Optimized Solid Phase-Assisted Synthesis of Dendrons Applicable as Scaffolds for Radiolabeled Bioactive Multivalent Compounds Intended for Molecular Imaging

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
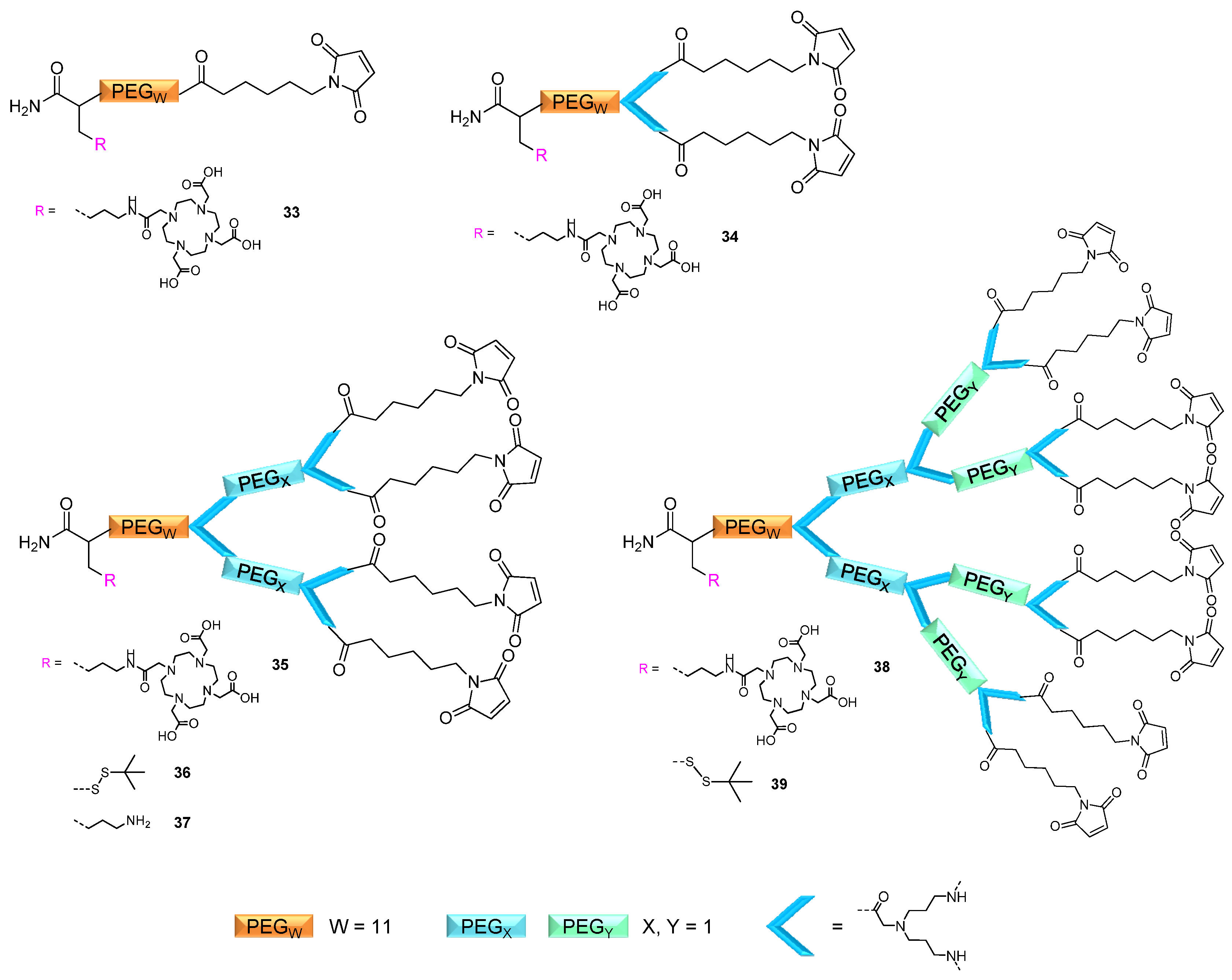
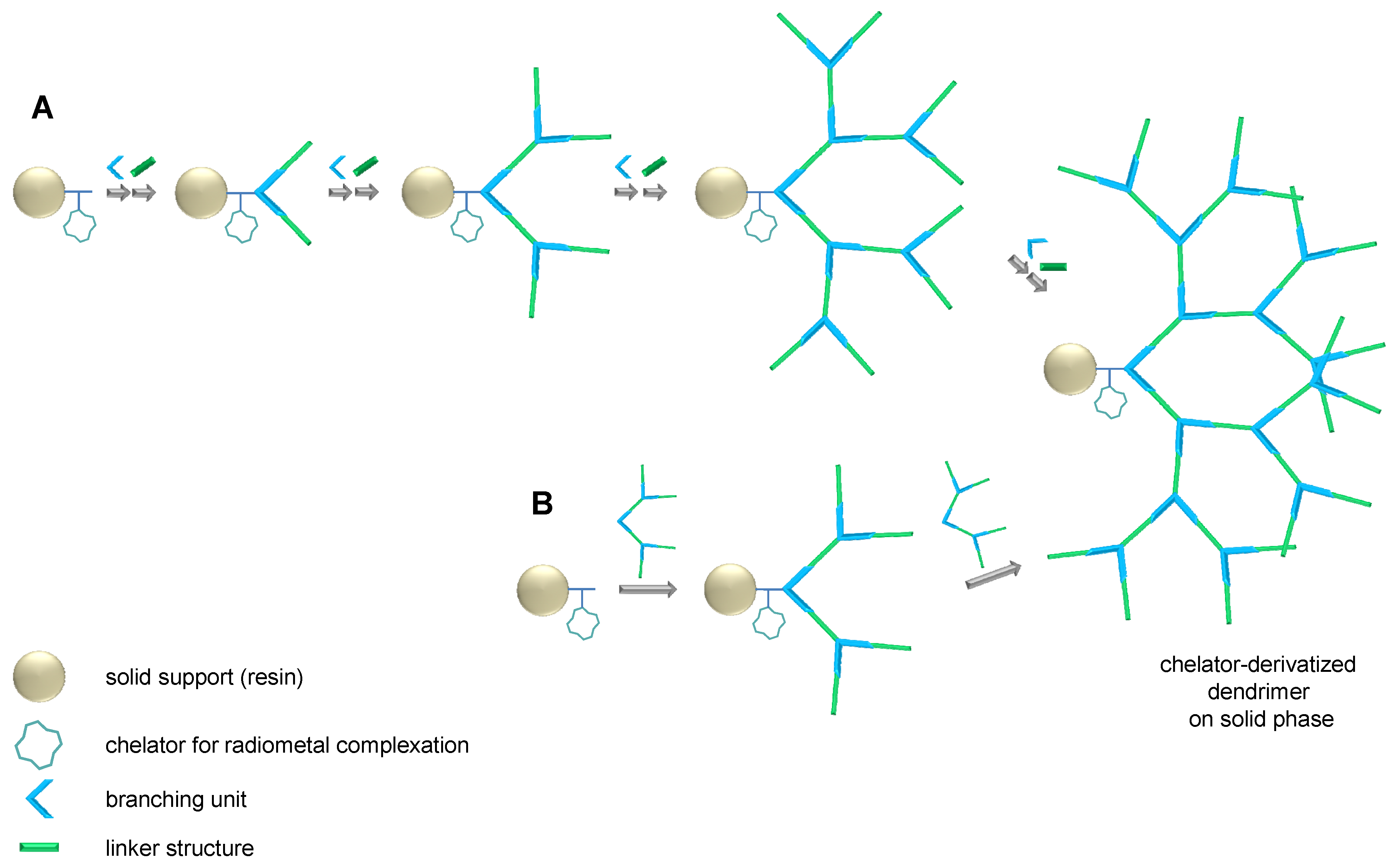
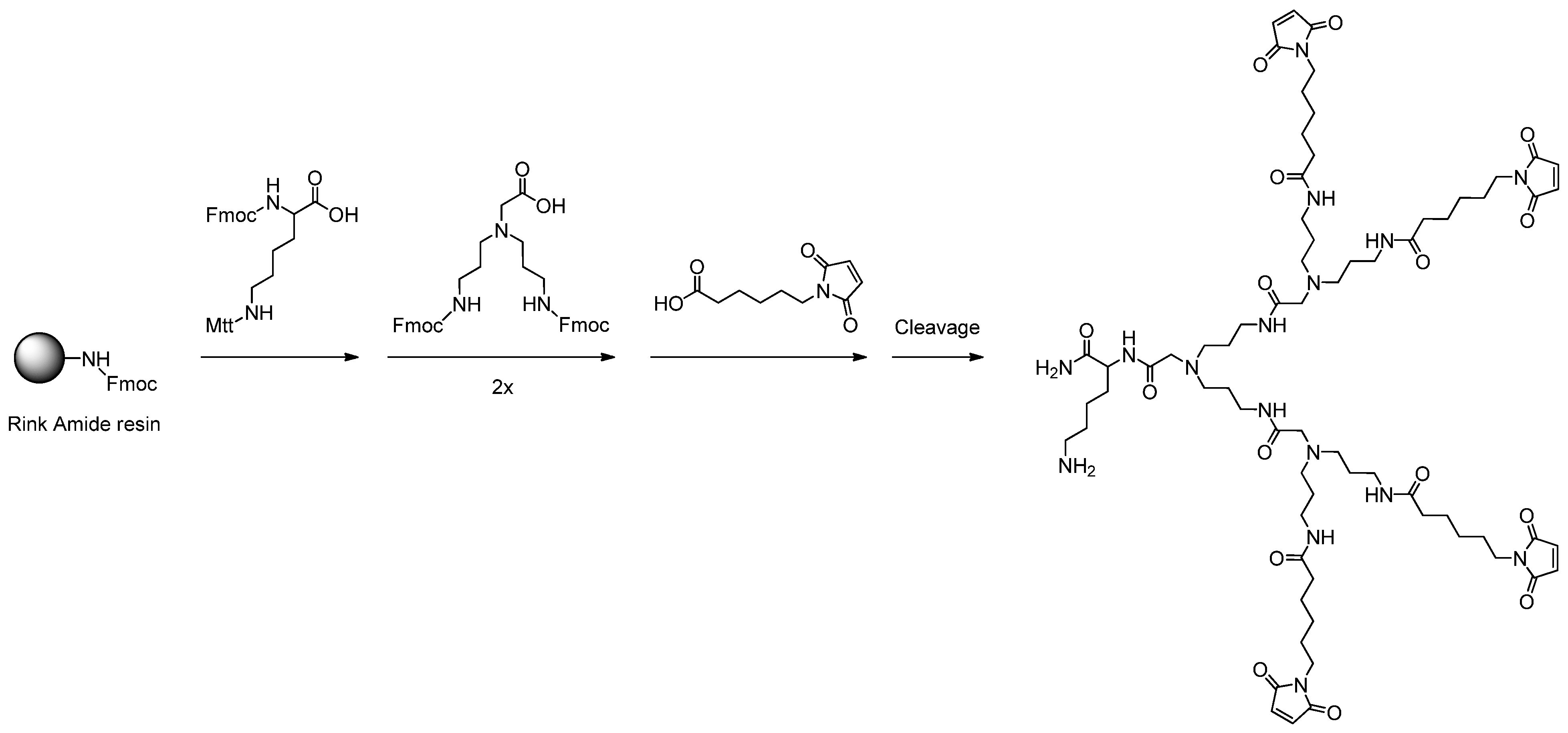
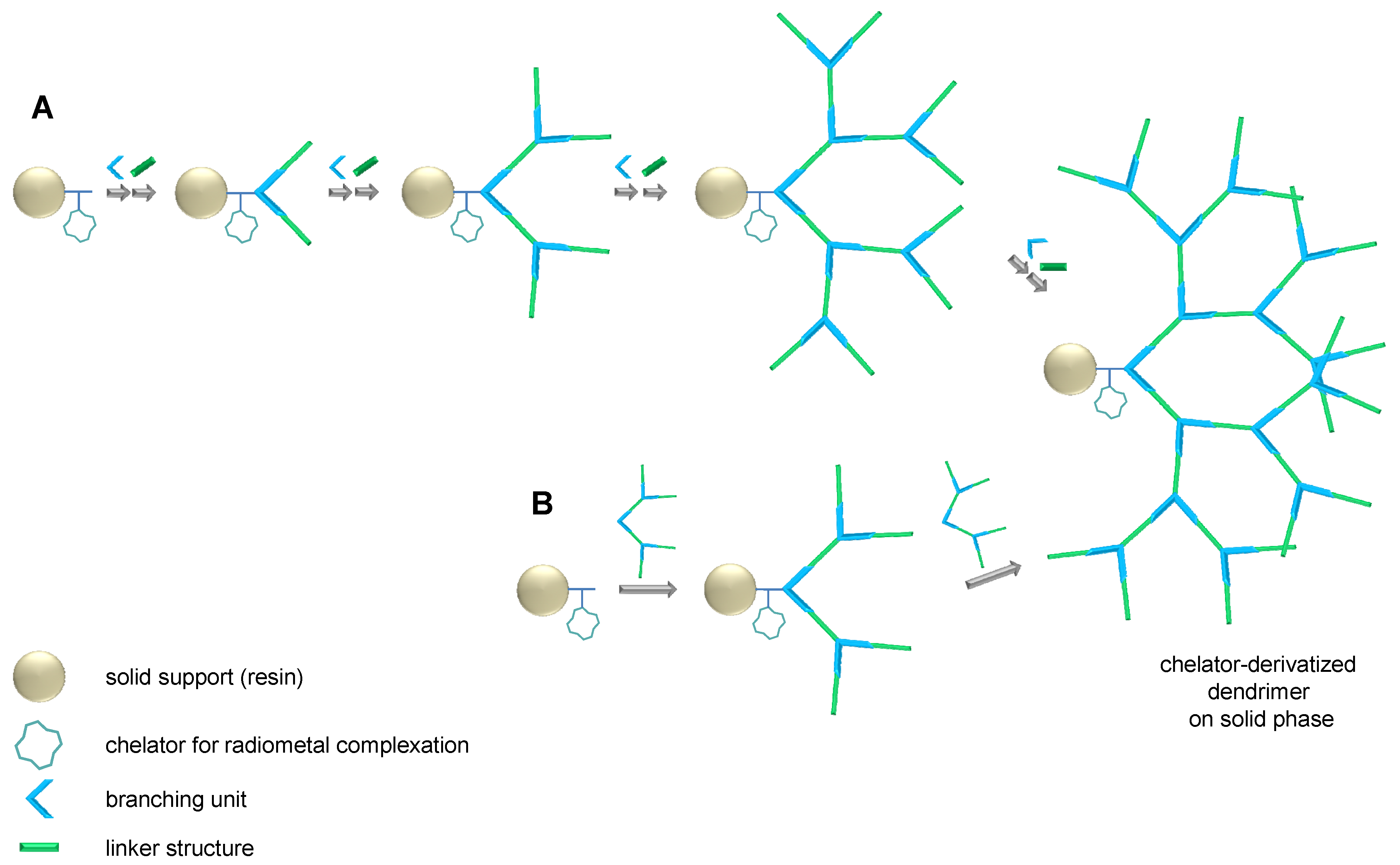
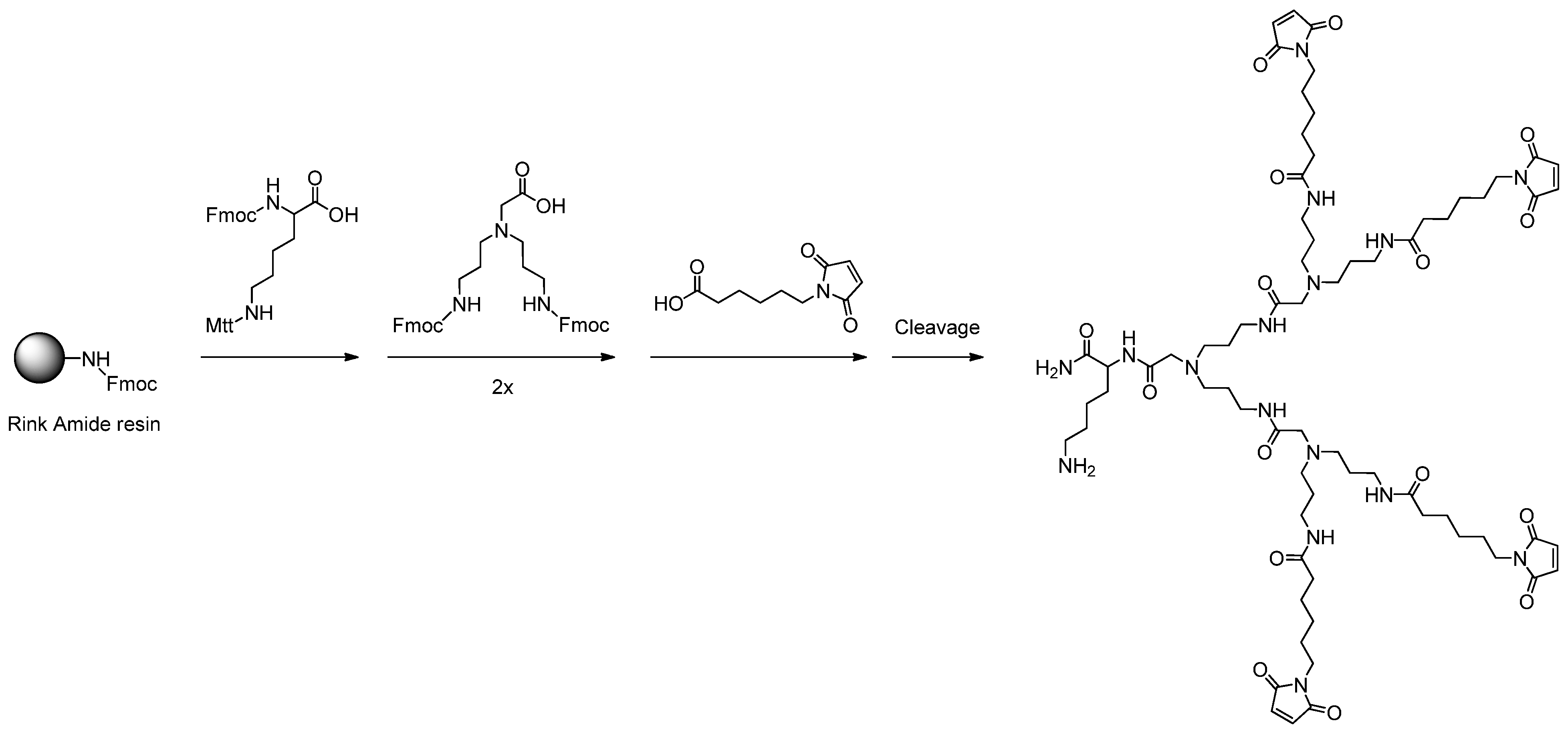
2.1. Dendron Scaffold Synthesis and Design

2.1.1. Influence of OEG Linkers on Product Purities and Yields
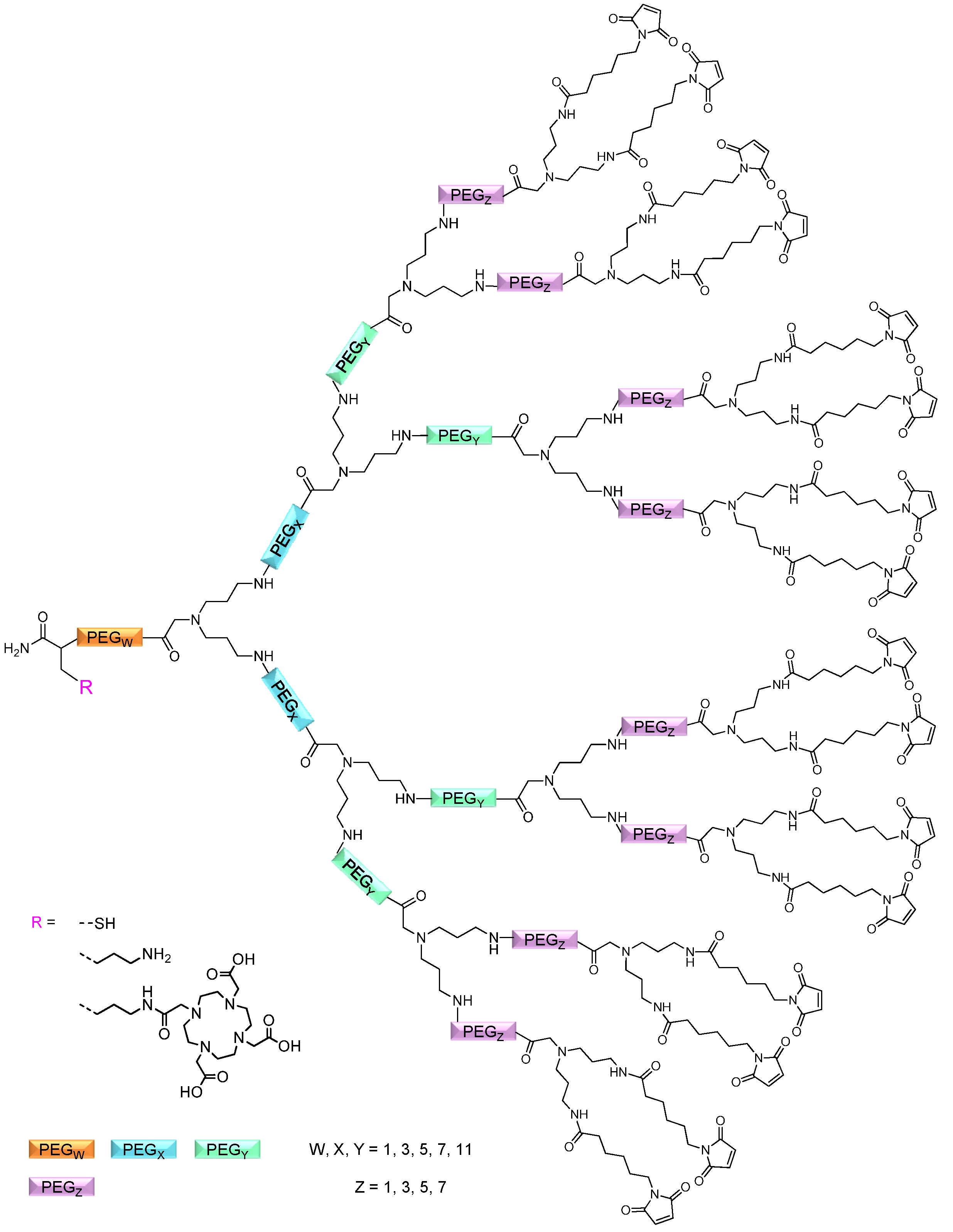
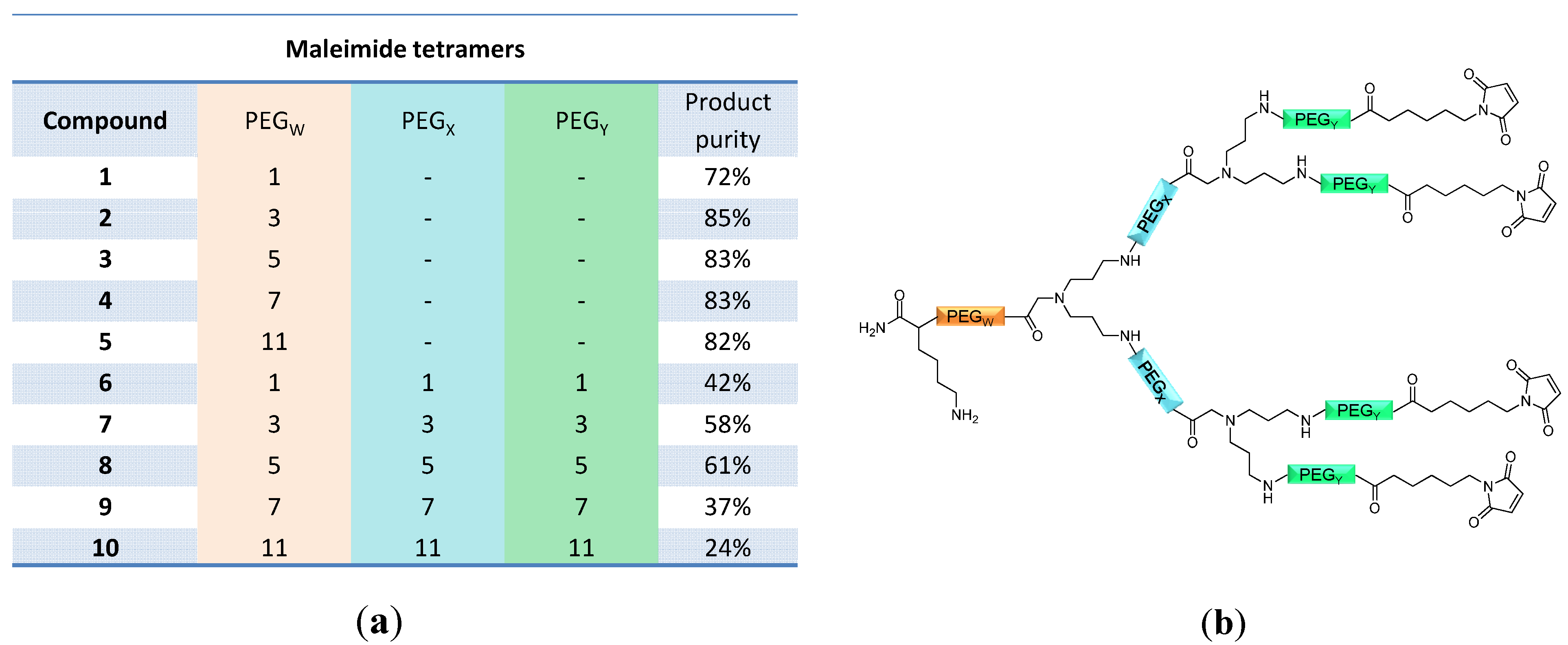
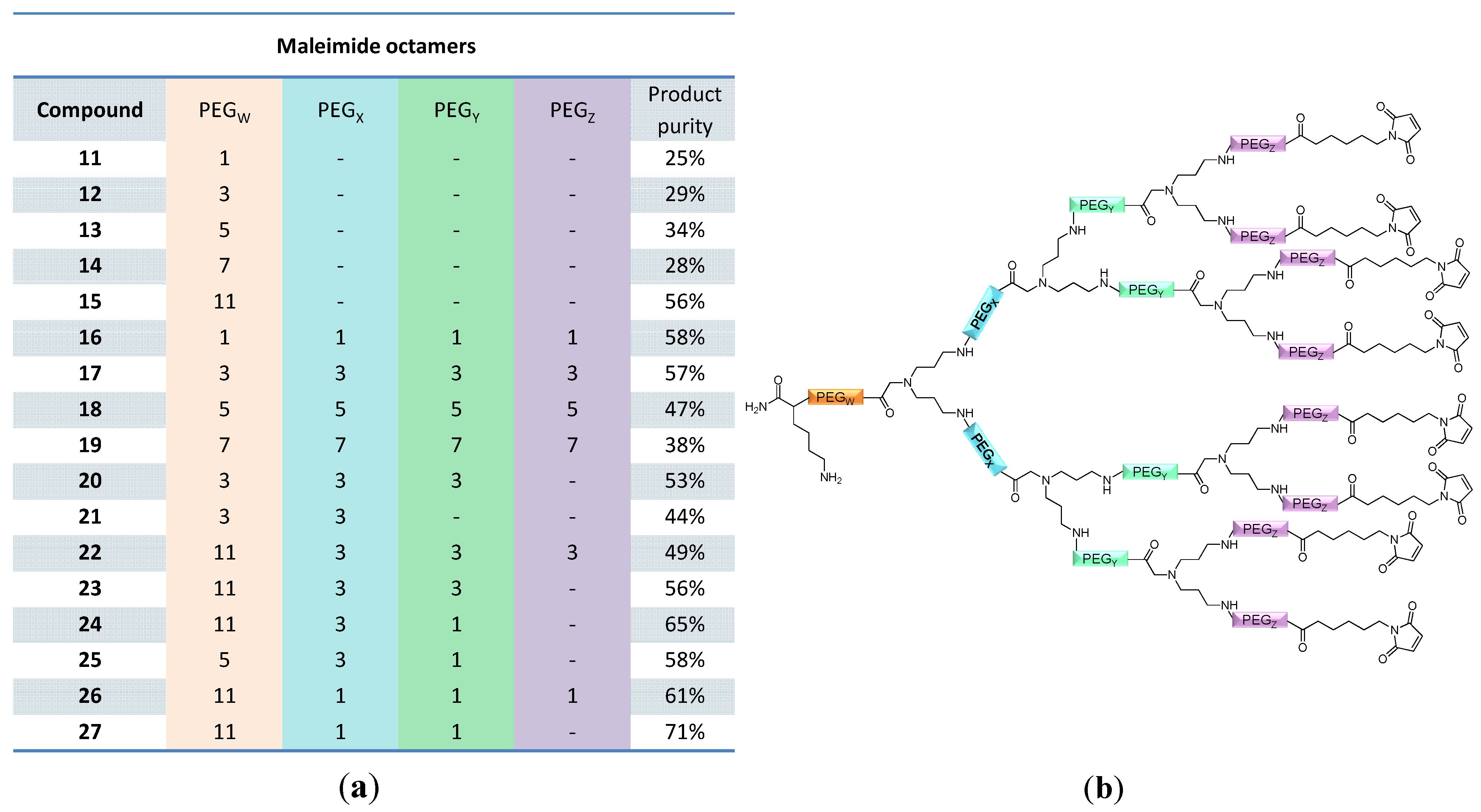
 linker, Figure 3 and Figure 4) was systematically optimized for the tetrameric and octameric maleimides using OEGs of different lengths (PEG1, PEG3, PEG5, PEG7 and PEG11; OEG linkers are denoted as “PEGs” due to their trade names). For the tetramers, the length of the applied OEG did not seem to have a crucial influence on the achievable product purities after cleavage as long as it exceeded the length of a PEG1 (1–5, Figure 3). linker (11–15, Figure 4). Thus, the importance to initially implement a long linker structure before assembling the dendron is of higher importance for larger constructs which can be attributed to their larger size and thus steric demand necessitating a larger distance to the resin for an efficient synthesis.
linker, Figure 3 and Figure 4) was systematically optimized for the tetrameric and octameric maleimides using OEGs of different lengths (PEG1, PEG3, PEG5, PEG7 and PEG11; OEG linkers are denoted as “PEGs” due to their trade names). For the tetramers, the length of the applied OEG did not seem to have a crucial influence on the achievable product purities after cleavage as long as it exceeded the length of a PEG1 (1–5, Figure 3). linker (11–15, Figure 4). Thus, the importance to initially implement a long linker structure before assembling the dendron is of higher importance for larger constructs which can be attributed to their larger size and thus steric demand necessitating a larger distance to the resin for an efficient synthesis. 

 ,
,  and
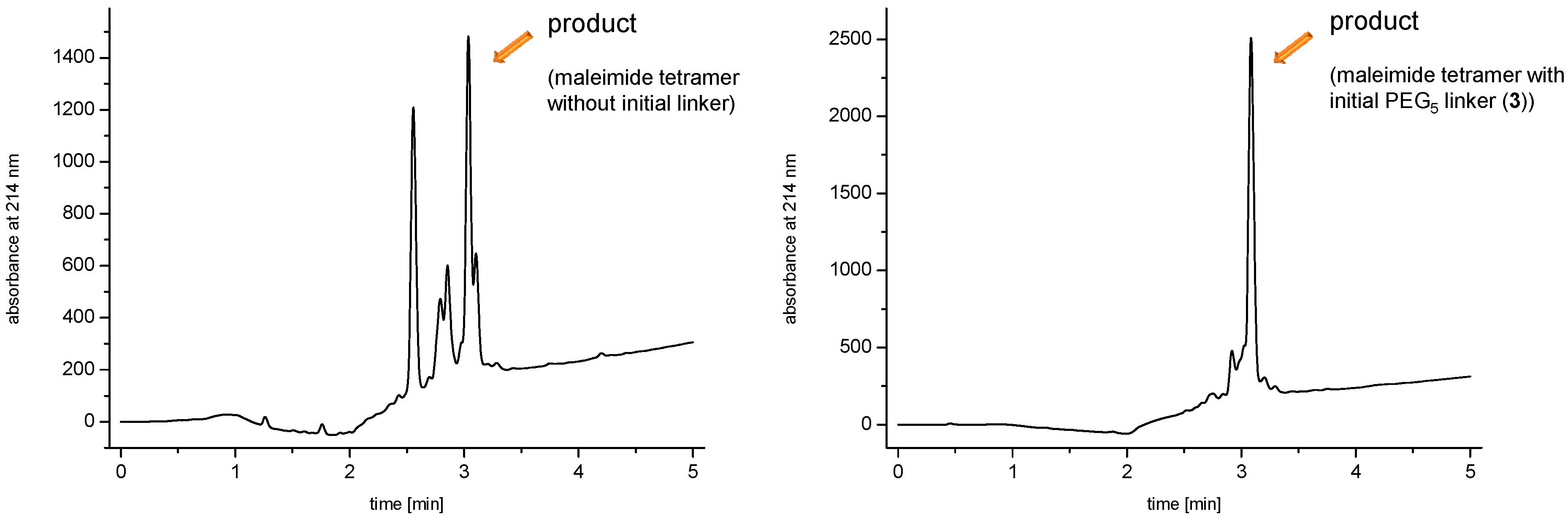
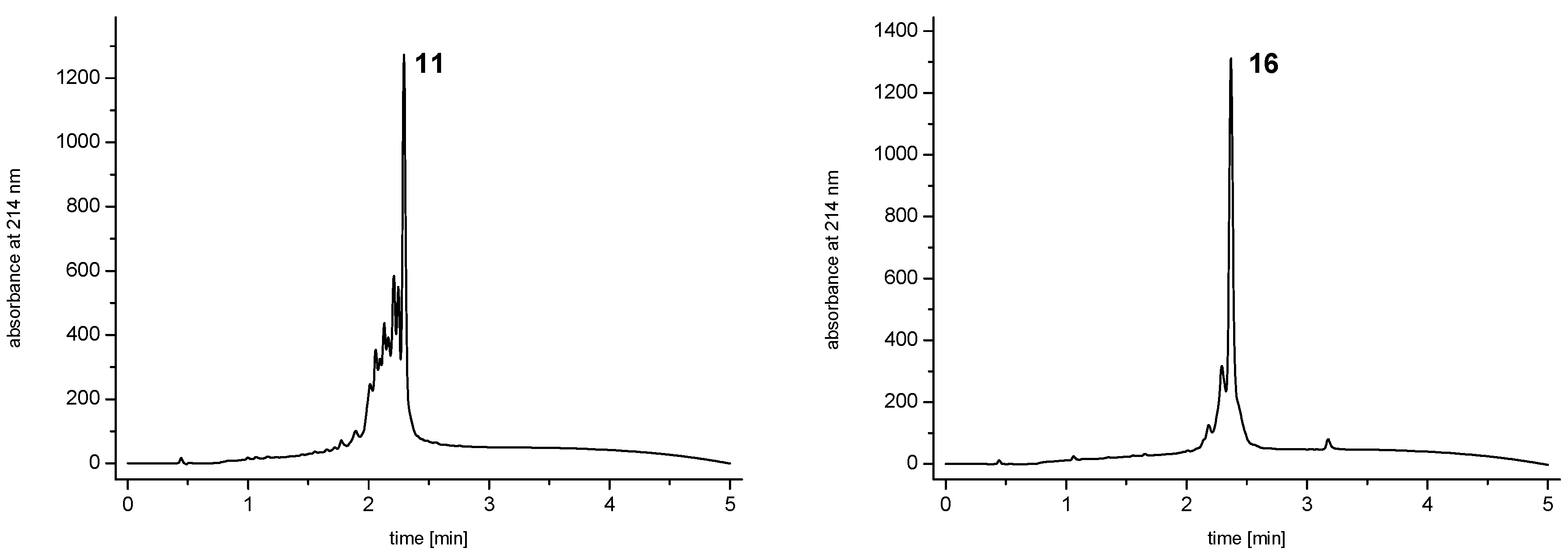
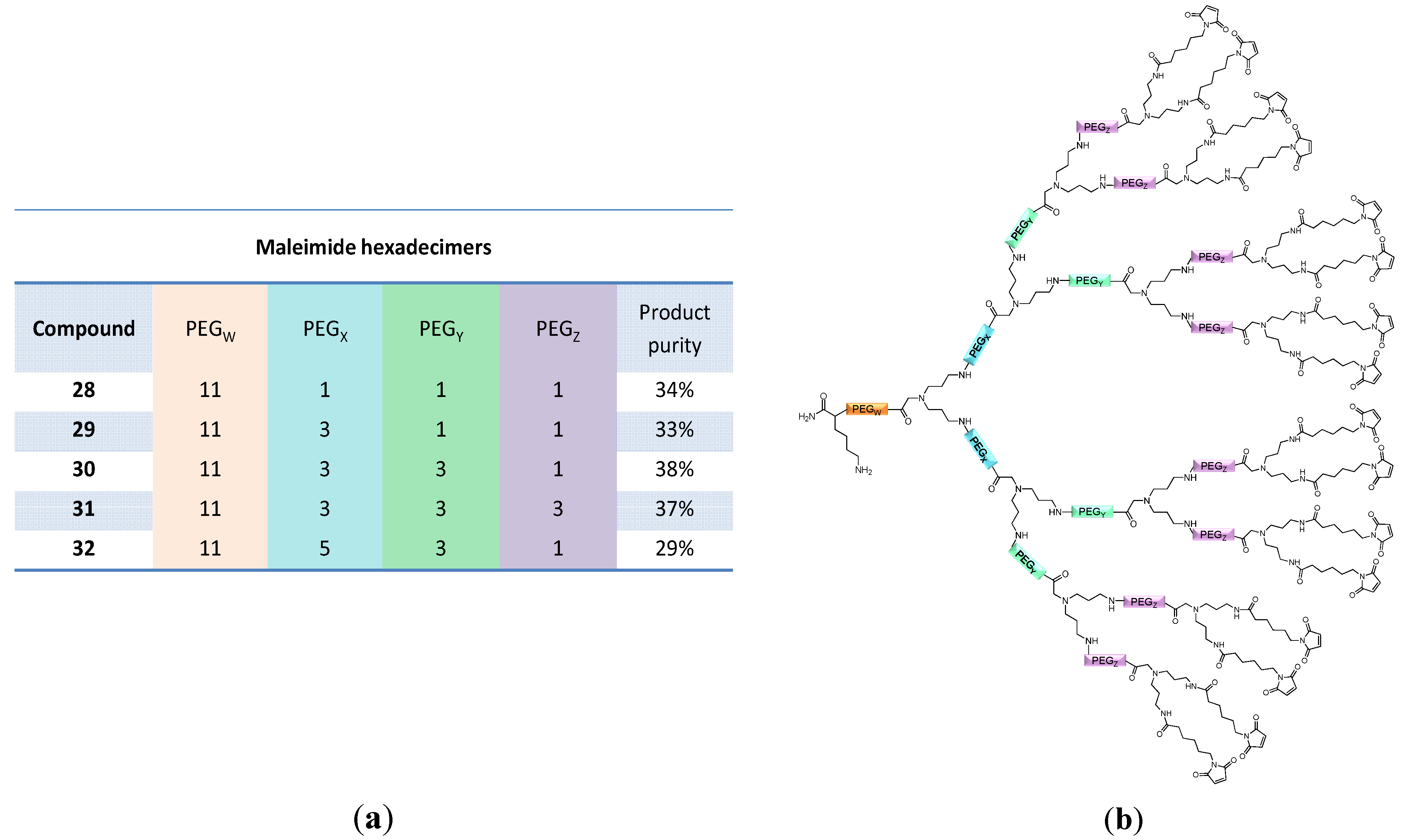
and  linkers, Figure 4), OEGs of different lengths were in the following not only implemented between the initial amino acid and the focal point of the dendrons ( linker), but also after each branching amino acid. Systematically investigating the influence of these additional OEG linkers, it was found that the tetrameric maleimide dendrons could not profit from the introduction of further ( and ) linkers (6–10, Figure 3). linker—the introduction of additional OEG ( , and ) linkers was however able to result in a considerable increase of product formation in case of octavalent maleimides (16–27, Figure 4). The positive influence however strongly depended on the linker lengths used. So could for example be shown that the product purities of the raw materials increased significantly when inserting an additional linker after each branching amino acid (comparing the raw product purities of 11 and 16 (Figure 5), 12 and 17, 13 and 18 as well as 14 and 19) but that PEG1-linkers gave the best results when using the same linker length in every position ( , and ; comparing product purities of 16, 17, 18 and 19).
linkers, Figure 4), OEGs of different lengths were in the following not only implemented between the initial amino acid and the focal point of the dendrons ( linker), but also after each branching amino acid. Systematically investigating the influence of these additional OEG linkers, it was found that the tetrameric maleimide dendrons could not profit from the introduction of further ( and ) linkers (6–10, Figure 3). linker—the introduction of additional OEG ( , and ) linkers was however able to result in a considerable increase of product formation in case of octavalent maleimides (16–27, Figure 4). The positive influence however strongly depended on the linker lengths used. So could for example be shown that the product purities of the raw materials increased significantly when inserting an additional linker after each branching amino acid (comparing the raw product purities of 11 and 16 (Figure 5), 12 and 17, 13 and 18 as well as 14 and 19) but that PEG1-linkers gave the best results when using the same linker length in every position ( , and ; comparing product purities of 16, 17, 18 and 19). 

2.1.2. Optimization of Other Reaction Parameters
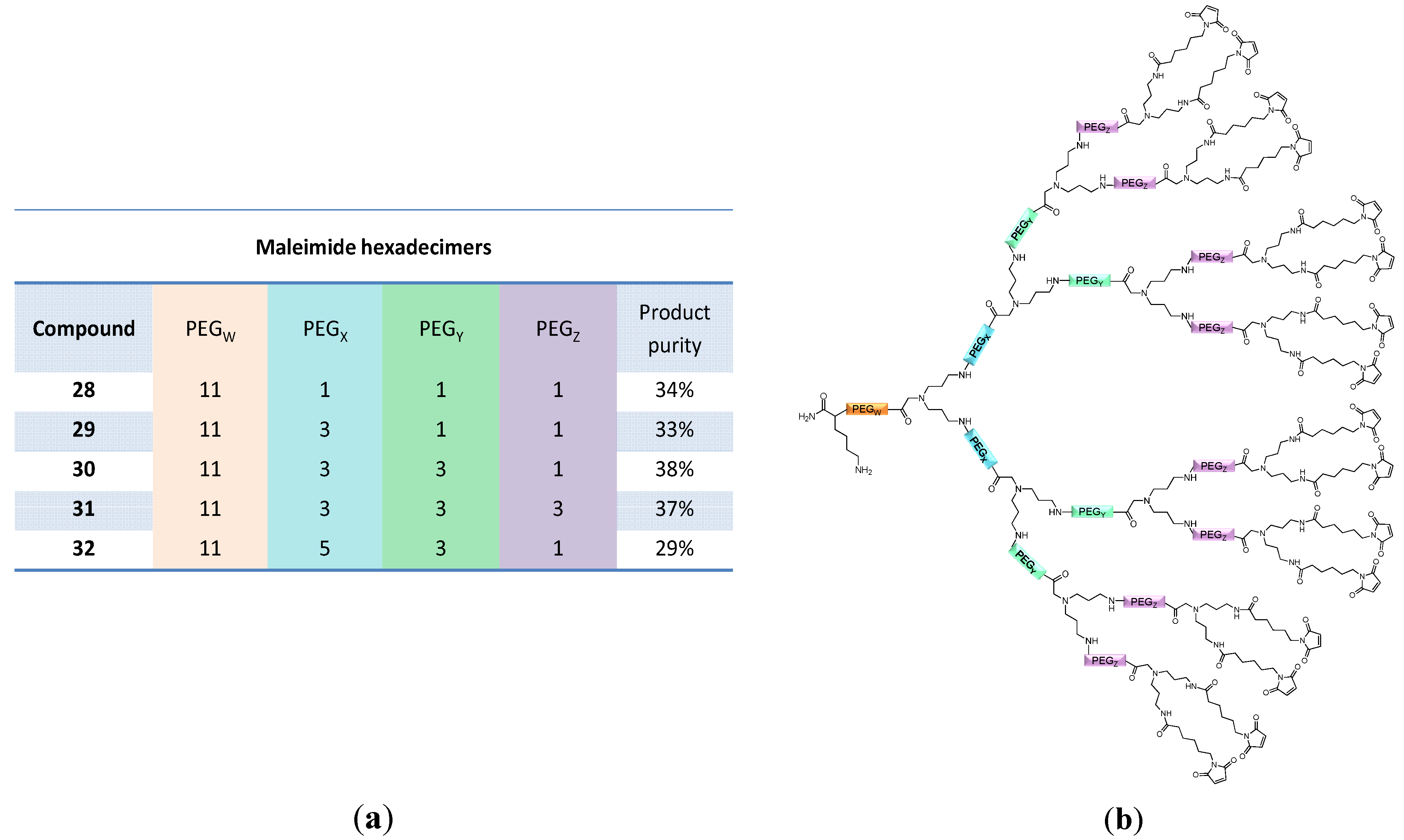
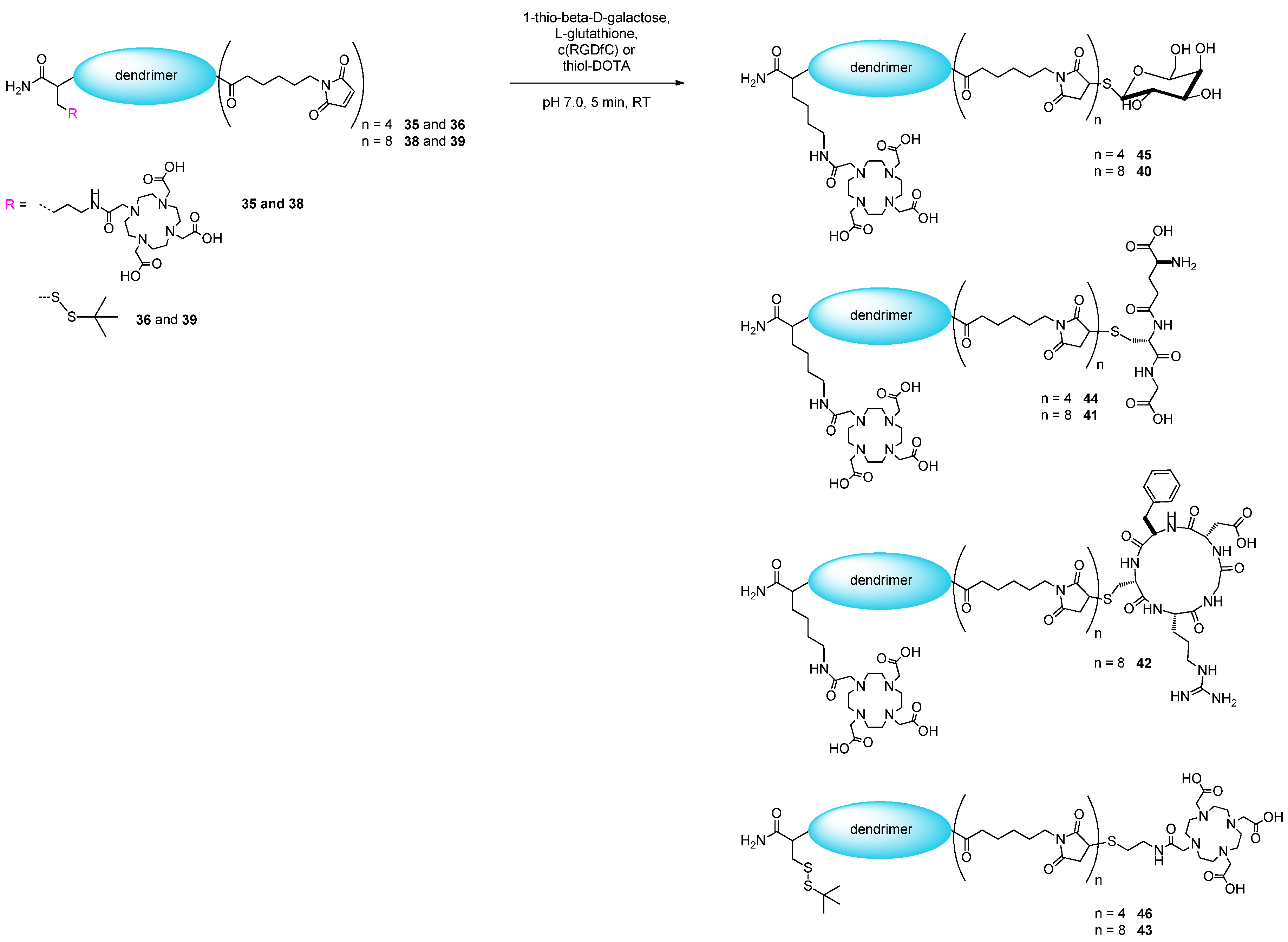
2.2. Applicability of the Multivalent Maleimides in Multimerization Reactions


2.3. 68Ga-Radiolabeling of the Octavalent Substances 40–43
3. Experimental
3.1. General Information
3.2. General Procedure for the Solid Phase-Assisted Synthesis of Dendron Scaffolds
3.3. General Procedure for the Conjugation of Thiol-Bearing Synthons to the Multivalent Maleimide Scaffolds
3.4. 68Ga-Radiolabeling of the DOTA-Comprising Multimers 40–43
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lo, S.T.; Kumar, A.; Hsieh, J.T.; Sun, X.K. Dendrimer nanoscaffolds for potential theranostics of prostate cancer with a focus on radiochemistry. Mol. Pharm. 2013, 10, 793–812. [Google Scholar] [CrossRef]
- Stanczyk, M.; Dziki, A.; Morawiec, Z. Dendrimers in therapy for breast and colorectal cancer. Curr. Med. Chem. 2012, 19, 4896–4902. [Google Scholar] [CrossRef]
- Yavuz, B.; Pehlivan, S.B.; Unlu, N. Dendrimeric systems and their applications in ocular drug delivery. Sci. World J. 2013, 732340:1–732340:13. [Google Scholar]
- Lebreton, S.; Monaghan, S.; Bradley, M. Solid-phase dendrimer chemistry: Synthesis and applications. Aldrichim. Acta 2001, 34, 75–83. [Google Scholar]
- Huang, A.Y.T.; Tsai, C.H.; Chen, H.Y.; Chen, H.T.; Lu, C.Y.; Lin, Y.T.; Kao, C.L. Concise solid-phase synthesis of inverse poly(amidoamine) dendrons using AB(2) building blocks. Chem. Commun. 2013, 49, 5784–5786. [Google Scholar]
- Wells, N.J.; Basso, A.; Bradley, M. Solid-phase dendrimer synthesis. Biopolymers 1998, 47, 381–396. [Google Scholar] [CrossRef]
- Swali, V.; Wells, N.J.; Langley, G.J.; Bradley, M. Solid-phase dendrimer synthesis and the generation of super-high-loading resin beads for combinatorial chemistry. J. Org. Chem. 1997, 62, 4902–4903. [Google Scholar] [CrossRef]
- Kowalczyk, W.; Monso, M.; de la Torre, B.G.; Andreu, D. Synthesis of multiple antigenic peptides (MAPs)-strategies and limitations. J. Pept. Sci. 2011, 17, 247–251. [Google Scholar]
- Kantchev, E.A.B.; Chang, C.C.; Cheng, S.F.; Roche, A.C.; Chang, D.K. Direct solid-phase synthesis and fluorescence labeling of large, monodisperse mannosylated dendrons in a peptide synthesizer. Org. Biomol. Chem. 2008, 6, 1377–1385. [Google Scholar] [CrossRef]
- Kantchev, E.A.B.; Chang, C.C.; Chang, D.K. Direct Fmoc/tert-Bu solid phase synthesis of octamannosyl polylysine dendrimer-peptide conjugates. Biopolymers 2006, 84, 232–240. [Google Scholar] [CrossRef]
- Darbre, T.; Reymond, J.L. Peptide dendrimers as artificial enzymes, receptors, and drug-delivery agents. Accounts Chem. Res. 2006, 39, 925–934. [Google Scholar] [CrossRef]
- Clouet, A.; Darbre, T.; Reymond, J.L. Combinatorial synthesis, selection, and properties of esterase peptide dendrimers. Biopolymers 2006, 84, 114–123. [Google Scholar] [CrossRef]
- Sanclimens, G.; Crespo, L.; Giralt, E.; Albericio, F.; Royo, M. Preparation of de novo globular proteins based on proline dendrimers. J. Org. Chem. 2005, 70, 6274–6281. [Google Scholar] [CrossRef]
- Sanclimens, G.; Crespo, L.; Giralt, E.; Royo, M.; Albericio, F. Solid-phase synthesis of second-generation polyproline dendrimers. Biopolymers 2004, 76, 283–297. [Google Scholar] [CrossRef]
- Crespo, L.; Sanclimens, G.; Royo, M.; Giralt, E.; Albericio, F. Branched poly(proline) peptides: An efficient new approach to the synthesis of repetitive branched peptides. Eur. J. Org. Chem. 2002, 2002, 1756–1762. [Google Scholar] [CrossRef]
- Tsushima, H.; Matsumoto, K.; Jikei, M. Solid-phase synthesis of aromatic polyamide dendrons. Polym. Advan. Technol. 2011, 22, 1292–1296. [Google Scholar] [CrossRef]
- Spasser, L.; Portnoy, M. Solid-phase synthesis of uniform linear oligoethers with repeating functional arms as multivalent spacers. J. Polym. Sci. Pol. Chem. 2010, 48, 6009–6013. [Google Scholar] [CrossRef]
- Dahan, A.; Portnoy, M. Synthesis of poly(aryl benzyl ether) dendrimers on solid support. Macromolecules 2003, 36, 1034–1038. [Google Scholar] [CrossRef]
- Basso, A.; Evans, B.; Pegg, N.; Bradley, M. Solid phase synthesis of aryl-ether dendrimers. Chem. Commun. 2001, 2001, 697–698. [Google Scholar]
- How, S.E.; Unciti-Broceta, A.; Sanchez-Martin, R.M.; Bradley, M. Solid-phase synthesis of a lysine-capped bis-dendron with remarkable DNA delivery abilities. Org. Biomol. Chem. 2008, 6, 2266–2269. [Google Scholar] [CrossRef]
- Lebreton, S.; How, S.E.; Buchholz, M.; Yingyongnarongkul, B.E.; Bradley, M. Solid-phase construction: High efficiency dendrimer synthesis using AB(3) isocyanate-type monomers. Tetrahedron 2003, 59, 3945–3953. [Google Scholar] [CrossRef]
- Lebreton, S.; Newcombe, N.; Bradley, M. Rapid synthesis of high-loading resins using triple branched protected monomer for dendrimer synthesis. Tetrahedron Lett. 2002, 43, 2475–2478. [Google Scholar] [CrossRef]
- Lindner, S.; Michler, C.; Wängler, B.; Bartenstein, P.; Fischer, G.; Schirrmacher, R.; Wängler, C. PESIN multimerization improves receptor avidities and in vivo tumor targeting properties to GRPR-overexpressing tumors. Bioconjugate Chem. 2014, 25, 489–500. [Google Scholar] [CrossRef]
- Gao, J.M.; Chen, P.M.; Singh, Y.; Zhang, X.P.; Szekely, Z.; Stein, S.; Sinko, P.J. Novel monodisperse PEGtide dendrons: Design, fabrication, and evaluation of mannose receptor-mediated macrophage targeting. Bioconjugate Chem. 2013, 24, 1332–1344. [Google Scholar] [CrossRef]
- Dijkgraaf, I.; Yim, C.B.; Franssen, G.M.; Schuit, R.C.; Luurtsema, G.; Liu, S.A.; Oyen, W.J.G.; Boerman, O.C. PET imaging of αvβ3 integrin expression in tumours with Ga-68-labelled mono-, di- and tetrameric RGD peptides. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 128–137. [Google Scholar] [CrossRef]
- Fournier, P.; Dumulon-Perreault, V.; Ait-Mohand, S.; Langlois, R.; Benard, F.; Lecomte, R.; Guerin, B. Comparative study of 64Cu/NOTA-[D-Tyr6,betaAla11,Thi13,Nle14]BBN(6–14) monomer and dimers for prostate cancer PET imaging. EJNMMI Res. 2012. [Google Scholar] [CrossRef]
- Jacobson, O.; Zhu, L.; Niu, G.; Weiss, I.D.; Szajek, L.P.; Ma, Y.; Sun, X.; Yan, Y.; Kiesewetter, D.O.; Liu, S.; et al. MicroPET imaging of integrin alphavbeta3 expressing tumors using 89Zr-RGD peptides. Mol. Imaging Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef]
- Lang, L.X.; Li, W.H.; Guo, N.; Ma, Y.; Zhu, L.; Kiesewetter, D.O.; Shen, B.Z.; Niu, G.; Chen, X.Y. Comparison study of [F-18]FAI-NOTA-PRGD2, [F-18]FPPRGD2, and [Ga-68]Ga-NOTA-PRGD2 for PET imaging of U87MG tumors in mice. Bioconjugate Chem. 2011, 22, 2415–2422. [Google Scholar] [CrossRef]
- Wängler, C.; Maschauer, S.; Prante, O.; Schafer, M.; Schirrmacher, R.; Bartenstein, P.; Eisenhut, M.; Wängler, B. Multimerization of cRGD peptides by click chemistry: Synthetic strategies, chemical limitations, and influence on biological properties. ChemBioChem 2010, 11, 2168–2181. [Google Scholar] [CrossRef]
- Mittra, E.S.; Goris, M.L.; Iagaru, A.H.; Kardan, A.; Burton, L.; Berganos, R.; Chang, E.; Liu, S.L.; Shen, B.; Chin, F.T.; et al. Pilot pharmacokinetic and dosimetric studies of F-18-FPPRGD2: A PET radiopharmaceutical agent for imaging αvβ3 integrin levels. Radiology 2011, 260, 182–191. [Google Scholar] [CrossRef]
- Oxboel, J.; Brandt-Larsen, M.; Schjoeth-Eskesen, C.; Myschetzky, R.; El-Ali, H.H.; Madsen, J.; Kjaer, A. Comparison of two new angiogenesis PET tracers Ga-68-NODAGA-E[c(RGDyK)](2) and Cu-64-NODAGA-E[c(RGDyK)](2); in vivo imaging studies in human xenograft tumors. Nucl. Med. Biol. 2014, 41, 259–267. [Google Scholar] [CrossRef]
- Liu, S. Radiolabeled cyclic RGD peptides as integrin αvβ3-targeted radiotracers: Maximizing binding affinity via bivalency. Bioconjugate Chem. 2009, 20, 2199–2213. [Google Scholar] [CrossRef]
- Gillies, E.R.; Dy, E.; Frechet, J.M.J.; Szoka, F.C. Biological evaluation of polyester dendrimer: Poly(ethylene oxide) "Bow-Tie" hybrids with tunable molecular weight and architecture. Mol. Pharmaceut. 2005, 2, 129–138. [Google Scholar] [CrossRef]
- Liu, Z.F.; Yan, Y.J.; Chin, F.T.; Wang, F.; Chen, X.Y. Dual integrin and gastrin-releasing peptide receptor targeted tumor imaging using F-18-labeled PEGylated RGD-bombesin heterodimer F-18-FB-PEG(3)-Glu-RGD-BBN. J. Med. Chem. 2009, 52, 425–432. [Google Scholar] [CrossRef]
- Wängler, C.; Moldenhauer, G.; Eisenhut, M.; Haberkorn, U.; Mier, W. Antibody-dendrimer conjugates: The number, not the size of the dendrimers, determines the immunoreactivity. Bioconjugate Chem. 2008, 19, 813–820. [Google Scholar] [CrossRef]
- Liu, S. Radiolabeled multimeric cyclic RGD peptides as integrin αvβ3 targeted radiotracers for tumor imaging. Mol. Pharmaceut. 2006, 3, 472–487. [Google Scholar] [CrossRef]
- Breeman, W.A.P.; de Jong, M.; de Blois, E.; Bernard, B.F.; Konijnenberg, M.; Krenning, E.P. Radiolabelling DOTA-peptides with Ga-68. Eur. J. Nuc. Med. Mol. Imaging 2005, 32, 478–485. [Google Scholar] [CrossRef]
- Cheng, C.; Pan, L.; Dimitrakopoulou-Strauss, A.; Schäfer, M.; Wängler, C.; Wängler, B.; Haberkorn, U.; Strauss, L.G. Comparison between 68Ga-bombesin (68Ga-BZH3) and the cRGD tetramer 68Ga-RGD4 studies in an experimental nude rat model with a neuroendocrine pancreatic tumor cell line. EJNMMI Res. 2011. [Google Scholar] [CrossRef]
- Wängler, C.; Wängler, B.; Eisenhut, M.; Haberkorn, U.; Mier, W. Improved syntheses and applicability of different DOTA building blocks for multiply derivatized scaffolds. Bioorg. Med. Chem. 2008, 16, 2606–2616. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available.
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fischer, G.; Wängler, B.; Wängler, C. Optimized Solid Phase-Assisted Synthesis of Dendrons Applicable as Scaffolds for Radiolabeled Bioactive Multivalent Compounds Intended for Molecular Imaging. Molecules 2014, 19, 6952-6974. https://doi.org/10.3390/molecules19066952
Fischer G, Wängler B, Wängler C. Optimized Solid Phase-Assisted Synthesis of Dendrons Applicable as Scaffolds for Radiolabeled Bioactive Multivalent Compounds Intended for Molecular Imaging. Molecules. 2014; 19(6):6952-6974. https://doi.org/10.3390/molecules19066952
Chicago/Turabian StyleFischer, Gabriel, Björn Wängler, and Carmen Wängler. 2014. "Optimized Solid Phase-Assisted Synthesis of Dendrons Applicable as Scaffolds for Radiolabeled Bioactive Multivalent Compounds Intended for Molecular Imaging" Molecules 19, no. 6: 6952-6974. https://doi.org/10.3390/molecules19066952