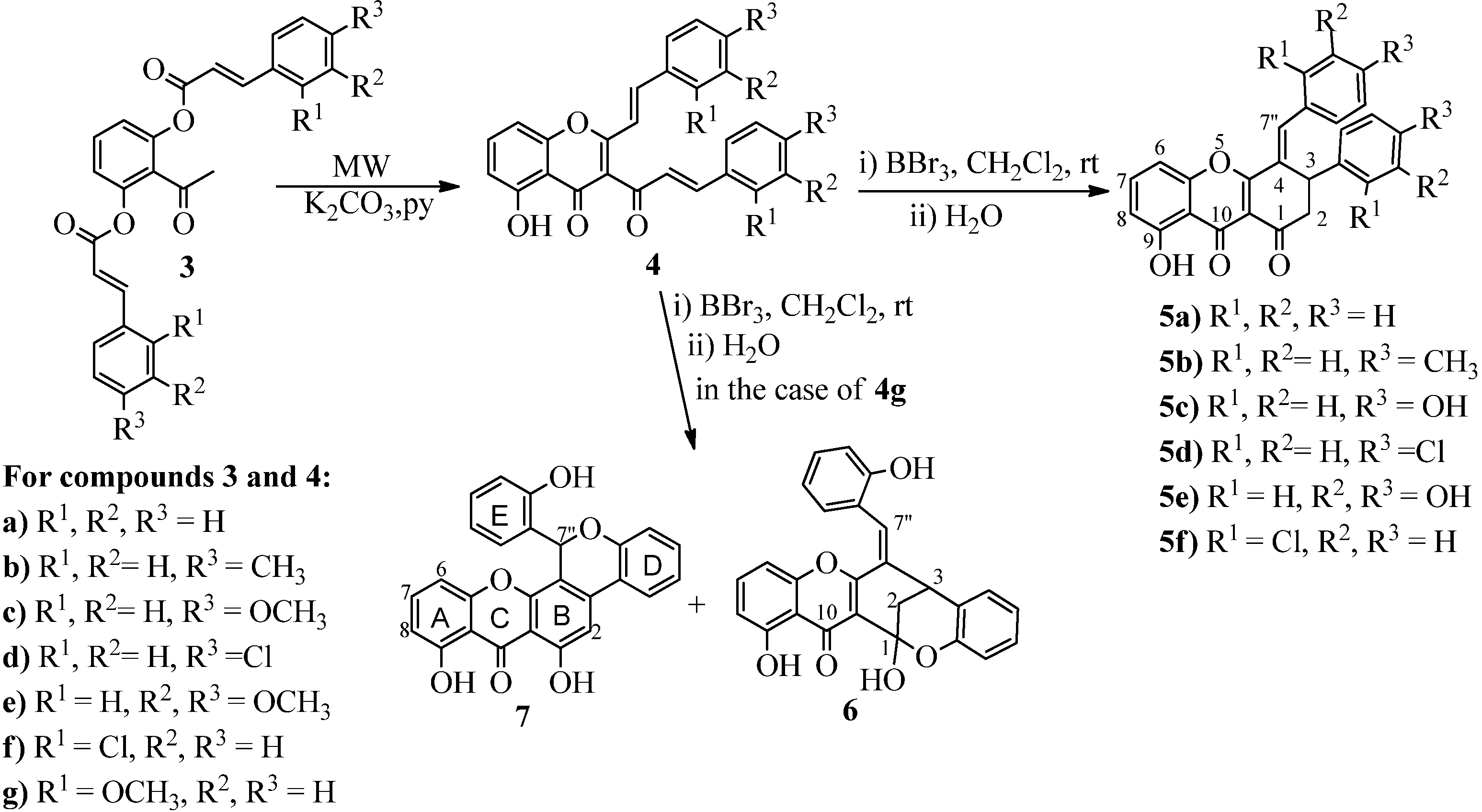
3.2. Synthesis
Compounds
3 and
4 were synthesised according to previously reported methodologies and the obtained NMR data were also identical to the data previously reported in the literature [
11,
12]. In the case of xanthenediones
5, although the synthetic methodology was previously reported only the characterization of derivatives
5c and
5d had been published [
11].
(E)-3-Aryl-4-benzylidene-8-hydroxy-3,4-dihydro-1H-xanthene-1,9(2H)-dione (5a). This compound was obtained by the reaction of (E,E)-3-cinnamoyl-5-hydroxy-2-styrylchromone (4a, 157.8 mg, 0.400 mmol) with BBr3 (2 mL of a 1 mol L−1 solution in CH2Cl2). After the slowly addition of BBr3 the mixture was stirred at room temp. for 3 h, then water (80 mL) was added. The resulting reaction mixture was stirred for 4 h and extracted with CH2Cl2 (3 × 80 mL). The combined extracts were evaporated and purified by TLC using CH2Cl2 as eluent. The desired compound 5a was obtained as yellow crystals (107.3 mg, 68%). M.p. 216–218 °C. 1H-NMR (300 MHz, CDCl3, TMS) δ (ppm): 3.08 (dd, 1H, J = 3.2 and 15.5 Hz, H-2trans); 3.15 (dd, 1H, J = 5.0 and 15.5 Hz, H-2cis); 4.76 (dd, 1H, J = 3.2 and 5.0 Hz, H-3); 6.82 (dd, 1H, J = 0.7 and 8.4 Hz, H-8); 7.02 (dd, 1H, J = 0.7 and 8.4 Hz, H-6); 7.24–7.30 (m, 3H, H-2',6' and H-4'); 7.31–7.36 (m, 2H, H-3',5'); 7.38 (br s, 5H, H-2",6", H-3",5" and H-4"); 7.57 (t, 1H, J = 8.4 Hz, H-7); 8.11 (s, 1H, H-7"); 12.63 (s, 1H, 9-OH). 13C-NMR (75 MHz, CDCl3, TMS) δ (ppm): 40.1 (C-3); 45.6 (C-2); 106.8 (C-6); 111.0 (C-9a); 112.7 (C-8); 113.4 (C-10a); 126.9 (C-2' and C-6'); 127.5 (C-4'); 128.9 (C-3" and C-5"); 129.2 (C-3' and C-5'); 129.5 (C-2" and C-6"); 129.7 (C-4); 129.9 (C-4"); 134.2 (C-1"); 136.0 (C-7); 139.0 (C-7"); 139.9 (C-1'); 154.7 (C-5a); 161.7 (C-9); 169.4 (C-4a); 180.1 (C-10); 191.3 (C-1). ESI-MS m/z 395 [M+H]+; 417 [M+Na]+; 433 [M+K]+. HR-ESIMS (positive ion): m/z calcd. for C26H19O4 [M+H]+, 395.1278; found, 395.1275.
(E)-8-Hydroxy-4-(4-methylbenzylidene)-3-(4-methylphenyl)-3,4-dihydro-1H-xanthene-1,9(2H)-dione (5b). This compound was obtained by the reaction of (E,E)-3-cinnamoyl-5-hydroxy-2-styrylchromone (4b, 172.8 mg, 0.409 mmol) with BBr3 (2 mL of a 1 mol L solution in CH2Cl2) and following the above described procedure. The desired compound 5b was obtained as yellow crystals (153.8 mg, 89%). M.p. 201–202 °C. 1H-NMR (300 MHz, CDCl3, TMS) δ (ppm): 2.29 (s, 3H, 4'-CH3); 2.37 (s, 3H, 4"-CH3); 3.05 (dd, 1H, J = 3.1 and 15.5 Hz, H-2trans); 3.12 (dd, 1H, J = 5.1 and 15.5 Hz, H-2cis); 4.74–4.76 (m, 1H, H-3); 6.82 (dd, 1H, J = 0.8 and 8.3 Hz, H-8); 7.02 (dd, 1H, J = 0.8 and 8.3 Hz, H-6); 7.10 (d, 2H, J = 8.0 Hz, H-3',5'); 7.13 (d, 1H, J = 8.0 Hz, H-2',6'); 7.18 (d, 2H, J = 8.1 Hz, H-3",5"); 7.29 (d, 2H, J = 8.1 Hz, H-2",6"); 7.56 (t, 1H, J = 8.3 Hz, H-7); 8.07 (s, 1H, H-7"); 12.67 (s, 1H, 9-OH). 13C-NMR (75 MHz, CDCl3, TMS) δ (ppm): 20.9 (4'-CH3); 21.4 (4"-CH3); 39.9 (C-3); 45.6 (C-2); 106.8 (C-6); 111.0 (C-9a); 112.6 (C-8); 113.3 (C-10a); 126.8 (C-2' and C-6'); 129.0 (C-4); 129.6 (C-3" and C-5"); 129.7 (C-3' and C-5'); 129.9 (C-2" and C-6"); 131.4 (C-1"); 135.9 (C-7); 136.8 (C-1'); 137.1 (C-4'); 139.0 (C-7"); 140.5 (C-4"); 154.7 (C-5a); 161.7 (C-9); 169.7 (C-4a); 180.2 (C-10); 191.5 (C-1). ESI-MS m/z 423 [M+H]+; 445 [M+Na]+. HR-ESIMS (positive ion): m/z calcd. for C28H22O4 [M]+, 423.1591; found, 423.1587.
(E)-8-Hydroxy-4-(3,4-dihydroxybenzylidene)-3-(3,4-dihydroxyphenyl)-3,4-dihydro-1H-xanthene-1,9-(2H)-dione (5e). This compound was obtained by the reaction of (E,E)-3-cinnamoyl-5-hydroxy-2-styrylchromone (4e, 205.8 mg, 0.400 mmol) with BBr3 (2 mL of a 1 mol L−1 solution in CH2Cl2) and following the above described procedure. The desired compound 5e was filtered off and obtained as orange crystals (183.2 mg, 89%). M.p. 275–281 °C. 1H-NMR (500 MHz, DMSO-d6, TMS) δ (ppm): 2.68 (dd, 1H, J = 2.7 and 14.8 Hz, H-2trans); 3.16 (dd, 1H, J = 5.7 and 14.8 Hz, H-2cis); 4.60 (dbr, 1H, J = 2.7 Hz, H-3); 6.57 (dd, 1H, J = 2.3 and 8.2 Hz, H-6'); 6.67 (d, 1H, J = 2.3 Hz, H-2'); 6.67 (d, 1H, J = 8.2 Hz, H-5');6.77 (d, 1H, J = 8.3 Hz, H-5"); 6.83 (d, 1H, J = 8.3 Hz, H-8); 6.87 (dd, 1H, J = 1.8 and 8.3 Hz, H-6"); 6.97 (d, 1H, J = 1.8 Hz, H-2"); 7.29 (d, 1H, J = 8.3 Hz, H-6); 7.71 (t, 1H, J = 8.3 Hz, H-7); 8.05 (s, 1H, H-7"); 8.87 (s, 1H, 4'-OH); 8.91 (s, 1H, 3'-OH); 9.19 (s, 1H, 3"-OH); 9.84 (s, 1H, 4"-OH); 12.77 (s, 1H, 9-OH). 13C-NMR (75 MHz, DMSO-d6, TMS) δ (ppm): 39.3 (C-3); 46.2 (C-2); 107.8 (C-6); 110.3 (C-9a); 111.9 (C-8); 112.4 (C-10a); 114.3 (C-2'); 115.8 (C-5'); 116.1 (C-5"); 117.9 (C-6' and C-2"); 123.5 (C-6"); 125.9 (C-4*); 126.0 (C-1"*); 131.4 (C-1'); 136.4 (C-7); 139.4 (C-7"); 144.3 (C-3'); 145.4 (C-4'**); 145.5 (C-3"**); 148.3 (C-4"); 154.5 (C-5a); 160.7 (C-9); 169.9 (C-4a); 179.7 (C-10); 191.4 (C-1). *It may be interchanged. **It may be interchanged. ESI-MS m/z 459 [M+H]+; 481 [M+Na]+. HR-ESIMS (positive ion): m/z calcd. for C26H18O8 [M]+, 458.1002; found, 458.1003.
(E)-4-(2-Chlorobenzylidene)-3-(2-chlorophenyl)-8-hydroxy-3,4-dihydro-1H-xanthene-1,9(2H)-dione (5f). This compound was obtained by the reaction of (E,E)-3-cinnamoyl-5-hydroxy-2-styrylchromone 4f (185.8 mg, 0.401 mmol) with BBr3 (2 mL of a 1 mol L−1 solution in CH2Cl2) and following the above described procedure. The desired compound 5f was obtained as yellow crystals (100.3 mg, 54%). D.p. 242 °C. 1H-NMR (300 MHz, CDCl3, TMS) δ (ppm): 2.97 (dd, 1H, J = 2.2 and 15.4 Hz, H-2trans); 3.11 (dd, 1H, J = 6.6 and 15.4 Hz, H-2cis); 4.91 (dd, 1H, J = 2.2 and 6.6 Hz, H-3); 6.82 (br d, 1H, J = 7.8 Hz, H-6"); 6.89 (br d, 1H, J = 8.2 Hz, H-8); 7.09 (br d, 1H, J = 8.2 Hz, H-6); 7.10–7.14 (m, 2H, H-5' and H-5"); 7.14–7.18 (m, 1H, H-6'); 7.23 (ddd, 1H, J = 2.1, 7.1 and 7.6 Hz, H-4'); 7.32 (ddd, 1H, J = 1.3, 7.4 and 8.1 Hz, H-4"); 7.42 (dd, 1H, J = 1.0 and 7.6 Hz, H-3'); 7.48 (dd, 1H, J = 0.6 and 8.1 Hz, H-3"); 7.63 (t, 1H, J = 8.2 Hz, H-7); 8.26 (s, 1H, H-7"); 12.59 (s, 1H, 9-OH). 13C-NMR (75 MHz, CDCl3, TMS) δ (ppm): 38.3 (C-3); 43.9 (C-2); 107.0 (C-6); 111.2 (C-9a); 113.0 (C-8); 114.1 (C-10a); 126.9 (C-6'); 127.6 (C-5' *); 127.7 (C-5" *); 129.0 (C-6"); 129.1 (C-4'); 130.0 (C-3"); 130.2 (C-4); 130.7 (C-3'); 131.0 (C-4"); 132.5 (C-1"); 132.9 (C-2'); 135.0 (C-2"); 136.3 (C-7); 136.5 (C-7"); 138.7 (C-1'); 154.8 (C-5a); 161.8 (C-9); 168.7 (C-4a); 180.1 (C-10); 191.3 (C-1). * May be interchanged. ESI-MS m/z 463 ([M+H]+, 35Cl); 465 ([M+H]+, 35Cl, 37Cl); 485 ([M+Na]+, 35Cl); 487 ([M+Na]+, 35Cl, 37Cl); 489 ([M+Na]+, 37Cl). HR-ESIMS (positive ion): m/z calcd. for C26H1635Cl2O4 ([M]+, 35Cl), 462.0426; found, 462.0406. Calcd. for C26H1635Cl37ClO4 ([M]+, 35Cl37Cl), 464.0396; found, 464.0374. Calcd. for C26H1637Cl2O4 ([M]+, 37Cl), 466.0367; found, 466.0384.
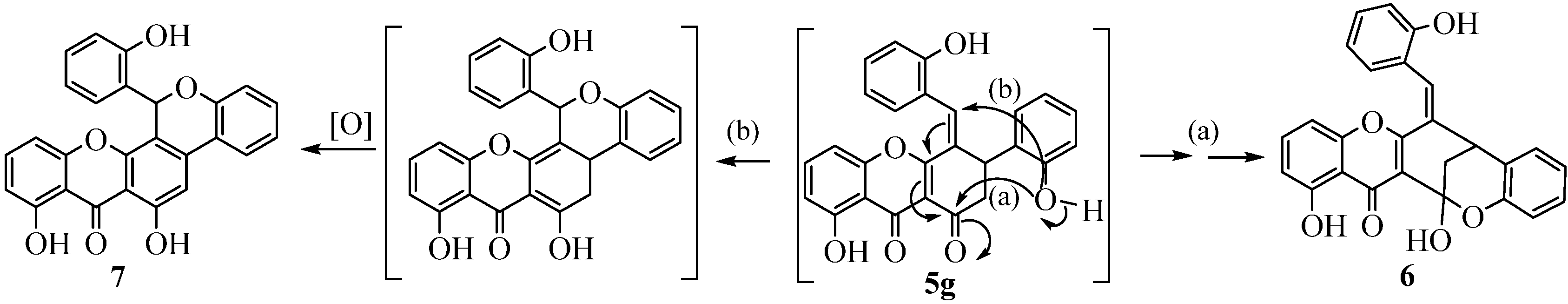
(E)-1,13-Dihydroxy-6-(2-hydroxybenzylidene)-6,7-dihydro-7,13-methanobenzo[7,8]oxocino[4,3-b]-chromen-14(13H)-one (6). This compound was obtained by the reaction of (E,E)-3-cinnamoyl-5-hydroxy-2-styrylchromone (4g, 181.8 mg, 0.400 mmol) with BBr3 (2 mL of a 1 mol L−1 solution in CH2Cl2). After the slowly addition of BBr3 the mixture was stirred at room temp. for 3 h, then water (80 mL) was added. The resulting reaction mixture was stirred for 4h and extracted with CH2Cl2 (3 × 80 mL). The combined extracts were evaporated and purified by TLC using CH2Cl2 as eluent. The desired compound 6 was obtained as orange crystals (74.5 mg, 41%). M.p. 195–198 °C. 1H-NMR (300 MHz, CDCl3, TMS) δ (ppm): 2.56 (d, 2H, J = 3.1 Hz, H-2); 4.05 (t, 1H, J = 3.1 Hz, H-3); 6.57 (dd, 1H, J = 0.8 and 8.1 Hz, H-6); 6.64 (dd, 1H, J = 0.8 and 8.1 Hz, H-8); 6.79 (br d, 1H, J = 8.0 Hz, H-3'); 6.85 (ddd, 1H, J = 1.1, 7.5 and 8.1 Hz, H-5'); 7.07–7.11 (m, 1H, H-4'); 7.12 (br d, 1H, J = 7.5 Hz, H-6'); 7.18–7.23 (m, 1H, H-5"); 7.27 (s, 1H, H-7"); 7.34 (dd, 1H, J = 1.1, 7.6 Hz, H-6"); 7.37 (t, 1H, J = 8.1 Hz, H-7); 7.37–7.40 (m, H, H-3"); 7.42 (m, 1H, H-4"); 13.00 (s, 1H, 9-OH). 13C-NMR (75 MHz, CDCl3, TMS) δ (ppm): 31.3 (C-2); 38.3 (C-3); 100.7 (C-1); 104.9 (C-10a); 107.1 (C-6); 108.5 (C-9a); 111.0 (C-8); 116.9 (C-3"); 118.1 (C-3'); 120.2 (C-1"); 121.4 (C-5'); 122.4 (C-1'); 124.9 (C-5"); 127.1 (C-6"); 128.0 (C-6'); 128.7 (C-4'); 129.1 (C-7"); 131.4 (C-4); 132.5 (C-4); 136.5 (C-7); 151.4 (C-2"); 152.1 (C-2'); 156.0 (C-5a); 158.6 (4a); 162.8 (C-9); 183.2 (C-10). ESI-MS m/z 409 [M−H2O+H]+; 431 [M−H2O+Na]+; 447 [M−H2O+Na]+. HR-ESIMS (positive ion): m/z calcd. for C26H19O6 [M+H]+, 427.1176; found, 427.1181; Calcd. for C26H17O5 [M-H2O+H]+, 409, 1070; found, 409.1072.
6,8-Dihydroxy-13-(2-hydroxyphenyl)chromeno[4,3-c]xanthen-7(13H)-one (7). This compound was obtained following the above described procedure for compound 6. The desired compound 7 was obtained as orange crystals (5.5 mg, 3%). D.p. 242 °C. 1H-NMR (300 MHz, CDCl3, TMS) δ (ppm): 6.43 (d, 1H, J = 1.2 and 7.9 Hz, H-6"); 6.61 (dt, 1H, J = 1.1 and 7.9 Hz, H-5"); 6.82 (dd, 2H, J = 0.8 and 8.3 Hz, H-8 and H-6); 6.94 (dd, 1H, J = 1.0 and 8.1 Hz, H-3"); 7.01 (dd, 1H, J = 1.1 and 8.1 Hz, H-3'); 7.08 (dt, 1H, J = 1.1 and 7.7 Hz, H-5'); 7.14 (s, 1H, H-7"); 7.14–7.17 (m, 1H, H-4"); 7.26 (s, 1H, H-2); 7.25–7.30 (m, 1H, H-4'); 7.57 (t, 1H, J = 8.3 Hz, H-7); 7.74 (dd, 1H, J = 1.5 and 7.7 Hz, H-6'); 11.80 (s, 1H, 9-OH); 12.01 (s, 1H, 1-OH). 13C-NMR (75 MHz, CDCl3, TMS) δ (ppm): 70.9 (C-7"); 104.9 (C-2); 106.7 (C-10a); 107.4 (C-6); 107.9 (C-9a); 108.9 (C-4); 111.4 (C-8); 116.8 (C-3"); 118.0 (C-3'); 120.2 (C-5"); 121.3 (C-1'); 123.3 (C-5' and C-1"); 124.7 (C-6'); 127.8 (C-6"); 130.5 (C-4"); 132.1 (C-4'); 137.6 (C-7); 139.2 (C-3); 152.0 (C-2'); 152.2 (C-4a); 155.7 (C-2"); 155.9 (C-5a); 161.4 (C-9); 161.7 (C-1); 185.5 (C-10). ESI-MS m/z 425 [M+H]+; 447 [M+Na]+; 463 [M+K]+.
1,8-Dihydroxy-4-(4-methylbenzyl)-3-(4-methylphenyl)-9H-xanthen-9-one (8b). This compound was obtained by the treatment of a solution of (E)-8-hydroxy-4-(4-methylbenzylidene)-3-(4-methylphenyl)-3,4-dihydro-1H-xanthene-1,9(2H)-dione (5b, 56.7 mg, 0.13 mmol) in DMSO (10 mL) with DBU (1.2 equiv, 24 µL, 0.16 mmol). The mixture was then irradiated in an Ethos SYNTH microwave (Milestone Inc., Sorisole, Italy) at 100 °C for 10 min. After this period the reaction mixture was poured onto ice (10 g) and the pH was adjusted to 3–4 with diluted HCl and extracted with chloroform. The desired compound 8b was obtained by TLC purification using a mixture of hexane/CH2Cl2 (3:1) as eluent, as orange crystals, 18% yield (10.2 mg). M.p. 155–157 °C; 1H-NMR (300 MHz, CDCl3, TMS) δ (ppm): 2.26 (s, 3H, 4"-CH3); 2.40 (s, 3H, 4'-CH3); 4.10 (s, 2H, H-7"); 6.77 (dd, 1H, J = 0.7 and 8.8 Hz, H-8); 6.79 (s, 1H, H-2); 6.81 (dd, 1H, J = 0.7 and 8.8 Hz, H-6); 6.90 (d, 2H, J = 8.1 Hz, H-2",6"); 6.99 (d, 2H, J = 8.1 Hz, H-3",5"); 7.17–7.19 (m, 2H, H-2',6'); 7.19–7.21 (m, 2H, H-3',5'); 7.55 (t, 1H, J = 8.8 Hz, H-7); 11.78 (s, 1H, 1-OH); 11.86 (s, 1H, 9-OH). 13C-NMR (75 MHz, CDCl3, TMS) δ (ppm): 20.9 (4"-CH3); 21.2 (4'-CH3); 31.7 (C-7"); 107.0 (C-10a); 107.2 (C-6); 107.7 (C-9a); 110.7 (C-8); 112.5 (C-2); 117.1 (C-4*); 127.8 (C-2" and C-6"); 128.6 (C-2' and C-6'); 128.9 (C-3', C-3", C-5' and C-5"); 135.2 (C-4"); 137.1 (C-1') 137.3 (C-7); 137.4 (C-3*); 137.7 (C-1"); 137.9 (C-4'); 152.3 (C-4a); 156.3 (C-5a); 158.8 (C-1); 161.2 (C-9); 186.1 (C-10). * May be interchanged. ESI-MS m/z 423 [M+H]+; 445 [M+Na]+. HR-ESIMS (positive ion): m/z calcd. for C28H22O4 [M]+, 422.1518; found, 422.1520.
1,8-Dihydroxy-4-(4-methylbenzoyl)-3-(4-methylphenyl)-9H-xanthen-9-one (9b). This compound was obtained by the treatment of a solution of (E)-8-hydroxy-4-(4-methylbenzylidene)-3-(4-methylphenyl)-3,4-dihydro-1H-xanthene-1,9(2H)-dione (5b, 66.9 mg, 0.16 mmol) in 1,2,4-trichlorobenzene (TCB) (10 mL) with DQQ (2.0 equiv, 72.0 mg, 0.32 mmol). The mixture was then irradiated in an Ethos SYNTH microwave (Milestone Inc.) at 170 °C for 30 min. After this the TCB was removed by column chromatography using hexane as eluent and the reaction mixture was eluted with CH2Cl2. The desired compound 9b was obtained by TLC purification using successively hexane and CH2Cl2 as eluents, as yellow solid, 28% yield (19.1 mg). M.p. 180–183 °C; 1H-NMR (300 MHz, CDCl3, TMS) δ (ppm): 2.28 (s, 3H, 4'-CH3); 2.36 (s, 3H, 4"-CH3); 2.98 (sb, 1H, 7"-OH); 6.02 (d, 1H, J = 9.0 Hz, H-7"); 6.60 (dd, 1H, J = 0.8 and 8.4 Hz, H-6); 6.77 (dd, 1H, J = 0.8 and 8.4 Hz, H-8); 6.91 (s, 1H, H-2); 7.06 (d, 2H, J = 8.2 Hz, H-3',5'); 7.15 (d, 2H, J = 8.2 Hz, H-3",5"); 7.22 (d, 2H, J = 8.2 Hz, H-2',6'); 7.50 (t, 1H, J = 8.4 Hz, H-7); 7.65 '(d, 2H, J = 8.2 Hz, H-2",6"); 11.71 (s, 1H, 9-OH); 12.06 (s, 1H, 1-OH). 13C-NMR (75 MHz, CDCl3, TMS) δ (ppm): 21.2 (4'-CH3); 21.7 (4"-CH3); 106.1 (C-10a); 107.4 (C-6); 107.7 (C-9a); 111.1 (C-8); 112.3 (C-2); 118.6 (C-4); 128.5 (C-2' and C-6'); 129.2 (C-3', C-5', C-3" and C-5"); 129.6 (C-2" and C-6"); 135.4 (C-1"); 135.6 (C-1'); 137.6 (C-7); 138.7 (C-4'); 144.4 (C-4"); 150.4 (C-3); 153.4 (C-4a); 155.9 (C-5a); 161.2 (C-9); 161.4 (C-1); 185.7 (C-10); 194.0 (C-7");. ESI-MS m/z 437 [M+H]+, 459 [M+Na]+, 475 [M+K]+. HR-ESIMS (positive ion): m/z calcd. for C28H20O5 [M]+, 436,1311; found, 436.1319.
6,8-Dihydroxy-2-methyl-13-(4-methylphenyl)indeno[1,2-c]xanthen-7(13H)-one (10b). This compound was obtained by the treatment of a solution of (E)-8-hydroxy-4-(4-methylbenzylidene)-3-(4-methylphenyl)-3,4-dihydro-1H-xanthene-1,9(2H)-dione (5b, 50.5 mg, 0.12 mmol) in dry 1,2,4-trichlorobenzene (TCB) (10 mL) and in the presence of 100 mg molecular sieves with DQQ (1.5 equiv, 40.6 mg, 0.18 mmol). The mixture was then irradiated in an Ethos SYNTH microwave (Milestone Inc.) at 170 °C for 30 min. After this the TCB was removed by column chromatography using hexane as eluent and the reaction mixture was eluted with CH2Cl2. The desired compound 10b was obtained by TLC purification using a mixture of hexane/CH2Cl2 (3:2) as eluent, as yellow crystals, 52% yield (26.0 mg). D.p. 195 °C; 1H-NMR (300 MHz, CDCl3, TMS) δ (ppm): 2.28 (s, 3H, 4"-CH3); 2.36 (s, 3H, 4'-CH3); 5.14 (s, 1H, H-7"); 6.64 (dd, 1H, J = 0.7 and 8.3 Hz, H-6); 6.70 (dd, 1H, J = 0.7 and 8.3 Hz, H-8); 7.01 (d, 2H, J = 8.2 Hz, H-2",6"); 7.06 (d, 2H, J = 8.2 Hz, H-3",5"); 7.13 (br s, 1H, H-3'); 7.14 (s, 1H, H-2); 7.20 (br d, 1H, J = 7.9 Hz, H-5'); 7.47 (t, 1H, J = 8.3 Hz, H-7); 7.67 (d, 1H, J = 7.9 Hz, H-6'); 11.90 (s, 1H, 9-OH); 12.05 (s, 1H, 1-OH). 13C-NMR (75 MHz, CDCl3, TMS) δ (ppm): 21.1 (4"-CH3); 21.8 (4'-CH3); 51.5 (C-7"); 102.1 (C-2); 106.4 (C-10a); 106.9 (C-6); 107.8 (C-9a); 110.7 (C-8); 121.1 (C-6'); 123.3 (C-4); 126.1 (C-3'); 128.0 (C-2" and C-6"); 128.6 (C- 5'); 129.2 (C-3" and C-5"); 136.4 (C-4"); 136.8 (C-1"*); 136.9 (C-7); 137.0 (C-1'*); 140.3 (C-4'); 150.7 (C-2'); 151.4 (C-4a); 152.0 (C-3); 155.9 (C-5a); 161.3 (C-9); 162.2 (C-1); 185.5 (C-10) * May be interchanged. ESI-MS m/z 421 [M+H]+; 443 [M+Na]+, 459 [M+K]+. HR-ESIMS (positive ion): m/z calcd. for C28H20O4 [M]+, 420.1362; found, 420.1364.
1,8-Dihydroxy-4-[(4-methylphenyl)hydroxymethyl]-3-(4-methylphenyl)-9H-xanthen-9-one (11b). This compound was obtained by the treatment of a solution of (E)-8-hydroxy-4-(4-methylbenzylidene)-3-(4-methylphenyl)-3,4-dihydro-1H-xanthene-1,9(2H)-dione (5b, 58.8 mg, 0.14 mmol) in dried 1,2,4-trichlorobenzene (TCB) (10 mL) and in the presence of 100 mg molecular sieves with DQQ (1.3 equiv, 42.0 mg, 0.18 mmol). The mixture was then irradiated in an Ethos SYNTH microwave (Milestone Inc.) at 100 °C for 30 min. After this the TCB was removed by column chromatography using hexane as eluent and the reaction mixture was eluted with CH2Cl2. The desired compound 11b was obtained by TLC purification using a mixture of hexane/CH2Cl2 (3:2) as eluent, as yellow crystals, 30% yield (18.6 mg). M.p. 134–136 °C; 1H-NMR (300 MHz, CDCl3, TMS) δ (ppm): 2.29 (s, 3H, 4"-CH3); 2.39 (s, 3H, 4'-CH3); 6.02 (d, 1H, J = 9.0 Hz, H-7"); 6.55 (br d, 1H, J = 8.2 Hz, H-6); 6.74 (dd, 1H, J = 0.7 and 8.2 Hz, H-8); 6.79 (s, 1H, H-2); 7.07 (d, 2H, J = 8.0 Hz, H-3",5"); 7.17 (d, 2H, J = 8.0 Hz, H-2",6"); 7.22 (d, 2H, J = 8.1 Hz, H-3',5'); 7.28 (d, 2H, J = 8.1 Hz, H-2',6'); 7.48 (t, 1H, J = 8.2 Hz, H-7); 11.70 (s, 1H, 9-OH); 11.84 (s, 1H, 1-OH). 13C-NMR (75 MHz, CDCl3, TMS) δ (ppm): 21.0 (4"-CH3); 21.2 (4'-CH3); 70.8 (C-7"); 106.7 (C-6); 107.4 (C-10a); 107.6 (C-9a); 111.1 (C-8); 112.6 (C-2); 119.5 (C-4); 125.1 (C-2" and C-6"); 128.6 (C-2' and C-6'); 128.8 (C-3" and C-5"); 129.2 (C-3' and C-5'); 136.4 (C-1' and C-4"); 137.4 (C-7); 138.3 (C-4'); 140.8 (C-1"); 151.9 (C-3); 154.6 (C-4a); 155.5 (C-5a); 159.8 (C-1); 161.2 (C-9); 185.8 (C-10). ESI-MS m/z 421 [M−18+H]+, 443 [M−18+Na]+, 459 [M−18+K]+. HR-ESIMS (positive ion): m/z calcd. for C28H20O5 [M]+, 438,1467; found, 438.1477.
6,8,13-Trihydroxy-2-methyl-13-(4-methyphenyl)indeno[1,2-c]xanthen-7(13H)-one (12b). This compound was obtained by the treatment of a solution of (E)-8-hydroxy-4-(4-methylbenzylidene)-3-(4-methylphenyl)-3,4-dihydro-1H-xanthene-1,9(2H)-dione (5b, 57.6 mg, 0.14 mmol) in dried 1,24-trichlorobenzene (TCB) (10 mL) and in the presence of 100 mg molecular sieves with chloranil (2.0 equiv, 68.0 mg, 0.28 mmol). The mixture was then irradiated in an Ethos SYNTH microwave (Milestone Inc.) at 170 °C for 30 min. After this the TCB was removed by column chromatography using hexane as eluent and a semi-purified fraction was eluted with CH2Cl2. The desired compound 12b was obtained by TLC purification using a mixture of hexane/CH2Cl2 (3:2) as eluent, as yellow crystals, 34% yield (20.7 mg). D.p. 145 °C; 1H-NMR (300 MHz, CDCl3, TMS) δ (ppm): 2.29 (s, 3H, 4"-CH3); 2.35 (s, 3H, 4'-CH3); 6.70 (dd, 1H, J = 0.8 and 8.3 Hz, H-6); 6.73 (dd, 1H, J = 0.8 and 8.3 Hz, H-8); 7.07 (s, 1H, H-2); 7.08 (d, 2H, J = 8.4 Hz, H-3",5"); 7.18 (d, 1H, J = 0.9 Hz, H-3'); 7.20–7.23 (m, 1H, H-5'); 7.33 (d, 2H, J = 8.4 Hz, H-2",6"); 7.50 (t, 1H, J = 8.3 Hz, H-7); 7.58 (d, 1H, J = 7.7 Hz, H-6'); 11.81 (s, 1H, 9-OH); 12.22 (s, 1H, 1-OH). 13C-NMR (75 MHz, CDCl3, TMS) δ (ppm): 21.1 (4"-CH3); 21.8 (4'-CH3); 83.1 (C-7"); 102.5 (C-2); 106.7 (C-10a); 107.2 (C-6); 107.6 (C-9a); 111.0 (C-8); 121.3 (C-6'); 125.1 (C-3', C-2" and C-6"); 128.9 (C-3" and C-5"); 130.1 (C-5'); 134.9 (C-1'); 137.0 (C-4"); 137.1 (C-7); 139.0 (C-4 and C-1"); 141.5 (C-4'); 150.2 (C-3 *); 151.5 (C-4a); 152.8 (C-2' *); 155.8 (C-5a); 161.2 (C-9); 163.7 (C-1); 185.3 (C-10) *May be interchanged. ESI-MS m/z 459 [M+Na]+. HR-ESIMS (positive ion): m/z calcd. for C28H21O5 [M+H]+, 437.1384; found, 437.1380.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


