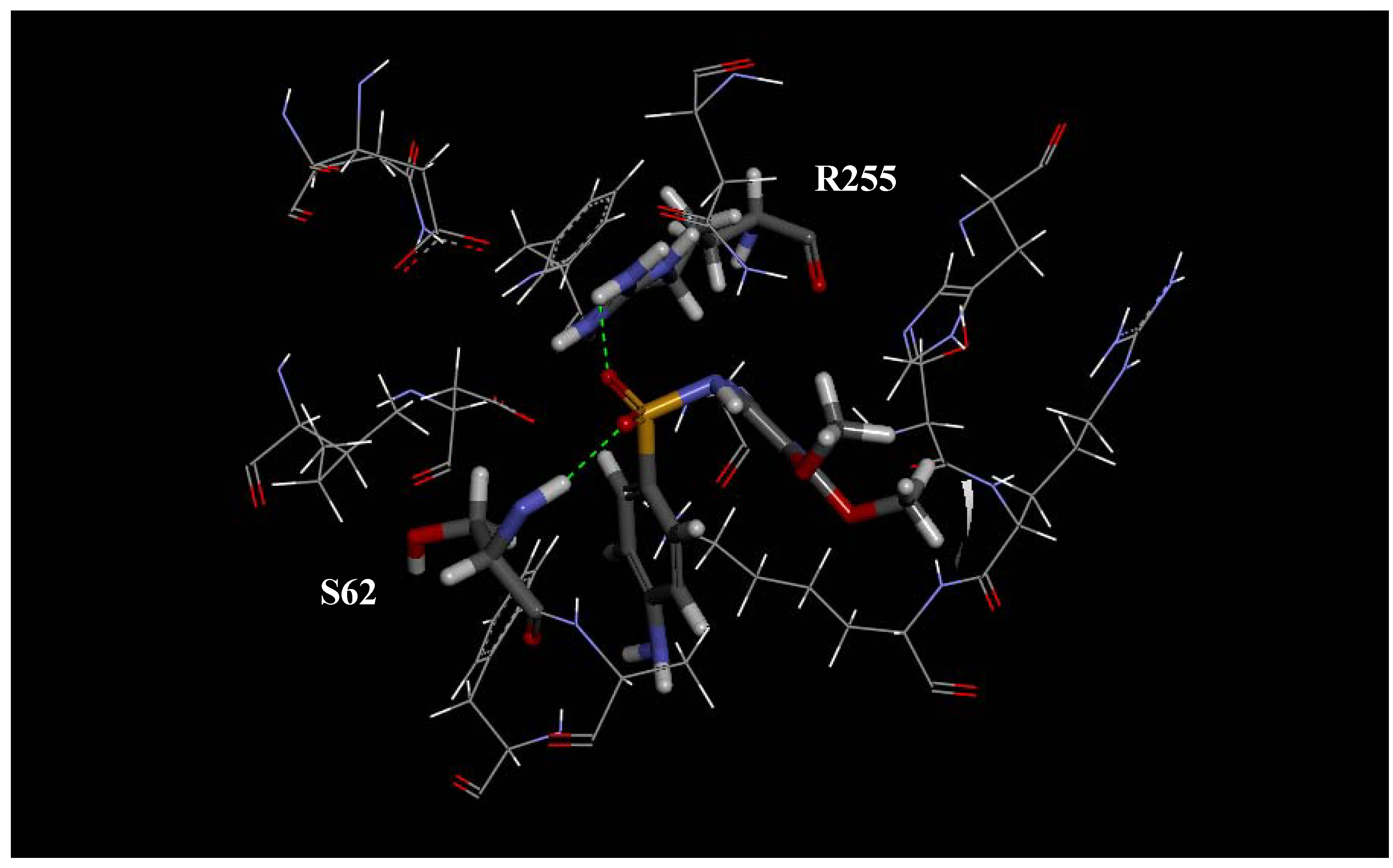
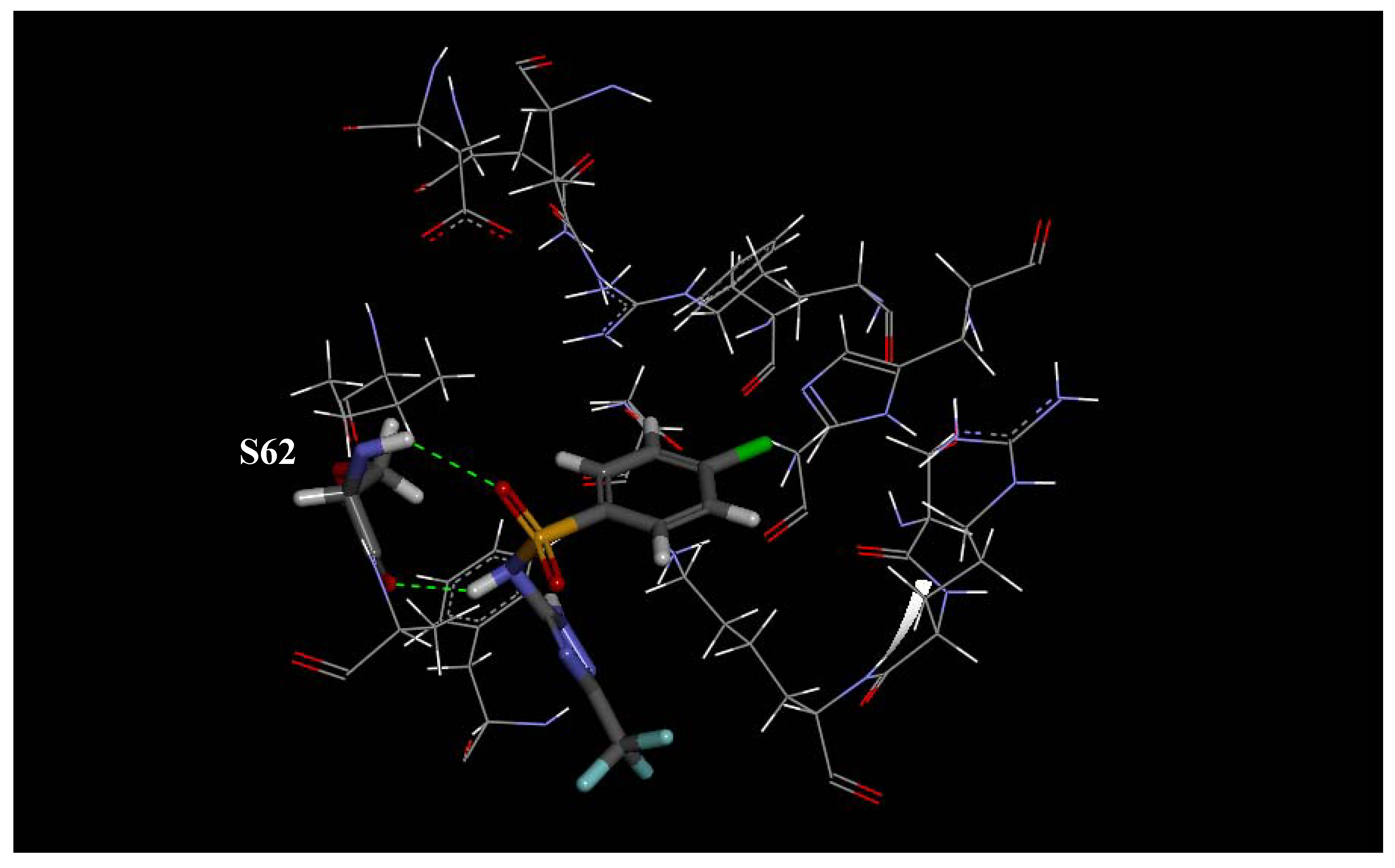
2.1. Molecular Modeling and Docking

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active site residues of DHPS | ||
|---|---|---|
| N°. | E. coli (PDB entry: 1AJ0) | P. falciparum model from site-direct mutagenesis from 1AJ0 |
| 22 | N | N |
| 62 | T | S |
| 63 | R | A |
| 96 | D | D |
| 115 | N | N |
| 117 | I | I |
| 185 | D | D |
| 190 | F | F |
| 215 | L | F |
| 217 | G | G |
| 219 | S | S |
| 220 | R | R |
| 221 | K | K |
| 255 | R | R |
| 257 | H | H |
| Ligand | Docking Score (kcal/mol) |
|---|---|
| 14 | −93.42 |
| 15 | −91.03 |
| 16 | −89.00 |
| 13 | −88.89 |
| 20 | −88.65 |
| 18 | −87.73 |
| 19 | −87.24 |
| 17 | −85.98 |
| 25 | −83.87 |
| 23 | −83.85 |
| 21 | −83.48 |
| sulfalene | −78.78 |
| sulfadoxine | −78.01 |
| 22 | −76.55 |
| 26 | −76.17 |
| 24 | −76.15 |
| 27 | −71.79 |
| sulfadiazine | −70.03 |



3. Experimental
3.1. General
3.2. General Procedure for Preparing 3-Amine-1H-1,2,4-triazoles 5–7
3.3. General Procedure for Preparing 1H-1,2,4-Triazol-3-yl benzenesulfonamides 13–27
4. Conclusions
Acknowledgements
- Samples Availability: Not available.
References
- World Health Organization (WHO). 10 Facts on Malaria. Available online: http://www.who.int/features/factfiles/malaria/en/index.html/ accessed on 10 March 2011.
- World Health Organization (WHO). Global report on antimalarial drug efficacy and drug resistance: 2000–2010. Available online: http://www.who.int/malaria/en accessed on 10 March 2011.
- Kaur, K.; Jain, M.; Reddy, R.P.; Jain, R. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 2010, 45, 3245–3264. [Google Scholar] [CrossRef]
- Biot, C.; Chibale, K. Novel approaches to antimalarial drug discovery. Infect. Disord.-Drug Targets 2006, 6, 173–204. [Google Scholar] [CrossRef]
- Welch, J.T.; Eswarakrishnan, S. Fluorine in Bioorganic Chemistry; John Wiley and Sons Ltd: New York, NY, USA, 1991. [Google Scholar]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; John Wiley and Sons Ltd: Chichester, UK, 2009. [Google Scholar]
- Elliott, A.J.M. Chemistry of Organic Fluorine Compounds. II. A Critical Review, ACS Monograph 187; Hudlicky, M., Pavlath, A.E., Eds.; American Chemical Society: Washington, DC, USA, 1995; pp. 1119–1125. [Google Scholar]
- Kirk, K.L. Fluorination in medicinal chemistry: Methods, strategies, and recent developments. Org. Process. Res. Dev. 2008, 12, 305–321. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluorine Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Park, B.K.; Kitteringham, N.R.; O’Neill, P.M. Metabolism of fluorine containing drugs. Ann. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. [Google Scholar] [CrossRef]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Boechat, N; Bastos, M.M. Trifluoromethylation of carbonyl compounds. Curr. Org. Synth. 2010, 7, 403–413. [Google Scholar] [CrossRef]
- Kgokong, J.L.; Smith, P.P.; Matsabisa, G.M. 1,2,4-Triazino-[5,6-b]indole derivatives: Effects of the trifluoromethyl group on in vitro antimalarial activity. Bioorg. Med. Chem. 2005, 13, 2935–2942. [Google Scholar] [CrossRef]
- Jonet, A.; Dassonville-Klimpt, A.; Do Nascimento, S.; Leger, J-M.; Guillon, J. First enantioselective synthesis of 4-aminoalcohol quinoline derivatives through a regioselective SN2 epoxide opening mechanism. Tetrahedron: Asymmetry 2011, 22, 138–148. [Google Scholar] [CrossRef]
- Kabri, Y.K.; Azas, N.; Dumetre, A.; Hutter, S.; Laget, M.; Verhaeghe, P.; Gellis, A.; Vanelle, P. Original quinazoline derivatives displaying antiplasmodial properties. Eur. J. Med. Chem. 2010, 45, 616–622. [Google Scholar] [CrossRef]
- Dias, L.C.; Dessoy, M.A.; Silva, J.J.N.; Thiemann, O.H.; Oliva, G.; Andricopulo, A.D. Quimioterapia da Doença de Chagas: Estado da Arte e Perspectivas no Desenvolvimento de Novos Fármacos. Quim. Nova 2009, 9, 2444–2457. [Google Scholar]
- Al-Abdely, H.M.; Graybill, J.R.; Loebenberg, D.; Melby, P.C. Efficacy of the triazole SCH 56592 against Leishmania amazonensis and Leishmania donovani in experimental murine cutaneous and visceral leishmaniases. Antimicrob. Agents Chemother. 1999, 43, 2910–2914. [Google Scholar]
- Guantai, E.M.; Ncokazi, K.; Egan, T.J.; Gut, J.; Rosenthal, P.J.; Smith, P.J.; Chibale, K. Design, synthesis and in vitro antimalarial evaluation of triazole-linked chalcone and dienone hybrid compounds. Bioorg. Med. Chem. 2010, 18, 8243–8256. [Google Scholar] [CrossRef]
- Hans, R.H.; Gut, J.; Rosenthal, P.J.; Chibale, K. Comparison of the antiplasmodial and falcipain-2 inhibitory activity of β-amino alcohol thiolactone-chalcone and isatin-chalcone hybrids. Bioorg. Med. Chem. Lett. 2010, 20, 2234–2237. [Google Scholar] [CrossRef]
- Vlahakis, J.Z.; Lazar, C.; Crandall, I.E.; Szarek, W.A. Anti-Plasmodium activity of imidazolium and triazolium salts. Bioorg. Med. Chem. 2010, 18, 6184–6196. [Google Scholar] [CrossRef]
- Mishra, N.; Arora, P.; Kumar, B.; Mishra, L.C.; Bhattacharya, A.; Awasthi, S.K.; Bhasin, V.K. Synthesis of novel substituted 1,3-diaryl propenone derivatives and their antimalarial activity in vitro. Eur. J. Med. Chem. 2008, 43, 1530–1535. [Google Scholar] [CrossRef]
- D’hooghe, M.; Kenis, S.; Vervisch, K.; Lategan, C.; Smith, P.J.; Chibale, K.; De Kimpe, N. Synthesis of 2-(aminomethyl)aziridines and their microwave-assisted ring opening to 1,2,3-triaminopropanes as novel antimalarial pharmacophores. Eur. J. Med. Chem. 2011, 46, 579–587. [Google Scholar]
- Krungkrai, J.; Krungkrai, S.R.; Supuran, C.T. Carbonic anhydrase inhibitors: Inhibition of Plasmodium falciparum carbonic anhydrase with aromatic/heterocyclic sulfonamides—In vitro and in vivo studies. Bioorg. Med. Chem. Lett. 2008, 18, 5466–5471. [Google Scholar] [CrossRef]
- Krungkrai, J.; Scozzafava, A.; Reungprapavut, S.; Krungkrai, S.R.; Rattanajak, R.; Kamchonwongpaisan, S.; Supuran, C.T. . Carbonic anhydrase inhibitors. Inhibition of Plasmodium falciparum carbonic anhydrase with aromatic sulfonamides: Towards antimalarials with a novel mechanism of action? Bioorg. Med. Chem. 2005, 13, 483–489. [Google Scholar] [CrossRef]
- Johann, L.; Pegraro, S.; Dormeyer, M.; Michael, L.; Aschenbrenner, A.; Karmer, B. Sulfonyl-phenyl-ureido benzamidines: A novel structural class of potent antimalarial agents. Bioorg. Med. Chem. Lett. 2004, 14, 1979–1982. [Google Scholar] [CrossRef]
- Domınguez, J.N.; Leon, C.; Rodrigues, J.; Domınguez, N.G.; Gut, J.; Rosenthal, P.J. Synthesis and antimalarial activity of sulfonamide chalcone derivatives. Il Farmaco 2005, 60, 307–311. [Google Scholar] [CrossRef]
- Ryckebusch, A.; Deprez-Poulain, R.; Debreu-Fontaine, M.A.; Vandaele, R.; Mouray, E.; Grellier, P.; Sergheraert, C. Parallel synthesis and anti-malarial activity of a sulfonamide library. Bioorg. Med. Chem. Lett. 2002, 12, 2595–2598. [Google Scholar] [CrossRef]
- Posner, G.H.; Maxwell, J.P.; O'Dowd, H.; Krasavin, M.; Xie, S.; Shapiro, T.A. Antimalarial sulfide, sulfone, and sulfonamide trioxanes. Bioorg. Med. Chem. 2000, 8, 1361–1370. [Google Scholar] [CrossRef]
- Parai, M.K.; Panda, G.; Srivastava, K.; Puri, S.K. Design, synthesis and antimalarial activity of benzene and isoquinoline sulfonamide derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 776–781. [Google Scholar] [CrossRef]
- Martyn, D.C.; Cortese, J.F.; Tyndall, E.; Dick, J.; Mazitschek, R.; Munoz, B.; Clardy, J. Antiplasmodial activity of piperazine sulfonamides. Bioorg. Med. Chem. Lett. 2010, 20, 218–221. [Google Scholar] [CrossRef]
- Ekoue-Kovi, K.; Yearick, K.; Iwaniuk, D.P.; Natarajan, J.K.; Alumasa, J.; Dios, A.C.; Roepe, P.D.; Wolf, C. Synthesis and antimalarial activity of new 4-amino-7-chloroquinolyl amides, sulfonamides, ureas and thioureas. Bioorg. Med. Chem. 2009, 17, 270–283. [Google Scholar]
- Kasekarn, W.; Sirawaraporn, R.; Chahomchuena, T.; Cowman, A.F.; Sirawaraporn, W. Molecular characterization of bifunctional hydroxymethyldihydropterin pyrophosphokinase-dihydropteroate synthase from Plasmodium falciparum. coli. Mol. Biochem. Parasitol. 2004, 137, 43–53. [Google Scholar] [CrossRef]
- Bzik, D.J.; Li, W.B.; Horii, T.; Inselburg, J. Molecular cloning and sequence analysis of the Plasmodium falciparum dihydrofolate reductase-thymidylate synthase gene. Proc Natl. Acad. Sci. USA 1987, 84, 8360–8364. [Google Scholar] [CrossRef]
- Biswas, S.; Escalante, A.; Chaiyaroj, S.; Angkasekwinai, P.; Lal, A.A. Prevalence of point mutations in the dihydrofolate re-ductase and dihydrofolate synthetase genes of Plasmodium falciparum isolates from India and Thailand: A molecular epidemiologic study. Top. Med. Int. Health 2000, 10, 737–743. [Google Scholar]
- Triglia, T.; Cowman, A.F. Primary structure and expression of the dihydropteroate synthetase gene of Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1994, 91, 7149–7153. [Google Scholar] [CrossRef]
- Paul, B.K.; Ghosh, S.N.; Bose, A.N.; Basu, U.P. Search for oral hypoglycemic agents. I. Synthesis and biological activity of some 5-arylsulfonamido-1,2,4-triazoles. Indian J. Chem. 1968, 6, 618–620. [Google Scholar]
- McColl, J.D.; Lee, C.F.; Hajdu, A. Effect of sulfonylurea derivatives in experimental ulcer formation in the rat. Arch. Int. Pharm. Ther. 1963, 141, 181–189. [Google Scholar]
- Joshi, K.C.; Singh, V.K. Potential organo-fluorine oral hypoglycemic agents. IV. Synthesis and hypoglycemic activity of some 5-arylsulfonamido-1,2,4-triazoles. J. Prakt. Chem. 1971, 313, 169–173. [Google Scholar] [CrossRef]
- Achari, A.; Somers, D.O.; Champness, J.N.; Bryant, P.K.; Rosemond, J.; Stammers, D.K. Crystal structure of the anti-bacterial sulfonamide drug target dihydropteroate synthase. Nat. Struct. Biol. 1997, 4, 490–497. [Google Scholar] [CrossRef]
- Molegro Virtual Docker/4.0 Molegro ApS: Aarhus, Denmark, 2009.
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2009, 37 (Suppl. 1), D26–D31. [Google Scholar]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M., Jr; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Boechat, N.; Pinheiro, L.C.S.; Santos-Filho, O.A.; Silva, I.C. Design and Synthesis of New N-(5-Trifluoromethyl)-1H-1,2,4-triazol-3-yl Benzenesulfonamides as Possible Antimalarial Prototypes. Molecules 2011, 16, 8083-8097. https://doi.org/10.3390/molecules16098083
Boechat N, Pinheiro LCS, Santos-Filho OA, Silva IC. Design and Synthesis of New N-(5-Trifluoromethyl)-1H-1,2,4-triazol-3-yl Benzenesulfonamides as Possible Antimalarial Prototypes. Molecules. 2011; 16(9):8083-8097. https://doi.org/10.3390/molecules16098083
Chicago/Turabian StyleBoechat, Nubia, Luiz C.S. Pinheiro, Osvaldo A. Santos-Filho, and Isabor C. Silva. 2011. "Design and Synthesis of New N-(5-Trifluoromethyl)-1H-1,2,4-triazol-3-yl Benzenesulfonamides as Possible Antimalarial Prototypes" Molecules 16, no. 9: 8083-8097. https://doi.org/10.3390/molecules16098083