
Catalytic Oxidation of NO over MnOx–CeO2 and MnOx–TiO2 Catalysts

Abstract
:1. Introduction
2. Results and Discussion
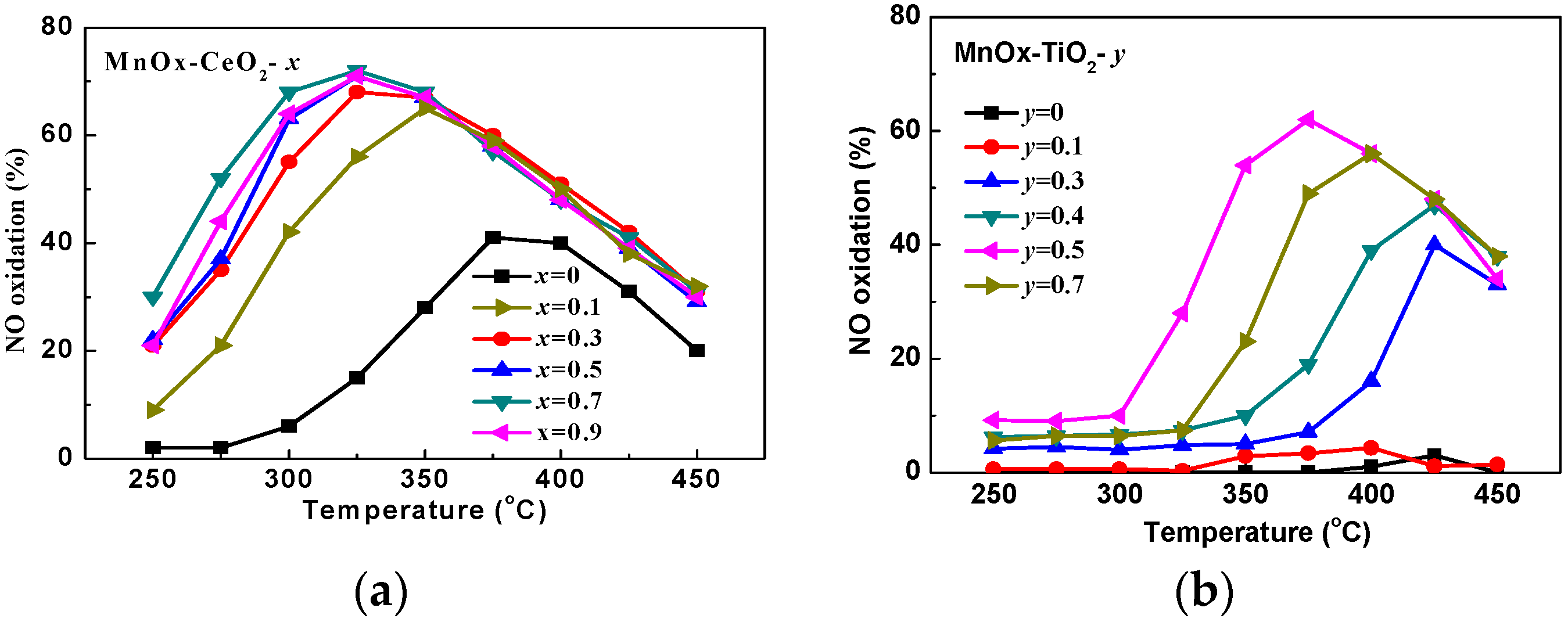
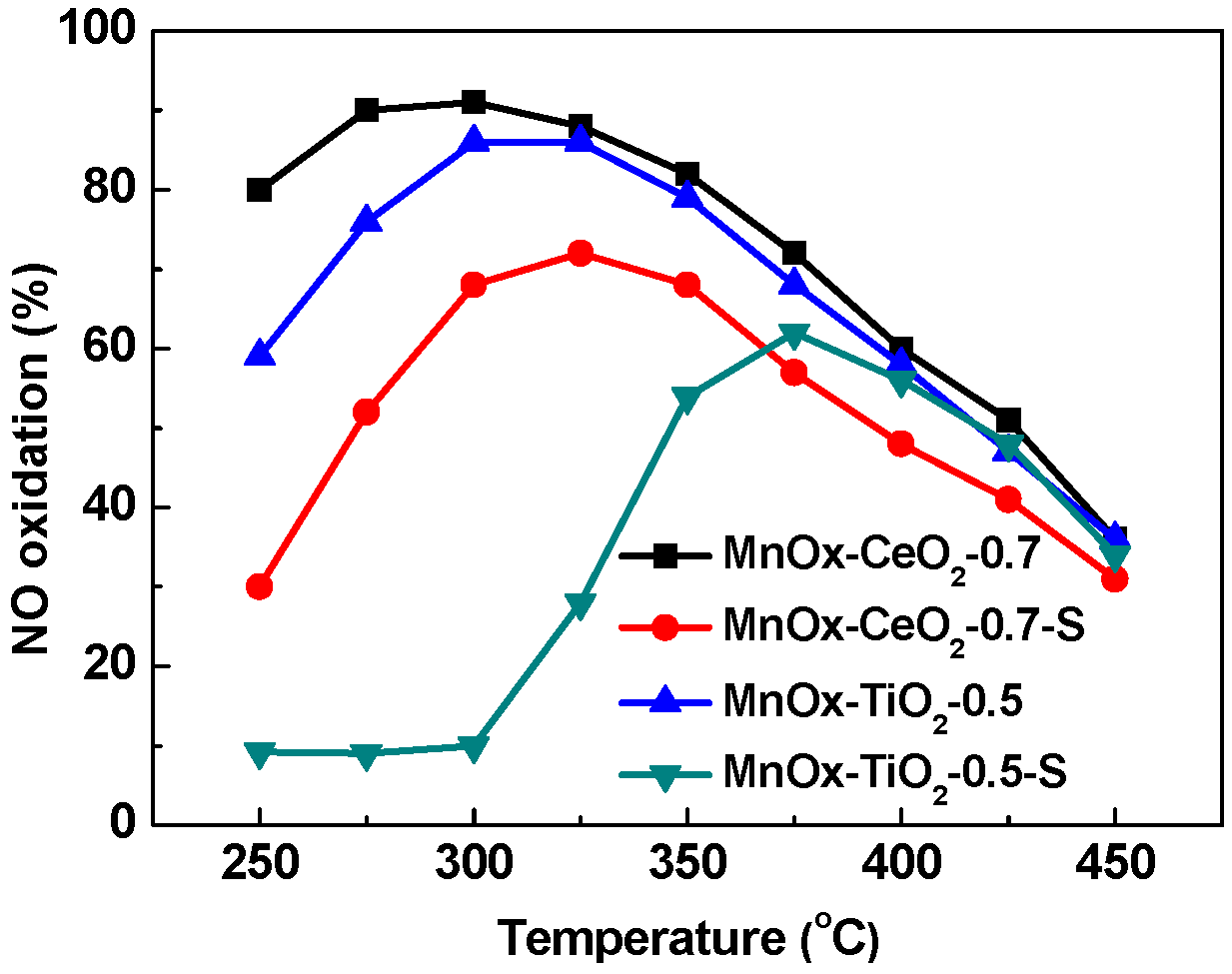
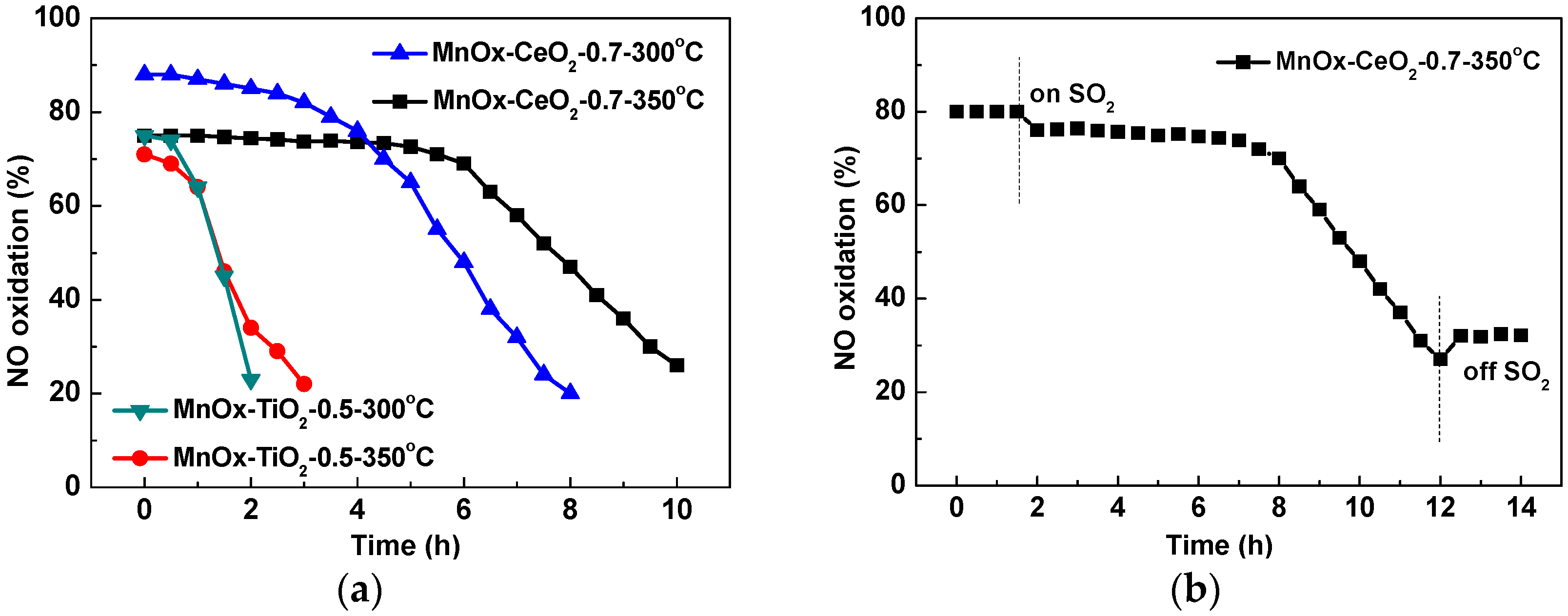
2.1. Catalytic Activity Tests
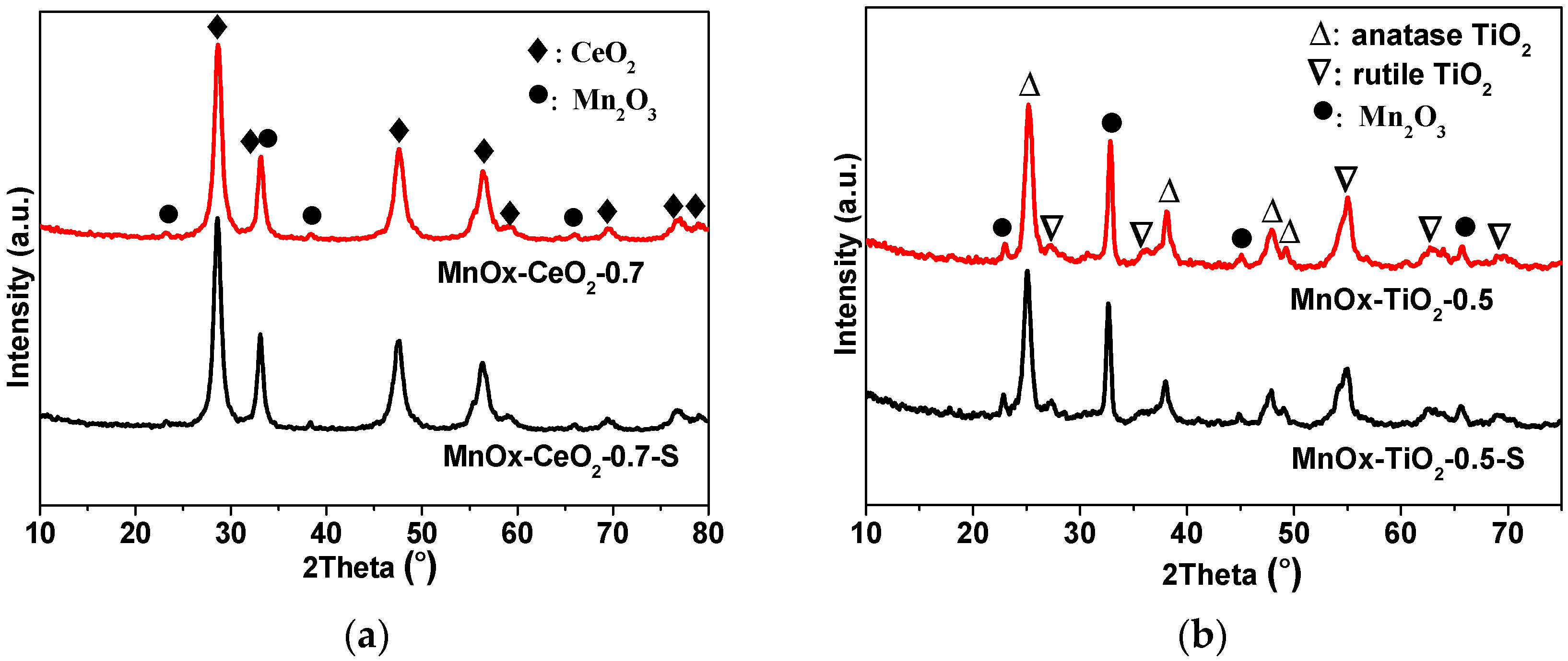
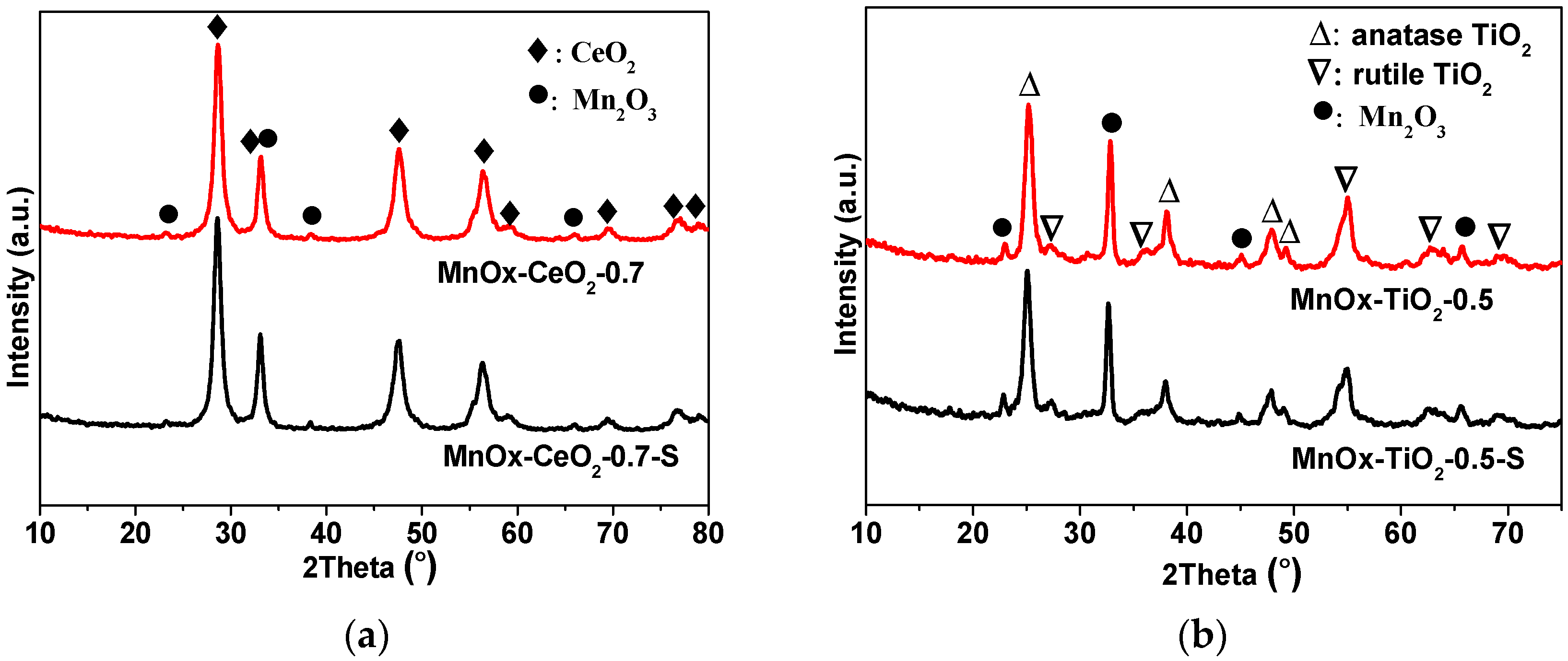
2.2. XRD and BET Characterizations
2.3. STEM-Mapping Analysis
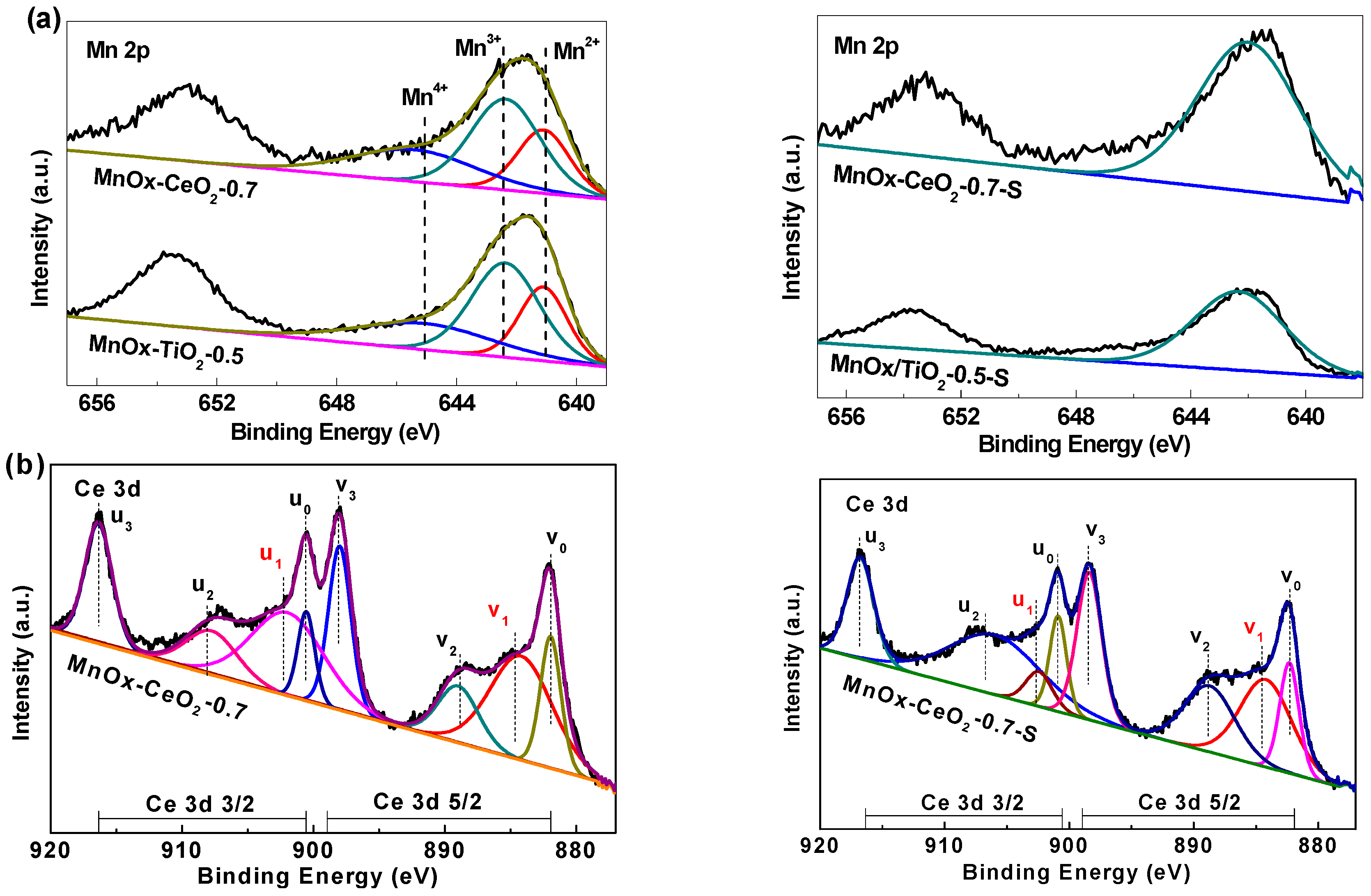
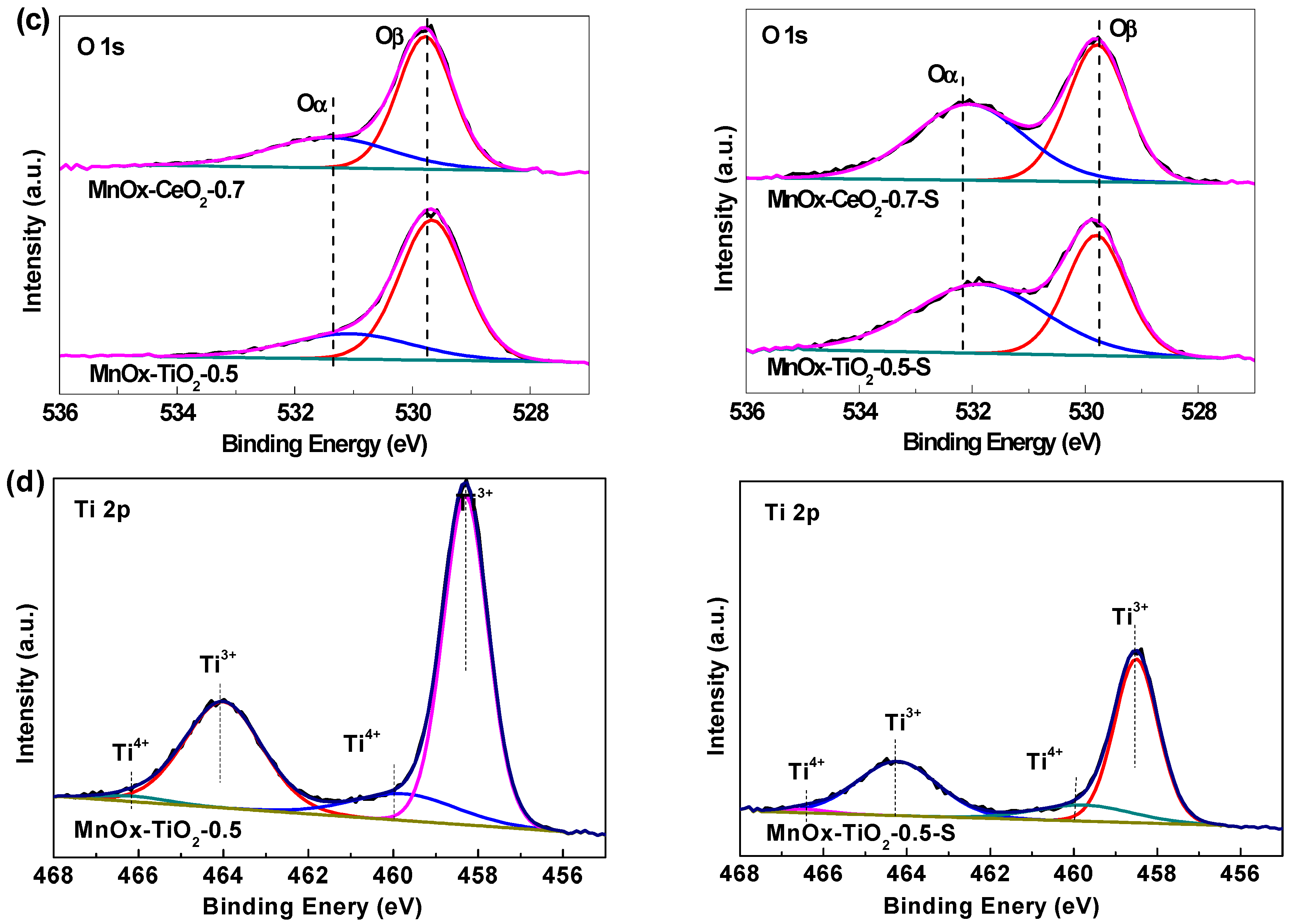
2.4. XPS Analysis
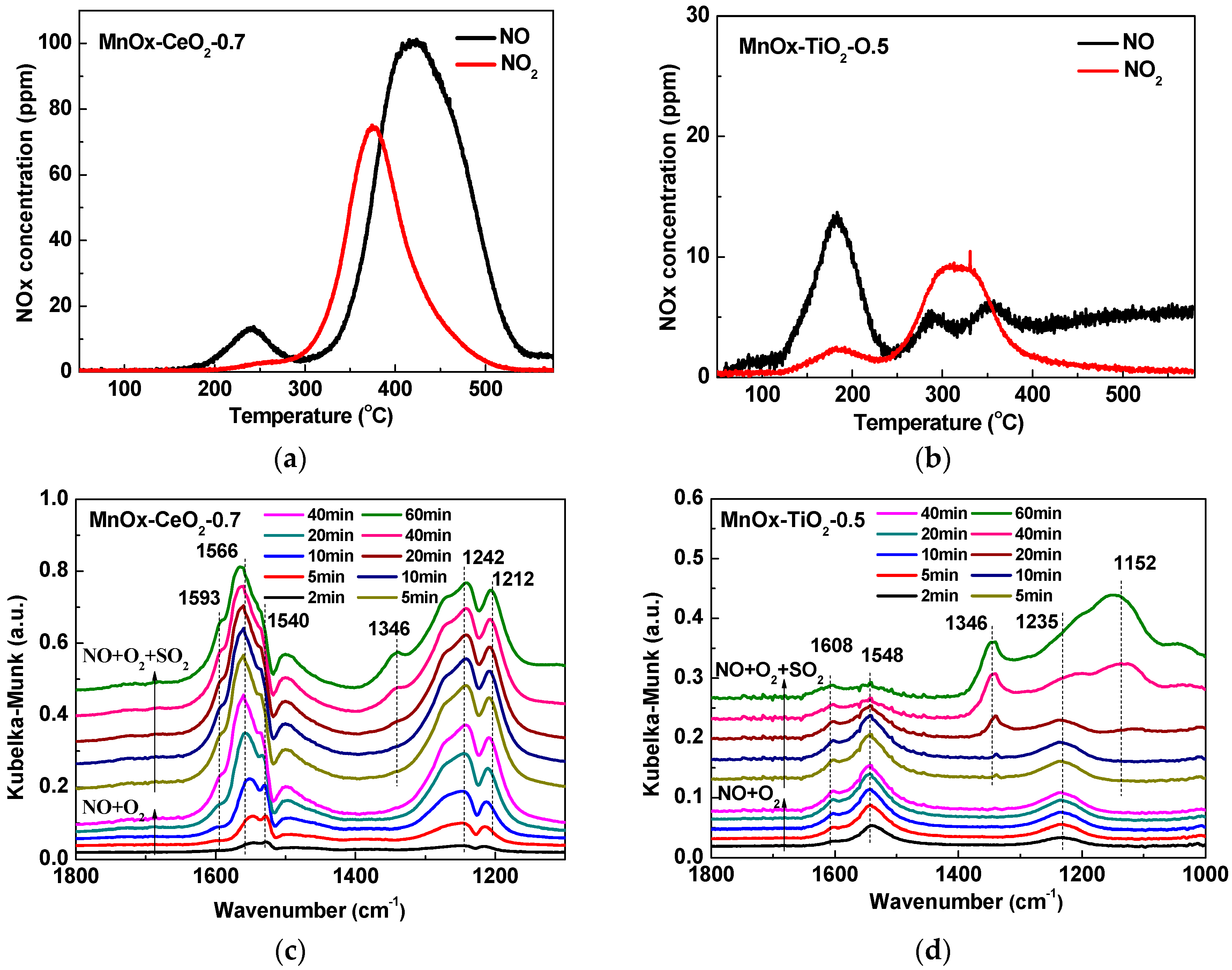
2.5. NO + O2-TPD and In Situ DRIFTS Analyses
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalytic Activity Measurement
3.3. Catalyst Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhu, J.; Thomas, A. Perovskite-Type Mixed Oxides as Catalytic Material for NO Removal. Appl. Catal. B Environ. 2009, 92, 225–233. [Google Scholar] [CrossRef]
- Skalska, K.; Miller, J.S.; Ledakowicz, S. Trends in NOx abatement: A review. Sci. Total Environ. 2010, 408, 3976–3989. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Lv, L.; Zhang, J.; Chen, X.; Tong, M.; Kang, W.; Zhou, Y.; Lu, J. Simultaneous removal of SO2 and NOx with ammonia combined with gas-phase oxidation of NO using ozone. Chem. Ind. Chem. Eng. Q. 2015, 21, 305–310. [Google Scholar] [CrossRef]
- Dawody, J.; Skoglundh, M.; Fridell, E. The effect of metal oxide additives (WO3, MoO3, V2O5, Ga2O3) on the oxidation of NO and SO2 over Pt/Al2O3 and Pt/BaO/Al2O3 catalysts. J. Mol. Catal. A Chem. 2004, 209, 215–225. [Google Scholar] [CrossRef]
- Crocoll, M.; Kureti, S.; Weisweiler, W. Mean field modeling of NO oxidation over Pt/Al2O3 catalyst under oxygen-rich conditions. J. Catal. 2005, 229, 480–489. [Google Scholar] [CrossRef]
- Fujii, T.; Sato, R. Characterization and activity of Pd-modified TiO2 catalysts for photocatalytic oxidation of NO in gas phase. J. Hazard. Mater. 2009, 164, 542–548. [Google Scholar]
- Kaneeda, M.; Iizuka, H.; Hiratsuka, T.; Shinotsuka, N.; Arai, M. Improvement of thermal stability of NO oxidation Pt/Al2O3 catalyst by addition of Pd. Appl. Catal. B 2009, 90, 564–569. [Google Scholar] [CrossRef]
- Boubnov, A.; Dahl, S.; Johnson, E.; Molina, A.P.; Simonsen, S.B.; Cano, F.M.; Helveg, S.; Lemus-Yegres, L.J.; Grunwaldt, J.D. Structure–activity relationships of Pt/Al2O3 catalysts for CO and NO oxidation at diesel exhaust conditions. Appl. Catal. B 2012, 126, 315–325. [Google Scholar] [CrossRef]
- Khosravi, M.; Sola, C.; Abedi, A.; Hayes, R.E.; Epling, W.S.; Votsmeier, M. Oxidation and selective catalytic reduction of NO by propene over Pt and Pt:Pd diesel oxidation catalysts. Appl. Catal. B 2014, 147, 264–274. [Google Scholar] [CrossRef]
- Berggrund, M.; Ingelsten, H.H.; Skoglundh, M.; Palmqvist, A.E.C. Influence of Synthesis Conditions for ZSM-5 on the Hydrothermal Stability of Cu-ZSM-5. Catal. Lett. 2009, 130, 79–85. [Google Scholar] [CrossRef]
- Karlsson, H.T.; Rosenberg, H.S. Flue gas denitrification. Selective catalytic oxidation of NO to NO2. Ind. Eng. Chem. Process Des. Dev. 1984, 23, 808–814. [Google Scholar] [CrossRef]
- Kantcheva, M.; Vakkasoglu, A.S. Cobalt supported on zirconia and sulfated zirconia I.: FT-IR spectroscopic characterization of the NOx species formed upon NO adsorption and NO/O2 coadsorption. J. Catal. 2004, 223, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Qiang, W.; Park, S.Y.; Jin, S.C.; Chung, J.S. Co/KxTi2O5 catalysts prepared by ion exchange method for NO oxidation to NO2. Appl. Catal. B 2008, 79, 101–107. [Google Scholar]
- Irfan, M.F.; Sang, D.K.; Goo, J.H. Co3O4 based catalysts for NO oxidation and NOx reduction in fast SCR process. Appl. Catal. B 2008, 78, 267–274. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Wu, Z.; Liu, Y. NO Catalytic Oxidation Behaviors over CoOx/TiO2 Catalysts Synthesized by Sol–Gel Method. Catal. Lett. 2010, 134, 295–302. [Google Scholar] [CrossRef]
- Shang, D.; Cai, W.; Zhao, W.; Bu, Y.; Zhong, Q. Catalytic Oxidation of NO to NO2 Over Co–Ce–Zr Solid Solutions: Enhanced Performance of Ce–Zr Solid Solution by Co. Catal. Lett. 2014, 144, 538–544. [Google Scholar] [CrossRef]
- Shang, D.; Zhong, Q.; Cai, W. Influence of the preparation method on the catalytic activity of Co/Zr1−xCexO2 for NO oxidation. J. Mol. Catal. A Chem. 2015, 399, 18–24. [Google Scholar] [CrossRef]
- Shang, D.; Zhong, Q.; Cai, W. High performance of NO oxidation over Ce–Co–Ti catalyst: The interaction between Ce and Co. Appl. Surf. Sci. 2015, 325, 211–216. [Google Scholar] [CrossRef]
- Yu, Y.; Zhong, Q.; Cai, W.; Ding, J. Promotional effect of N-doped CeO2 supported CoOx catalysts with enhanced catalytic activity on NO oxidation. J. Mol. Catal. A Chem. 2015, 398, 344–352. [Google Scholar] [CrossRef]
- Wu, Z.; Tang, N.; Ling, X.; Yue, L.; Wang, H. MnOx/TiO2 composite nanoxides synthesized by deposition-precipitation method as a superior catalyst for NO oxidation. J. Colloid Interface Sci. 2010, 352, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Chin, S.; Jeong, J.; Jurng, J. Low-temperature NO oxidation over Mn/TiO2 nanocomposite synthesized by chemical vapor condensation: Effects of Mn precursor on the surface Mn species. Micropor. Mesopor. Mater. 2012, 163, 96–101. [Google Scholar] [CrossRef]
- An, Z.; Zhuo, Y.; Xu, C.; Chen, C. Influence of the TiO2 crystalline phase of MnOx/TiO2 catalysts for NO oxidation. Chin. J. Catal. 2014, 35, 120–126. [Google Scholar] [CrossRef]
- Li, X.; Zhang, S.; Jia, Y.; Liu, X.; Zhong, Q. Selective catalytic oxidation of NO with O2 over Ce-doped MnOx catalysts. J. Nat. Gas Chem. 2012, 21, 17–24. [Google Scholar] [CrossRef]
- Zhang, M.; Li, C.; Qu, L.; Fu, M.; Zeng, G.; Fan, C.; Ma, J.; Zhan, F. Catalytic oxidation of NO with O2 over FeMnOx/TiO2: Effect of iron and manganese oxides loading sequences and the catalytic mechanism study. Appl. Surf. Sci. 2014, 300, 58–65. [Google Scholar] [CrossRef]
- Liu, L.; Gao, X.; Zheng, C.; Zhu, X.; Luo, Z.; Ni, M.; Cen, K.; Liu, L.; Ni, M.; Cen, K. Study of the Promotion Effect of Iron on Supported Manganese Catalysts for NO Oxidation. Aerosol Air Qual. Res. 2014, 14, 1038–1046. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, J.; Li, J.; Ma, L.; Woo, S.I. Novel Mn–Ce–Ti mixed-oxide catalyst for the selective catalytic reduction of NOx with NH3. ACS Appl. Mater. Interfaces 2014, 6, 14500–14508. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Zhang, D.; Maitarad, P.; Shi, L.; Rungrotmongkol, T.; Li, H.; Zhang, J.; Cao, W. Morphology-Dependent Properties of MnOx/ZrO2–CeO2 Nanostructures for the Selective Catalytic Reduction of NO with NH3. J. Phys. Chem. C 2013, 117, 10502–10511. [Google Scholar] [CrossRef]
- Luo, S.; Zhou, W.; Xie, A.; Wu, F.; Yao, C.; Li, X.; Zuo, S.; Liu, T. Effect of MnO2 polymorphs structure on the selective catalytic reduction of NOx with NH3 over TiO2-Palygorskite. Chem. Eng. J. 2016, 286, 291–299. [Google Scholar] [CrossRef]
- Hu, H.; Cai, S.; Li, H.; Huang, L.; Shi, L.; Zhang, D. Mechanistic aspects of deNOx processing over TiO2 supported Co-Mn oxide catalysts: Structure-activity relationships and in situ DRIFTs analysis. ACS Catal. 2015, 5, 6069–6077. [Google Scholar] [CrossRef]
- Cai, S.; Hu, H.; Li, H.; Shi, L.; Zhang, D. Design of multi-shell Fe2O3@MnOx@CNTs for the selective catalytic reduction of NO with NH: Improvement of catalytic activity and SO tolerance. Nanoscale 2016, 8, 3588–3598. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, J.; Li, H.; Cai, S.; Hu, H.; Fang, C.; Shi, L.; Zhang, D. Rational design and in situ fabrication of MnO2@NiCo2O4 nanowire arrays on Ni foam as high-performance monolith deNOx catalysts. J. Mater. Chem. A 2015, 3, 11543–11553. [Google Scholar] [CrossRef]
- Jin, R.; Yue, L.; Yan, W.; Cen, W.; Wu, Z.; Wang, H.; Weng, X. The role of cerium in the improved SO2 tolerance for NO reduction with NH3 over Mn–Ce/TiO2 catalyst at low temperature. Appl. Catal. B 2014, 148–149, 582–588. [Google Scholar] [CrossRef]
- Lu, X.; Song, C.; Jia, S.; Tong, Z.; Tang, X.; Teng, Y. Low-temperature selective catalytic reduction of NOx with NH3 over cerium and manganese oxides supported on TiO2-graphene. Chem. Eng. J. 2015, 260, 776–784. [Google Scholar] [CrossRef]
- Atribak, I.; Bueno-López, A.; García-García, A.; Navarro, P.; Frías, D.; Montes, M. Catalytic activity for soot combustion of birnessite and cryptomelane. Appl. Catal. B 2010, 93, 267–273. [Google Scholar] [CrossRef]
- Cimino, A.; Indovina, V. Catalytic activity of Mn3+ and Mn4+ ions dispersed in MgO for CO oxidation. J. Catal. 1974, 33, 493–496. [Google Scholar] [CrossRef]
- Xiong, Y.; Tang, C.; Yao, X.; Zhang, L.; Li, L.; Wang, X.; Deng, Y.; Gao, F.; Dong, L. Effect of metal ions doping (M = Ti4+, Sn4+) on the catalytic performance of MnOx/CeO2 catalyst for low temperature selective catalytic reduction of NO with NH3. Appl. Catal. A Gen. 2015, 495, 206–216. [Google Scholar] [CrossRef]
- Larsson, P.O.; Andersson, A. Complete Oxidation of CO, Ethanol, and Ethyl Acetate over Copper Oxide Supported on Titania and Ceria Modified Titania. J. Catal. 1998, 179, 72–89. [Google Scholar] [CrossRef]
- Wei, C.; Qin, Z.; Wei, Z. Solvent effects on formation of Cr-doped Ce0.2Zr0.8O2 synthesized with cinnamic acid and their catalysis in oxidation of NO. Chem. Eng. J. 2014, 246, 328–336. [Google Scholar]
- Jing, L.; Xu, Z.; Sun, X.; Shang, J.; Cai, W. The surface properties and photocatalytic activities of ZnO ultrafine particles. Appl. Surf. Sci. 2001, 180, 308–314. [Google Scholar] [CrossRef]
- Wu, X.D.; Li, H.R.; Liu, S.; Weng, D. Sulfur poisoning and regeneration of MnOx–CeO2–Al2O3 catalyst for soot oxidation. J. Rare Earths 2012, 30, 659–664. [Google Scholar] [CrossRef]
- Waqif, M.; Pieplu, A.; Saur, O.; Lavalley, J.C.; Blanchard, G. Use of CeO2–Al2O3 as a SO2 sorbent. Solid State Ion. 1997, 95, 163–167. [Google Scholar] [CrossRef]
- Yang, N.Z.; Guo, R.T.; Tian, Y.; Pan, W.G.; Chen, Q.L.; Wang, Q.S.; Lu, C.Z.; Wang, S.X. The enhanced performance of ceria by HF treatment for selective catalytic reduction of NO with NH3. Fuel 2016, 179, 305–311. [Google Scholar] [CrossRef]
- Wang, W.H.; Li, W.; Guo, R.T.; Chen, Q.L.; Wang, Q.S.; Pan, W.G.; Hu, G.X. A CeFeOx catalyst for catalytic oxidation of NO to NO2. J. Rare Earths 2016, 34, 876–881. [Google Scholar] [CrossRef]
- Zhao, B.; Ran, R.; Wu, X.; Weng, D.; Wu, X.; Huang, C. Comparative study of Mn/TiO2 and Mn/ZrO2 catalysts for NO oxidation. Catal. Commun. 2014, 56, 36–40. [Google Scholar] [CrossRef]
- Shen, M.; Zhao, Z.; Chen, J.; Su, Y.; Wang, J.; Wang, X. Effects of calcium substitute in LaMnO3 perovskites for NO catalytic oxidation. J. Rare Earths 2013, 31, 119–123. [Google Scholar] [CrossRef]
- Liu, F.; He, H.; Ding, Y.; Zhang, C. Effect of manganese substitution on the structure and activity of iron titanate catalyst for the selective catalytic reduction of NO with NH3. Appl. Catal. B 2009, 93, 3760–3769. [Google Scholar] [CrossRef]
- And, G.Q.; Yang, R.T. Characterization and FTIR Studies of MnOx–CeO2 Catalyst for Low-Temperature Selective Catalytic Reduction of NO with NH3. J. Phys. Chem. B 2004, 108, 15738–15747. [Google Scholar]
- Jiang, B.Q.; Wu, Z.B.; Liu, Y.; Lee, S.C.; Ho, W.K. DRIFT Study of the SO2 Effect on Low-Temperature SCR Reaction over Fe-Mn/TiO2. J. Phys. Chem. C 2010, 114, 4961–4965. [Google Scholar] [CrossRef]
- Tang, N.; Liu, Y.; Wang, H.; Wu, Z. Mechanism Study of NO Catalytic Oxidation over MnOx/TiO2 Catalysts. J. Phys. Chem. C 2011, 115, 8214–8220. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Surface Area (m2/g) |
|---|---|
| MnOx–CeO2-0.7 | 93.17 |
| MnOx–CeO2-0.7-S | 67.92 |
| MnOx–TiO2-0.5 | 60.21 |
| MnOx–TiO2-0.5-S | 39.71 |
| Catalysts | Atomic Concentration (%) | Surface Atomic Ratio (%) | ||||
|---|---|---|---|---|---|---|
| Mn | Ce or Ti | O | Mn3+/(Mn2+ + Mn3+ + Mn4+) | Ce3+/(Ce3+ + Ce4+) or Ti3+/(Ti3+ + Ti4+) | Oα/(Oα + Oβ) | |
| MnOx–CeO2-0.7 | 5.6 | 26.4 | 68.0 | 46.42 | 41.46 | 33.4 |
| MnOx–CeO2-0.7-S | 5.4 | 19.7 | 70.6 | - | 25.2 | 50.3 |
| MnOx–TiO2-0.5 | 13.6 | 18.8 | 67.6 | 47.31 | 87.9 | 25.6 |
| MnOx–TiO2-0.5-S | 9.4 | 13.3 | 74.4 | - | 87.1 | 55.8 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Huo, X.; Zhu, T.; Hong, X.; Sun, Y. Catalytic Oxidation of NO over MnOx–CeO2 and MnOx–TiO2 Catalysts. Molecules 2016, 21, 1491. https://doi.org/10.3390/molecules21111491
Zeng X, Huo X, Zhu T, Hong X, Sun Y. Catalytic Oxidation of NO over MnOx–CeO2 and MnOx–TiO2 Catalysts. Molecules. 2016; 21(11):1491. https://doi.org/10.3390/molecules21111491
Chicago/Turabian StyleZeng, Xiaolan, Xiaoyue Huo, Tianle Zhu, Xiaowei Hong, and Ye Sun. 2016. "Catalytic Oxidation of NO over MnOx–CeO2 and MnOx–TiO2 Catalysts" Molecules 21, no. 11: 1491. https://doi.org/10.3390/molecules21111491