Subnanosecond Charge Recombination Dynamics in P3HT/PC61BM Films

Abstract
:1. Introduction
2. Results and Discussion
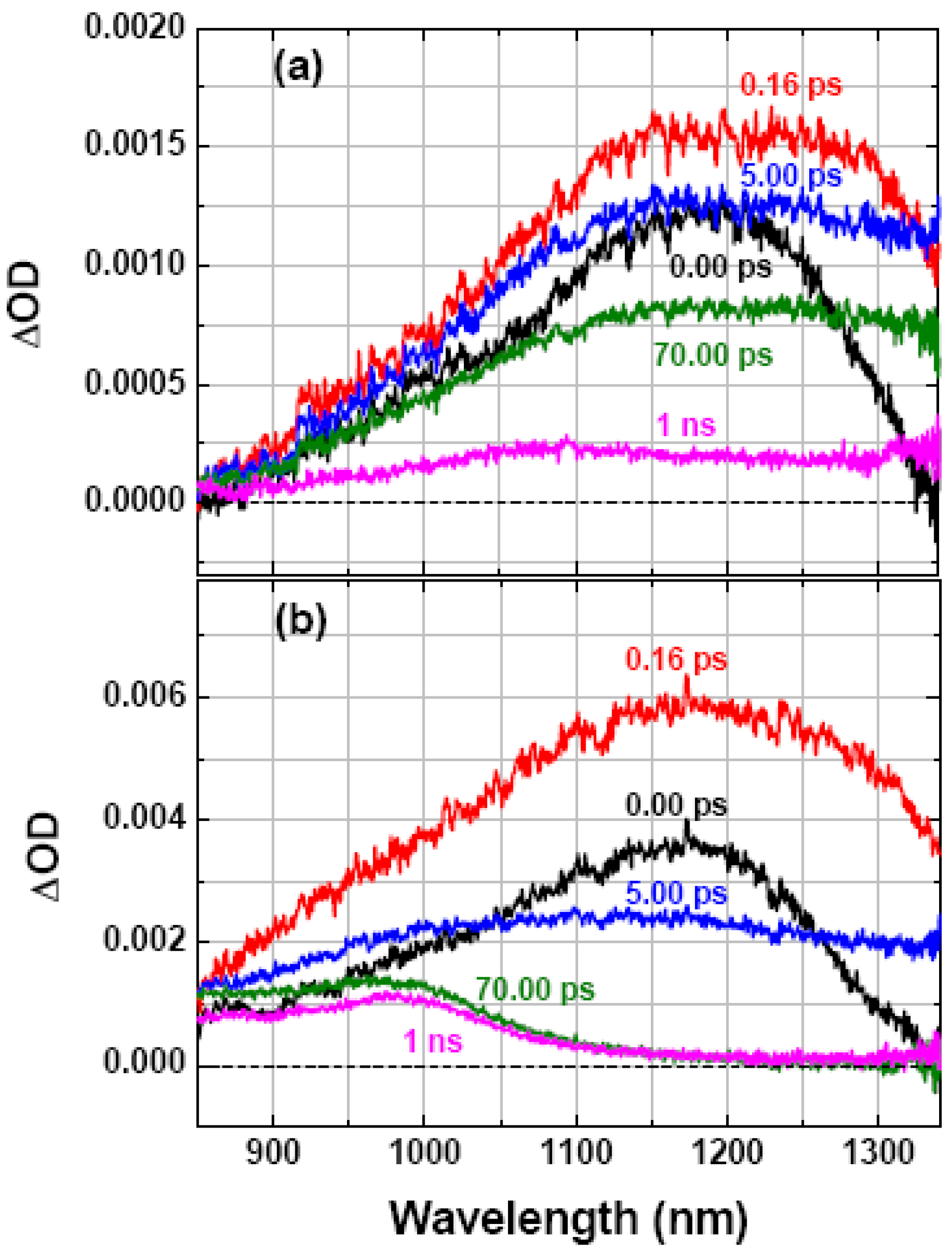
2.1. Characterization of the Hole Dynamics in Subnanosecond Timeframe

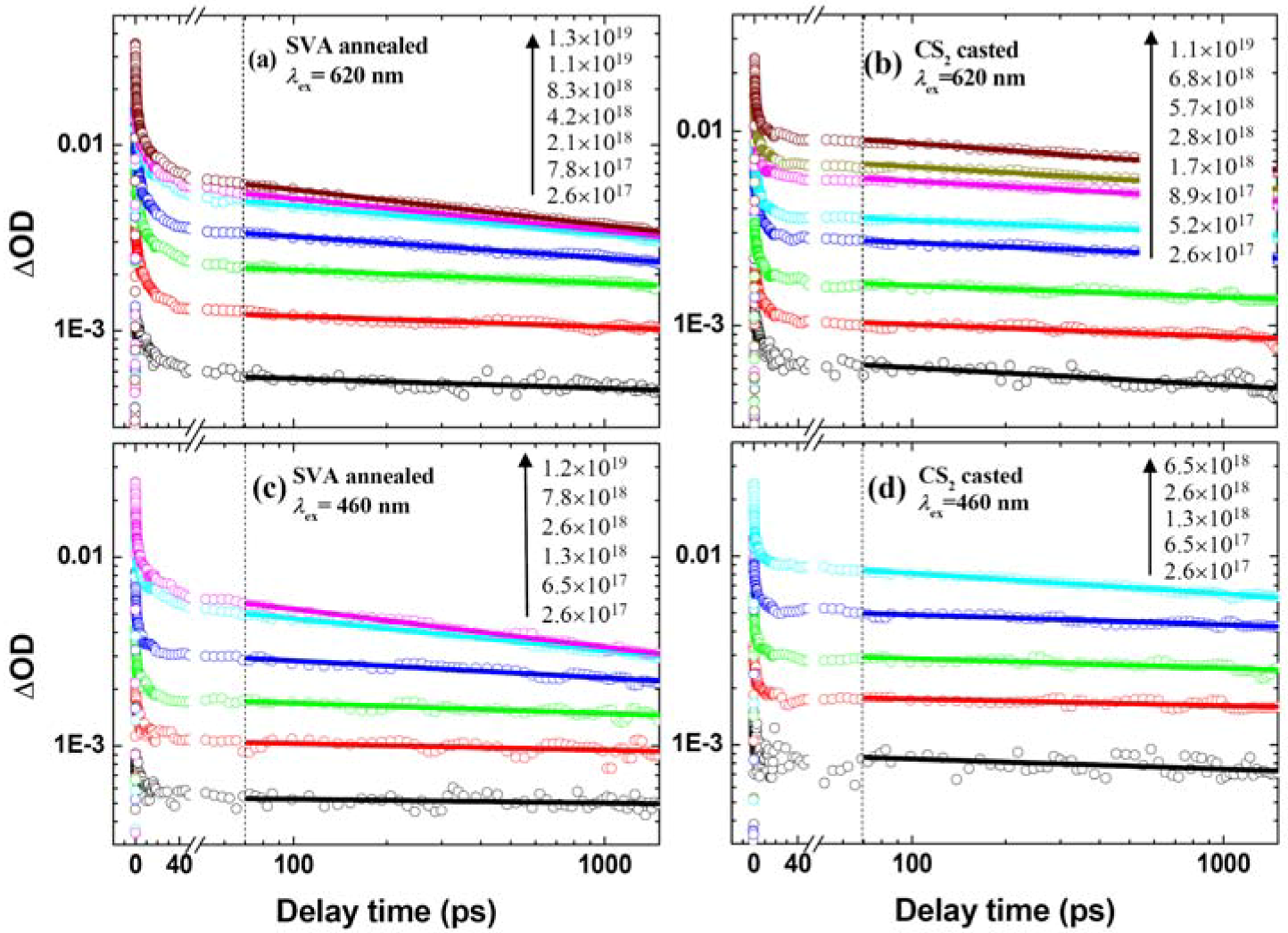
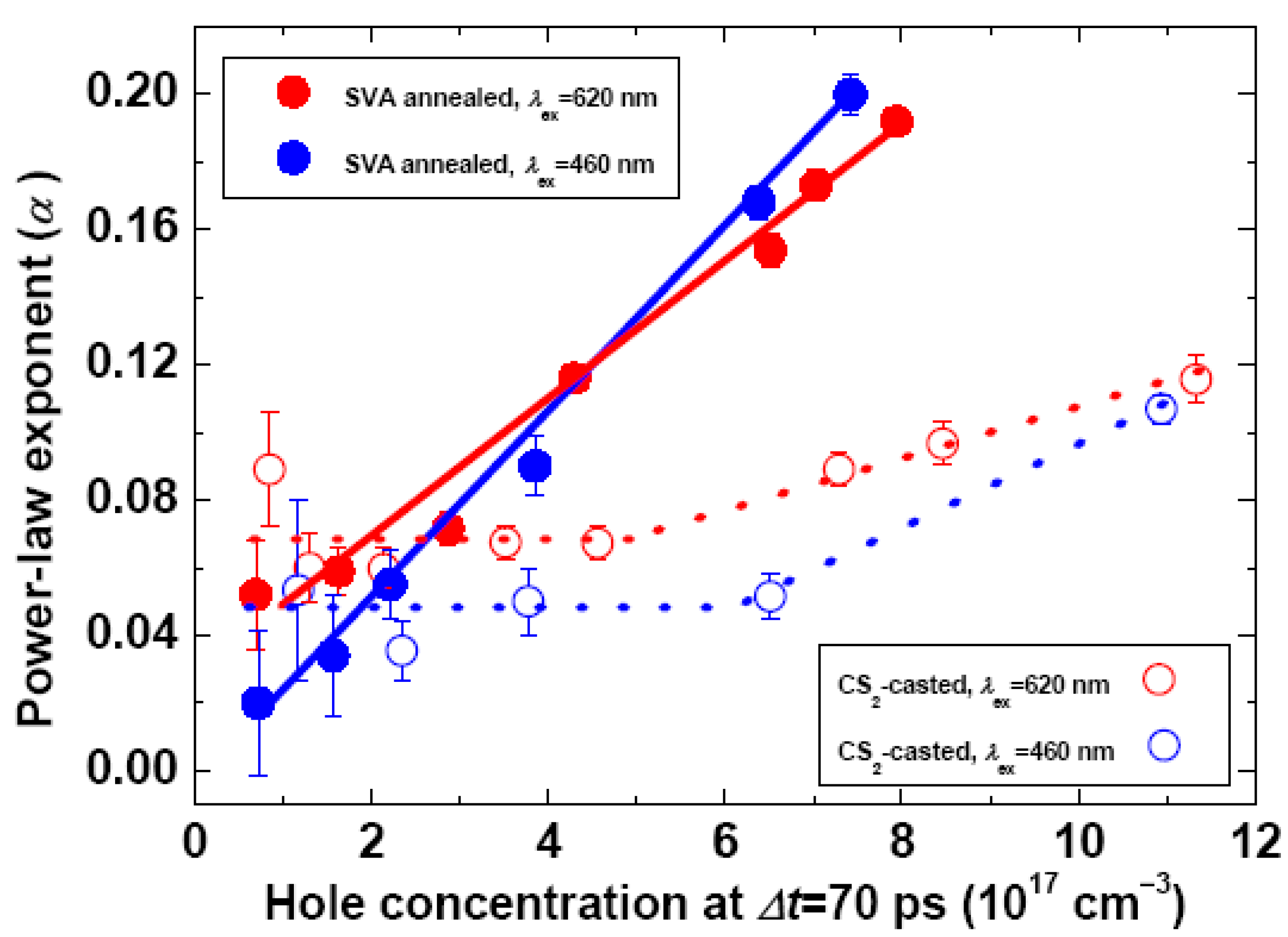
2.2. Effects of Film Morphology and Excitation Photon Energy/Fluence on the Hole Dynamics


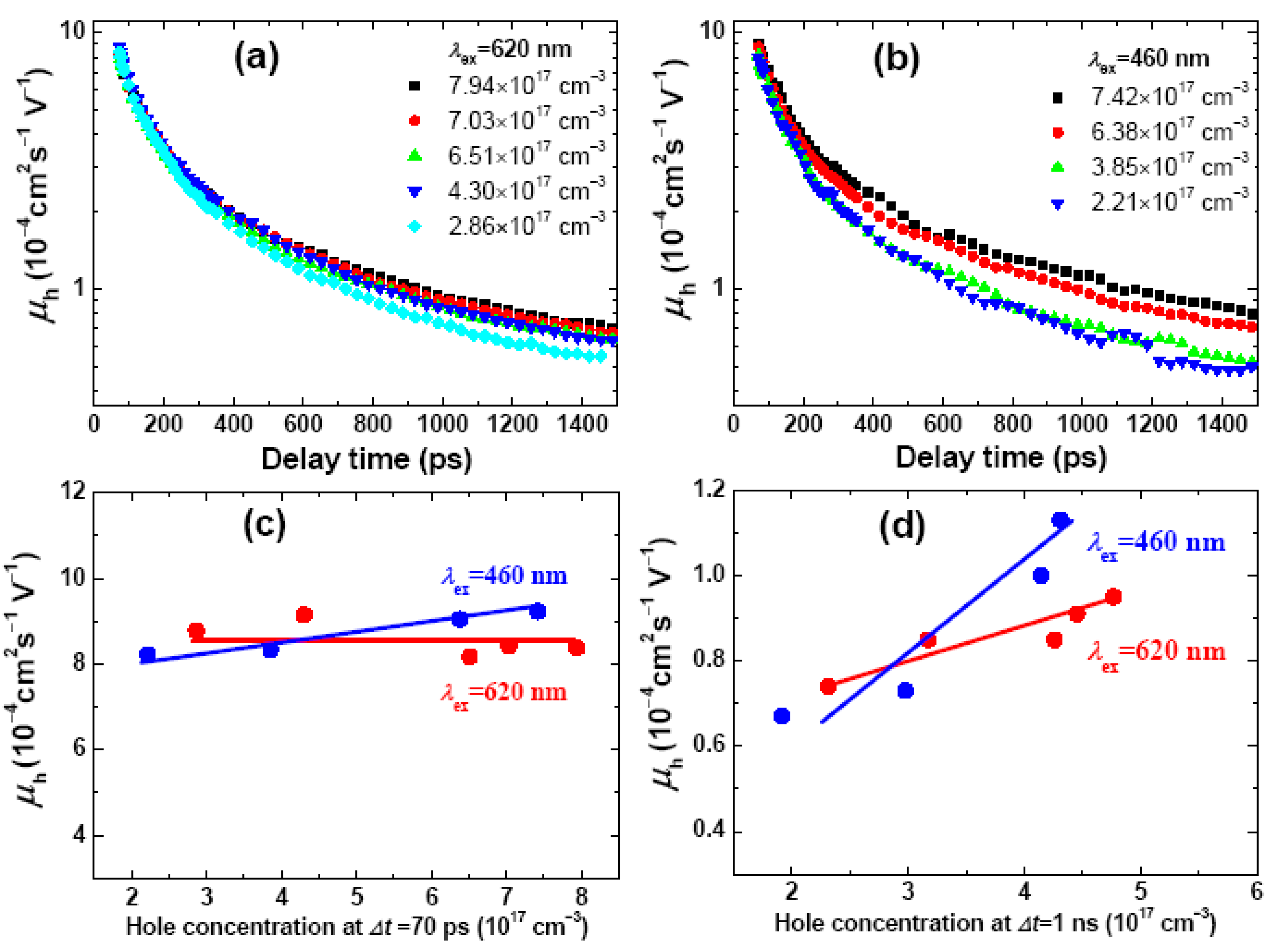
2.3. Concentration, Photon Energy and Time Dependence of Hole Mobility and Bimolecular Recombination



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λex | SVA annealed P3HT/PC61BM film | CS2-casted P3HT/PC61BM film | ||||
|---|---|---|---|---|---|---|
| (nm) | μh (cm2·s−1·V−1) | γbi (cm3·s−1) | μh (cm2·s−1·V−1) | γbi (cm3·s−1) | ||
| 620 | 8.6 × 10−4 | 4.5 × 10−10 | 3.8 × 10−4 | 2.0 × 10−10 | ||
| (8.6 × 10−5) | (4.5 × 10−11) | (3.4 × 10−5) | (1.7 × 10−11) | |||
| 460 | 8.7 × 10−4 | 4.5 × 10−10 | 3.1 × 10−4 | 1.6 × 10−10 | ||
| (8.8 × 10−5) | (4.6 × 10−11) | (2.8 × 10−5) | (1.4 × 10−11) | |||
3. Experimental
4. Conclusions
Supplementary Materials
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References and Notes
- Chen, H.; Hou, J.; Zhang, S.; Liang, Y.; Yang, G.; Yang, Y.; Yu, L.; Wu, Y.; Li, G. Polymer solar cells with enhanced open-circuit voltage and efficiency. Nat. Photonics 2009, 3, 649–653. [Google Scholar]
- Liang, Y.; Xu, Z.; Xia, J.; Tsai, S.T.; Wu, Y.; Li, G.; Ray, C.; Yu, L. For the bright future—Bulk heterojunction polymer solar cells with power conversion efficiency of 7.4%. Adv. Mater. 2010, 22, E135–E138. [Google Scholar] [CrossRef]
- Green, M.A.; Emery, K.; Hishikawa, Y.; Warta, W. Solar cell efficiency tables (version 37). Prog. Photovolt. Res. Appl. 2011, 19, 84–92. [Google Scholar] [CrossRef]
- Ma, W.; Yang, C.; Gong, X.; Lee, K.; Heeger, A.J. Thermally Stable, efficient polymer solar cells with nanoscale control of the interpenetrating network morphology. Adv. Funct. Mater. 2005, 15, 1617–1622. [Google Scholar] [CrossRef]
- Reyes-Reyes, M.; Kim, K.; Carroll, D.L. High-efficiency photovoltaic devices based on annealed poly(3-hexylthiophene) and 1-(3-methoxycarbonyl)-propyl-1-phenyl-(6,6)C61 blends. Appl. Phys. Lett. 2005, 87, 083506. [Google Scholar] [CrossRef]
- Irwin, M.D.; Buchholz, D.B.; Hains, A.W.; Chang, R.P.H.; Marks, T.J. p-Type semiconducting nickel oxide as an efficiency-enhancing anode interfacial layer in polymer bulk-heterojunction solar cells. Proc. Natl. Acad. Sci. USA 2008, 105, 2783–2787. [Google Scholar]
- Österbacka, R.; An, C.P.; Jiang, X.M.; Vardeny, Z.V. Two-dimensional electronic excitations in self-assembled conjugated polymer nanocrystals. Science 2000, 287, 839–842. [Google Scholar] [CrossRef]
- Jiang, X.M.; Österbacka, R.; Korovyanko, O.; An, C.P.; Horovitz, B.; Janssen, R.A.J.; Vardeny, Z.V. Spectroscopic studies of photoexcitations in regioregular and regiorandom polythiophene films. Adv. Funct. Mater. 2002, 12, 587–597. [Google Scholar] [CrossRef]
- Kanemoto, K.; Yasui, M.; Kosumi, D.; Ogata, A.; Sugisaki, M.; Karasawa, T.; Akai, I.; Hashimoto, H. Morphology-dependent carrier and exciton generations in regioregular poly(3-hexylthiophene) polymer diodes as revealed by bleaching spectroscopy. Phys. Rev. Lett. 2009, 103, 187402. [Google Scholar]
- Yu, G.; Gao, J.; Hummelen, J.C.; Wudl, F.; Heeger, A.J. Polymer photovoltaic cells: Enhanced efficiencies via a network of internal donor-acceptor heterojunctions. Science 1995, 270, 1789–1791. [Google Scholar]
- Po, R.; Maggini, M.; Camaioni, N. Polymer solar cells: Recent approaches and achievements. J. Phys. Chem. C 2010, 114, 695–706. [Google Scholar] [CrossRef]
- Li, G.; Shrotriya, V.; Huang, J.; Yao, Y.; Moriarty, T.; Emery, K.; Yang, Y. High-efficiency solution processable polymer photovoltaic cells by self-organization of polymer blends. Nat. Mater. 2005, 4, 864–868. [Google Scholar] [CrossRef]
- He, Y.; Chen, H.; Hou, J.; Li, Y. Indene-C60 bisadduct: A new acceptor for high-performance polymer solar cells. J. Am. Chem. Soc. 2010, 132, 1377–1382. [Google Scholar] [CrossRef]
- Guo, J.; Ohkita, H.; Benten, H.; Ito, S. Charge generation and recombination dynamics in poly(3-hexylthiophene)/fullerene blend films with different regioregularities and morphologies. J. Am. Chem. Soc. 2010, 132, 6154–6164. [Google Scholar]
- Howard, I.A.; Mauer, R.; Meister, M.; Laquai, F. Effect of morphology on ultrafast free carrier generation in polythiophene:fullerene organic solar cells. J. Am. Chem. Soc. 2010, 132, 14866–14876. [Google Scholar] [CrossRef]
- Clarke, T.M.; Jamieson, F.C.; Durrant, J.R. Transient absorption studies of bimolecular recombination dynamics in polythiophene/fullerene blend Films. J. Phys. Chem. C 2009, 113, 20934–20941. [Google Scholar]
- Koster, L.J.A.; Smits, E.C.P.; Mihailetchi, V.D.; Blom, P.W.M. Device model for the operation of polymer/fullerene bulk heterojunction solar cells. Phys. Rev. B 2005, 72, 085205. [Google Scholar] [CrossRef]
- Koster, L.J.A.; Mihailetchi, V.D.; Blom, P.W.M. Bimolecular recombination in polymer/fullerene bulk heterojunction solar cells. Appl. Phys. Lett. 2006, 88, 052104. [Google Scholar]
- Shuttle, C.G.; O’Regan, B.; Ballantyne, A.M.; Nelson, J.; Bradley, D.D.C.; Durrant, J.R. Bimolecular recombination losses in polythiophene:fullerene solar cells. Phys. Rev. B 2008, 78, 113201. [Google Scholar]
- Juska, G.; Sliauzys, G.; Genevicius, K.; Arlauskas, K.; Pivrikas, A.; Scharber, M.; Dennler, G.; Sariciftci, N.S.; Österbacka, R. Charge-carrier transport and recombination in thin insulating films studied via extraction of injected plasma. Phys. Rev. B 2006, 74, 115314. [Google Scholar]
- Pivrikas, A.; Juska, G.; Mozer, A.J.; Scharber, M.; Arlauskas, K.; Sariciftci, N.S.; Stubb, H.; Österbacka, R. Bimolecular recombination coefficient as a sensitive testing parameter for low-mobility solar-cell materials. Phys. Rev. Lett. 2005, 94, 176806. [Google Scholar]
- Montanari, I.; Nogueira, A.F.; Nelson, J.; Durrant, J.R.; Winder, C.; Loi, M.A.; Sariciftci, N.S.; Brabec, C. Transient optical studies of charge recombination dynamics in a polymer/fullerene composite at room temperature. Appl. Phys. Lett. 2002, 81, 3001–3003. [Google Scholar]
- Nogueira, A.F.; Montanari, I.; Nelson, J.; Durrant, J.R.; Winder, C.; Sariciftci, N.S.; Brabec, C. Charge recombination in conjugated polymer/fullerene blended films studied by transient absorption spectroscopy. J. Phys. Chem. B 2003, 107, 1567–1573. [Google Scholar] [CrossRef]
- Guo, J.; Ohkita, H.; Yokoya, S.; Benten, H.; Ito, S. Bimodal polarons and hole transport in poly(3-hexylthiophene): Fullerene blend films. J. Am. Chem. Soc. 2010, 132, 9631–9637. [Google Scholar] [CrossRef]
- Eng, M.P.; Barnes, P.R.F.; Durrant, J.R. Concentration-dependent hole mobility and recombination coefficient in bulk heterojunctions determined from transient absorption spectroscopy. J. Phys. Chem. Lett. 2010, 1, 3096–3100. [Google Scholar] [CrossRef]
- Soon, Y.W.; Clarke, T.M.; Zhang, W.; Agostinelli, T.; Kirkpatrick, J.; Smith, C.D.; McCulloch, I.; Nelson, J.; Durrant, J.R. Energy versus electron transfer in organic solar cells: a comparison of the photophysics of two indenofluorene: fullerene blend films. Chem. Sci. 2011, 2, 1111–1120. [Google Scholar] [CrossRef]
- Piris, J.; Dykstra, T.E.; Bakulin, A.A.P.; van Loosdrecht, H.M.; Knulst, W.; Trinh, M.T.; Schins, J.M.; Siebbeles, L.D.A. Photogeneration and ultrafast dynamics of excitons and charges in P3HT/PCBM blends. J. Phys. Chem. C 2009, 113, 14500–14506. [Google Scholar]
- Hwang, I.; Moses, D.; Heeger, A.J. Photoinduced carrier generation in P3HT/PCBM bulk heterojunction materials. J. Phys. Chem. C 2008, 112, 4350–4354. [Google Scholar] [CrossRef]
- Kraabel, B.; Hummelen, J.C.; Vacar, D.; Moses, D.; Sariciftci, N.S.; Heeger, A.J. Subpicosecond photoinduced electron transfer from conjugated polymers to functionalized fullerenes. J. Chem. Phys. 1996, 104, 4267–4273. [Google Scholar] [CrossRef]
- Kraabel, B.; McBranch, D.; Sariciftci, N.S.; Moses, D.; Heeger, A.J. Ultrafast spectroscopic studies of photoinduced electron transfer from semiconducting polymers to C60. Phys. Rev. B 1994, 50, 18543–18552. [Google Scholar] [CrossRef]
- Westerling, M.; Aarnio, H.; Österbacka, R.; Stubb, H.; King, S.M.; Monkman, A.P.; Andersson, M.R. Photoexcitation dynamics in an alternating polyfluorene copolymer. Phys. Rev. B 2007, 75, 224306. [Google Scholar]
- De, S.; Pascher, T.; Maiti, M.; Jespersen, K.G.; Kesti, T.; Zhang, F.; Inganäs, O.; Yartsev, A.; Sundström, V. Geminate charge recombination in alternating polyfluorene copolymer/fullerene blends. J. Am. Chem. Soc. 2007, 129, 8466–8472. [Google Scholar]
- Pal, S.K.; Kesti, T.; Maiti, M.; Zhang, F.; Inganäs, O.; Hellström, S.; Andersson, M.R.; Oswald, F.; Langa, F.; Österman, T.; et al. Geminate charge recombination in polymer/fullerene bulk heterojunction films and implications for solar cell function. J. Am. Chem. Soc. 2010, 132, 12440–12451. [Google Scholar]
- Westenhoff, S.; Beenken, W.J.D.; Friend, R.H.; Greenham, N.C.; Yartsev, A.; Sundström, V. Anomalous energy transfer dynamics due to torsional relaxation in a conjugated polymer. Phys. Rev. Lett. 2006, 97, 166804. [Google Scholar]
- Scheblykin, I.G.; Yartsev, A.; Pullerits, T.; Gulbinas, V.; Sundström, V. Excited state and charge photogeneration dynamics in conjugated polymers. J. Phys. Chem. B 2007, 111, 6303–6321. [Google Scholar]
- Gulbinas, V.; Zaushitsyn, Y.; Bässler, H.; Yartsev, A.; Sundström, V. Dynamics of charge pair generation in ladder-type poly(para-phenylene) at different excitation photon energies. Phys. Rev. B 2004, 70, 035215. [Google Scholar]
- The time origin is defined as the time at the maximum of the cross-correlation trace between the pump and the probe pulses.
- Guo, J.; Ohkita, H.; Benten, H.; Ito, S. Near-IR femtosecond transient absorption spectroscopy of ultrafast polaron and triplet exciton formation in polythiophene films with different regioregularities. J. Am. Chem. Soc. 2009, 131, 16869–16880. [Google Scholar]
- Zhang, W.; Hu, R.; Li, D.; Huo, M.-M.; Ai, X.-C.; Zhang, J.-P. Primary dynamics of exciton and charge photogeneration in solvent vapor annealed P3HT/PCBM films. J. Phys. Chem. C 2012, 116, 4298–4310. [Google Scholar]
- Nelson, J. Diffusion-limited recombination in polymer-fullerene blends and its influence on photocurrent collection. Phys. Rev. B 2003, 67, 155209. [Google Scholar] [CrossRef]
- Ballantyne, A.M.; Chen, L.; Nelson, J.; Bradley, D.D.C.; Astuti, Y.; Maurano, A.; Shuttle, C.G.; Durrant, J.R.; Heeney, M.; Duffy, W.; et al. Studies of highly regioregular poly(3-hexylselenophene) for photovoltaic applications. Adv. Mater. 2007, 19, 4544–4547. [Google Scholar] [CrossRef]
- Clarke, T.M.; Durrant, J.R. Charge photogeneration in organic solar cells. Chem. Rev. 2010, 110, 6736–6767. [Google Scholar] [CrossRef]
- Shuttle, C.G.; Hamilton, R.; O’Regan, B.C.; Nelson, J.; Durrant, J.R. Charge-density-based analysis of the current–voltage response of polythiophene/fullerene photovoltaic devices. Proc. Natl. Acad. Sci. USA 2010, 107, 16448–16452. [Google Scholar]
- Burkhard, G.F.; Hoke, E.T.; Scully, S.R.; McGehee, M.D. Incomplete exciton harvesting from fullerenes in bulk heterojunction solar cells. Nano Lett. 2009, 9, 4037–4041. [Google Scholar] [CrossRef]
- Dicker, G.; de Haas, M.P.; Siebbeles, L.D.A.; Warman, J.M. Electrodeless time-resolved microwave conductivity study of charge-carrier photogeneration in regioregular poly(3-hexylthiophene) thin films. Phys. Rev. B 2004, 70, 045203. [Google Scholar]
- Kim, Y.; Cook, S.; Tuladhar, S.M.; Choulis, S.A.; Nelson, J.; Durrant, J.R.; Bradley, D.D.C.; Giles, M.; Mcculloch, I.; Ha, C.; et al. A strong regioregularity effect in self-organizing conjugated polymer films and high-efficiency polythiophene:fullerene solar cells. Nat. Mater. 2006, 5, 197–203. [Google Scholar] [CrossRef]
- Kim, Y.; Cook, S.; Choulis, S.A.; Nelson, J.; Durrant, J.R.; Bradley, D.D.C. Organic photovoltaic devices based on blends of regioregular poly(3-hexylthiophene) and poly(9,9-dioctylfluorene-co-benzothiadiazole). Chem. Mater. 2004, 16, 4812–4818. [Google Scholar] [CrossRef]
- Mihailetchi, V.D.; Xie, H.; de Boer, B.; Koster, L.J.A.; Blom, P.W.M. Charge transport and photocurrent generation in poly(3-hexylthiophene): Methanofullerene bulk-heterojunction solar cells. Adv. Funct. Mater. 2006, 16, 699–708. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, W.; Zhao, N.-J.; Huo, M.-M.; Fu, L.-M.; Ai, X.-C.; Zhang, J.-P. Subnanosecond Charge Recombination Dynamics in P3HT/PC61BM Films. Molecules 2012, 17, 13923-13936. https://doi.org/10.3390/molecules171213923
Zhang W, Zhao N-J, Huo M-M, Fu L-M, Ai X-C, Zhang J-P. Subnanosecond Charge Recombination Dynamics in P3HT/PC61BM Films. Molecules. 2012; 17(12):13923-13936. https://doi.org/10.3390/molecules171213923
Chicago/Turabian StyleZhang, Wei, Ning-Jiu Zhao, Ming-Ming Huo, Li-Min Fu, Xi-Cheng Ai, and Jian-Ping Zhang. 2012. "Subnanosecond Charge Recombination Dynamics in P3HT/PC61BM Films" Molecules 17, no. 12: 13923-13936. https://doi.org/10.3390/molecules171213923