
Synthesis of Bisindole Alkaloids from the Apocynaceae Which Contain a Macroline or Sarpagine Unit: A Review




Abstract
:1. Introduction
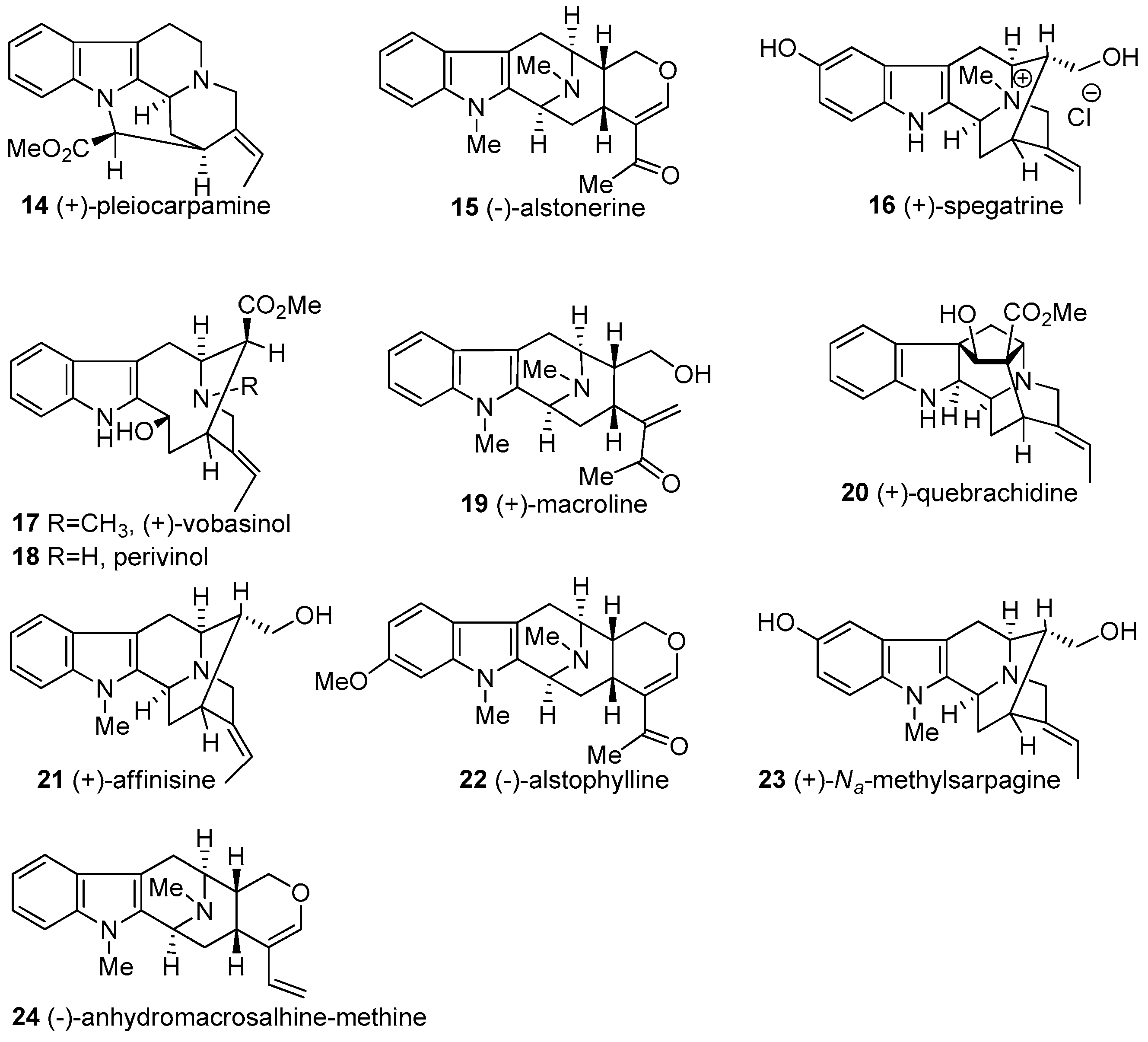
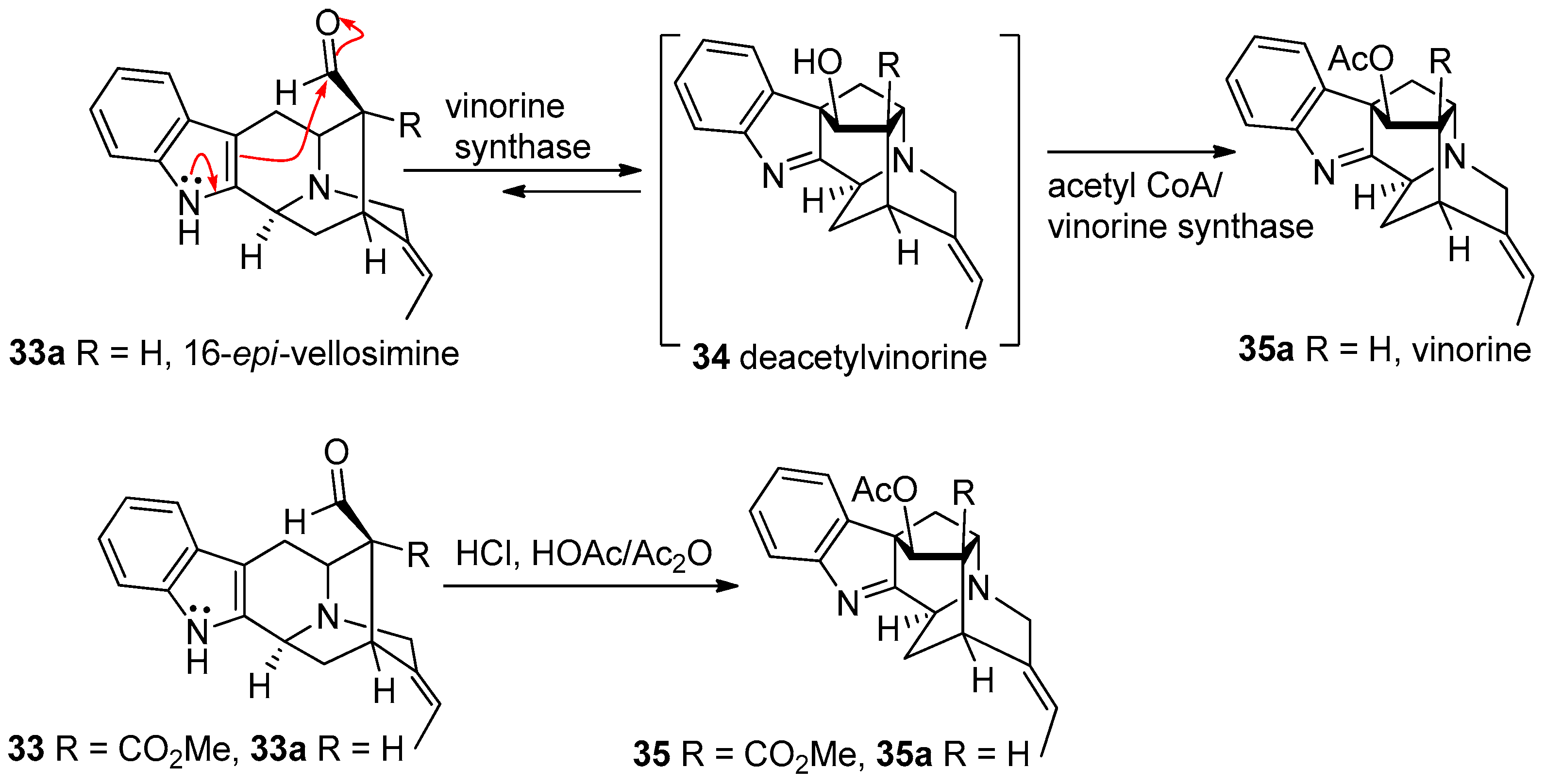
2. Biogenetic Relationship between Sarpagine/Macroline/Ajmaline Alkaloids
3. General Strategy to Access the Tetracyclic Core 27 of the Sarpagine/Macroline/Ajmaline Related Indole Alkaloids
4. Partial and Total Synthesis of Bisindole Alkaloids
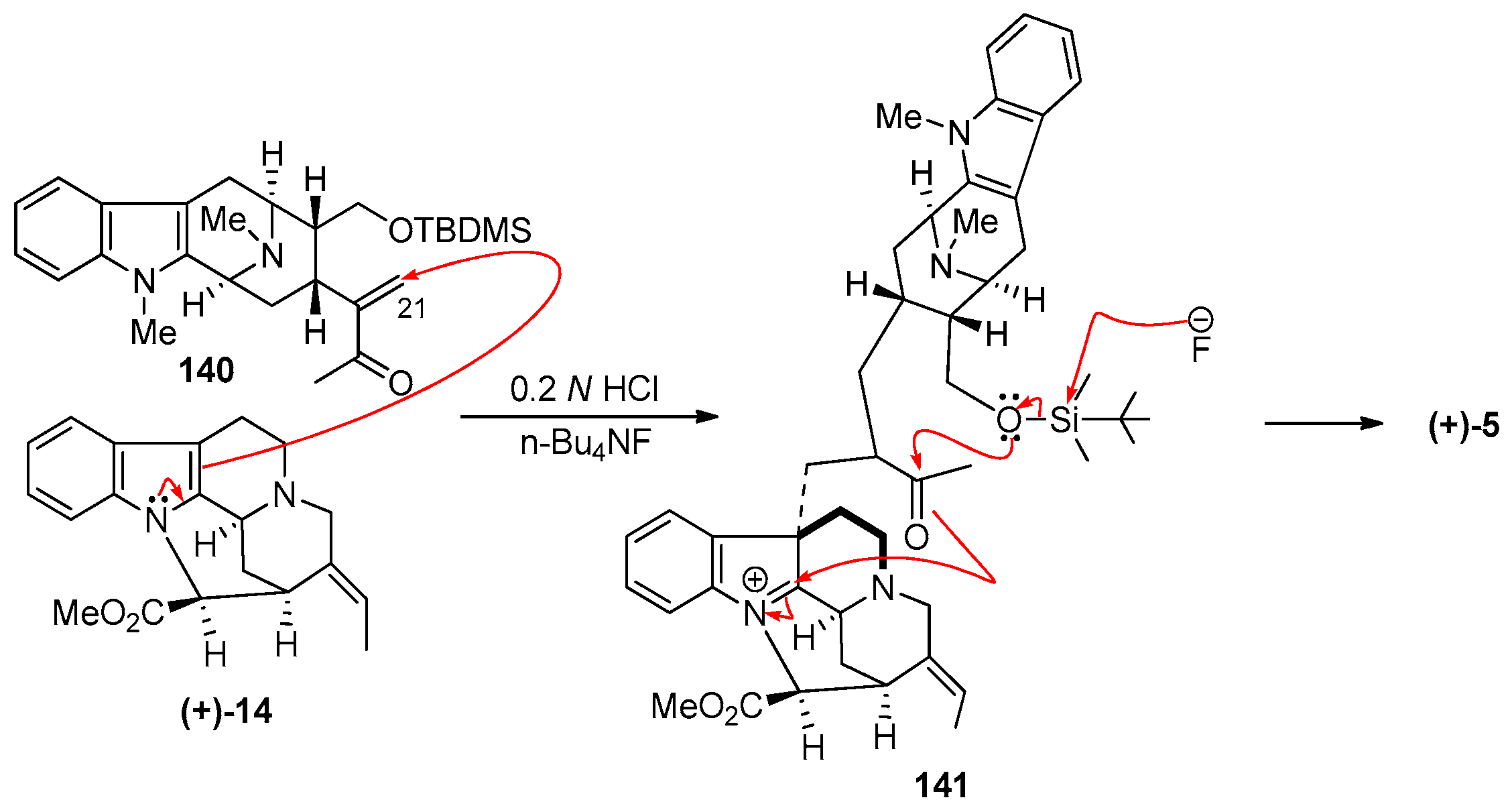
4.1. (−)-Alstonisidine
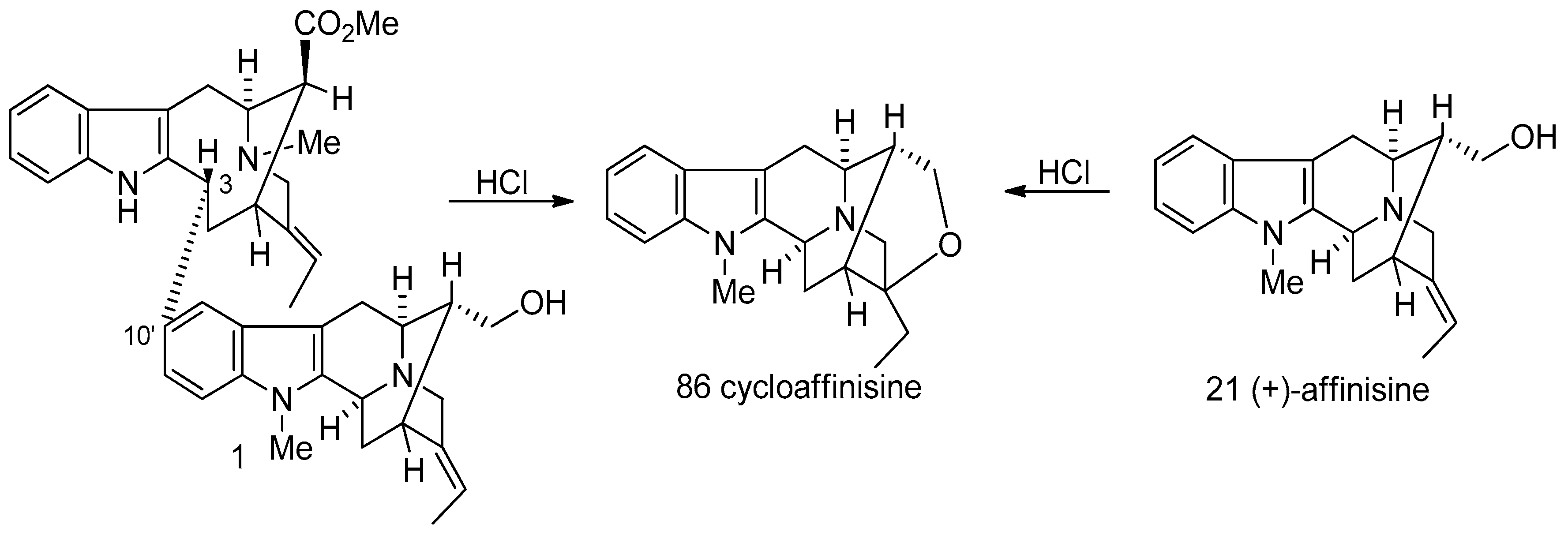
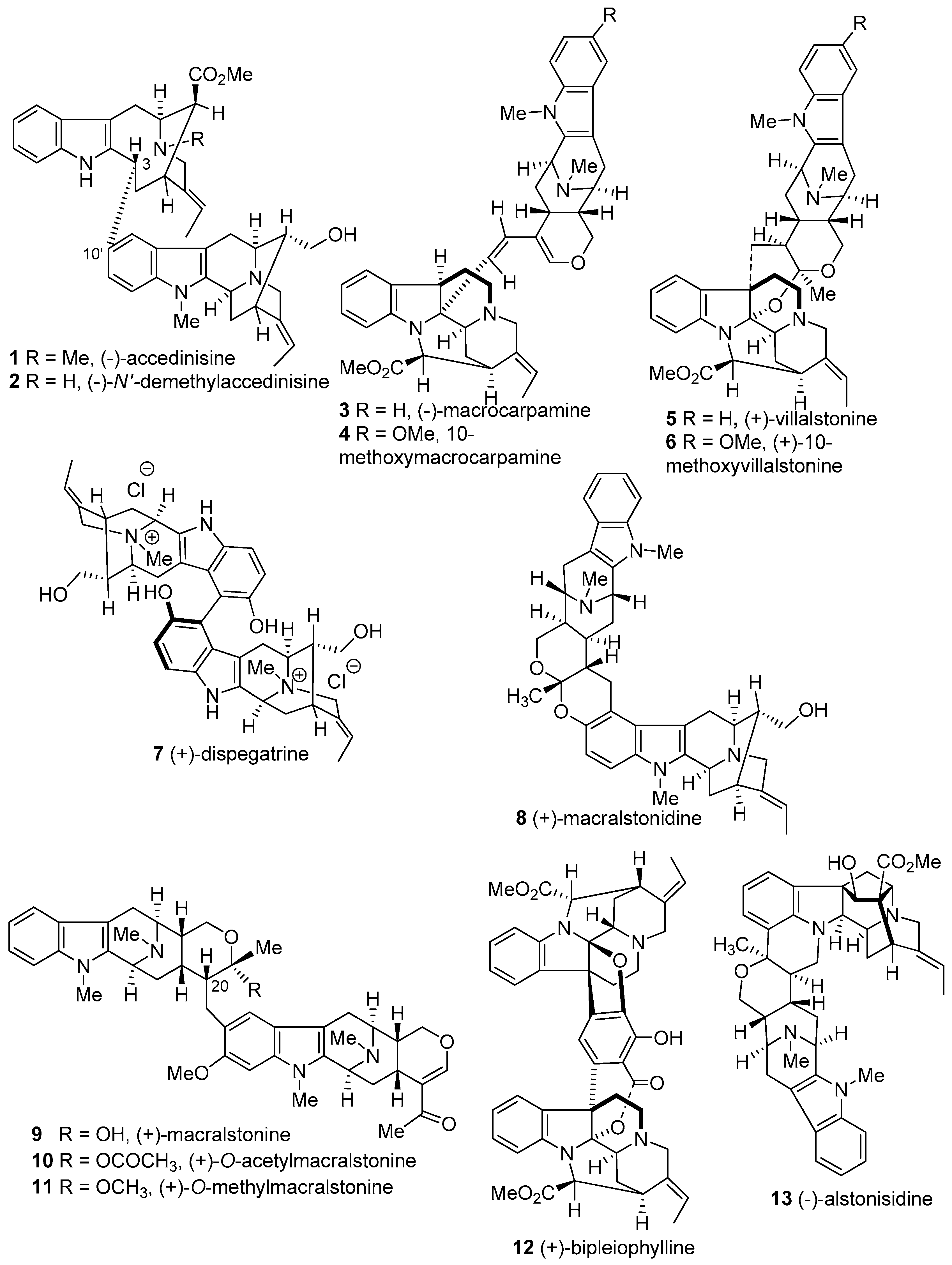
4.2. (−)-Accedinisine and (−)-N′-demethylaccedinisine
4.3. (+)-Macralstonine
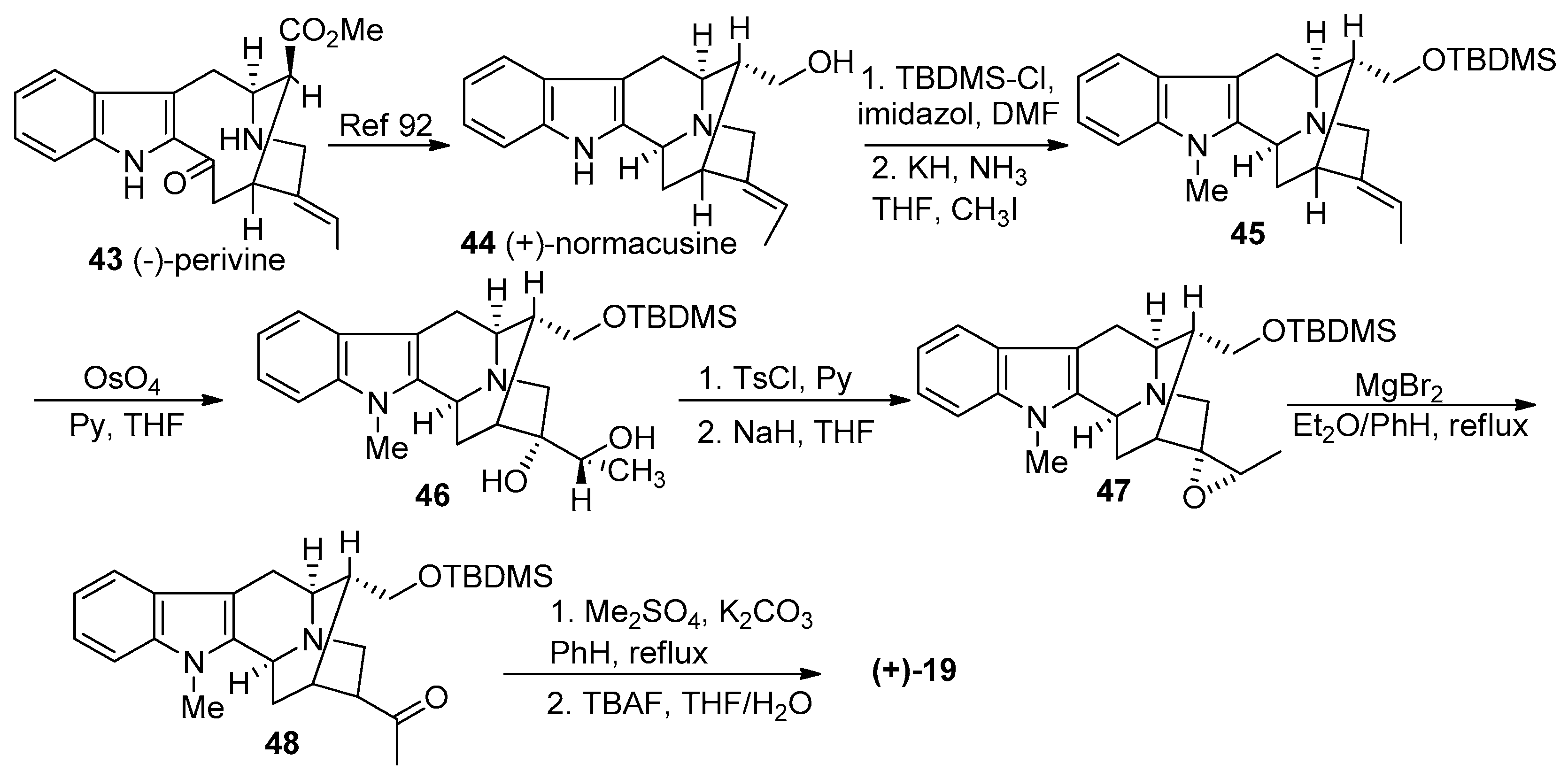
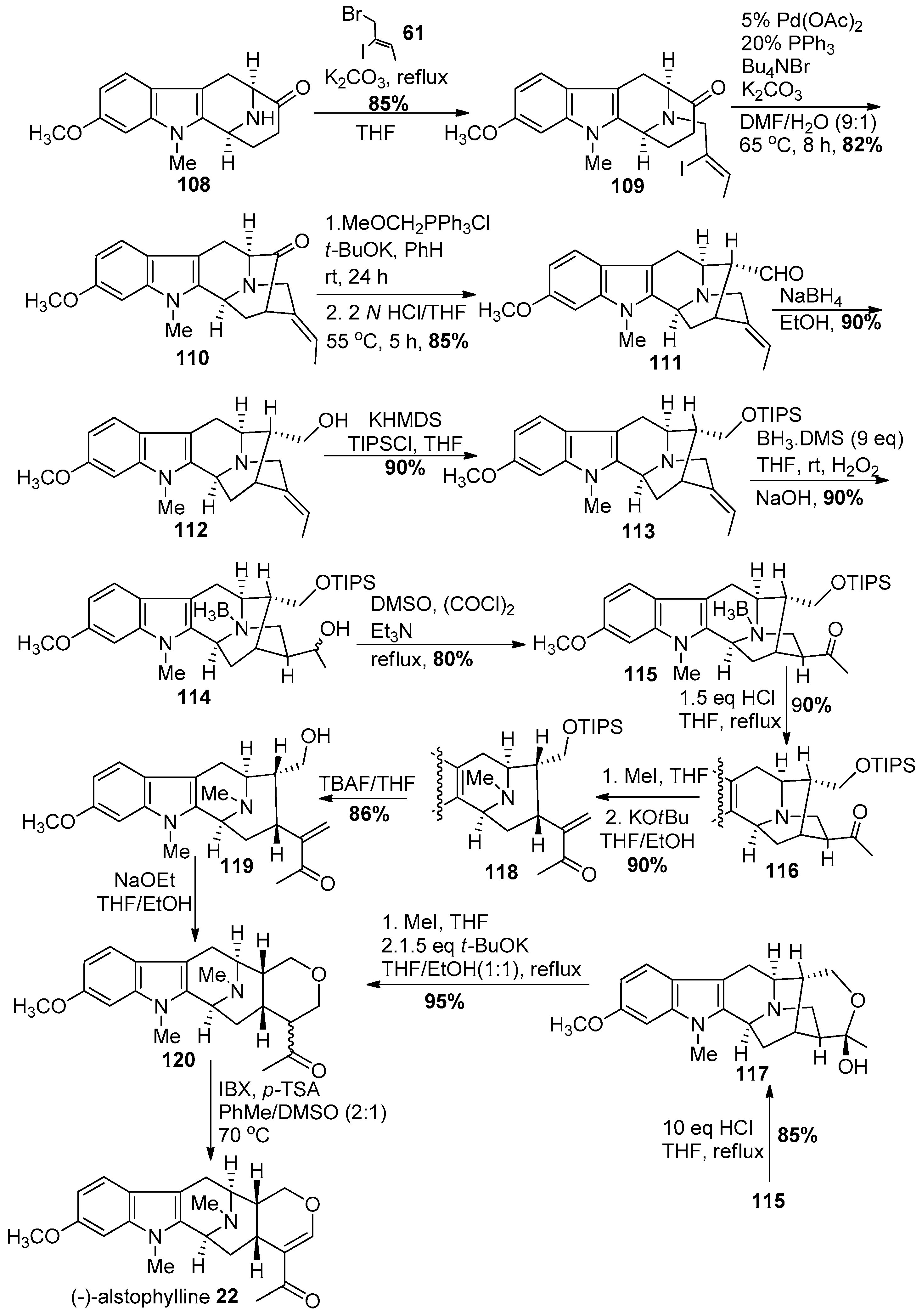
4.4. (+)-Macralstonidine
4.5. (+)-Villalstonine
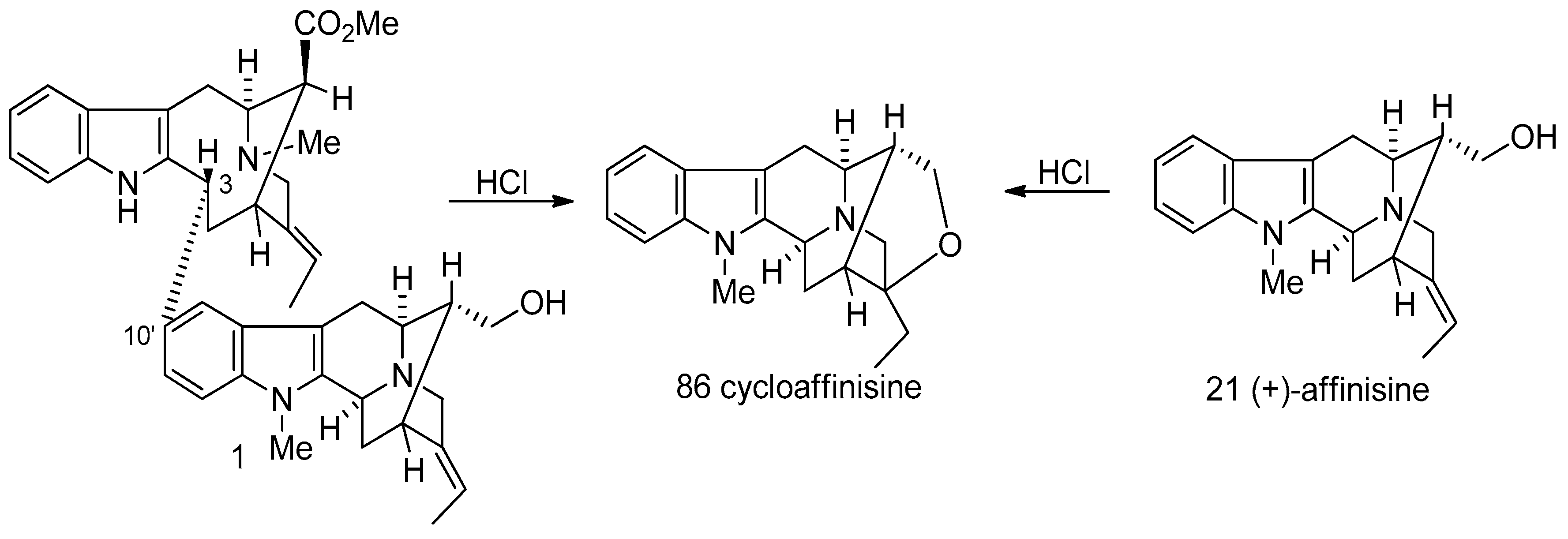
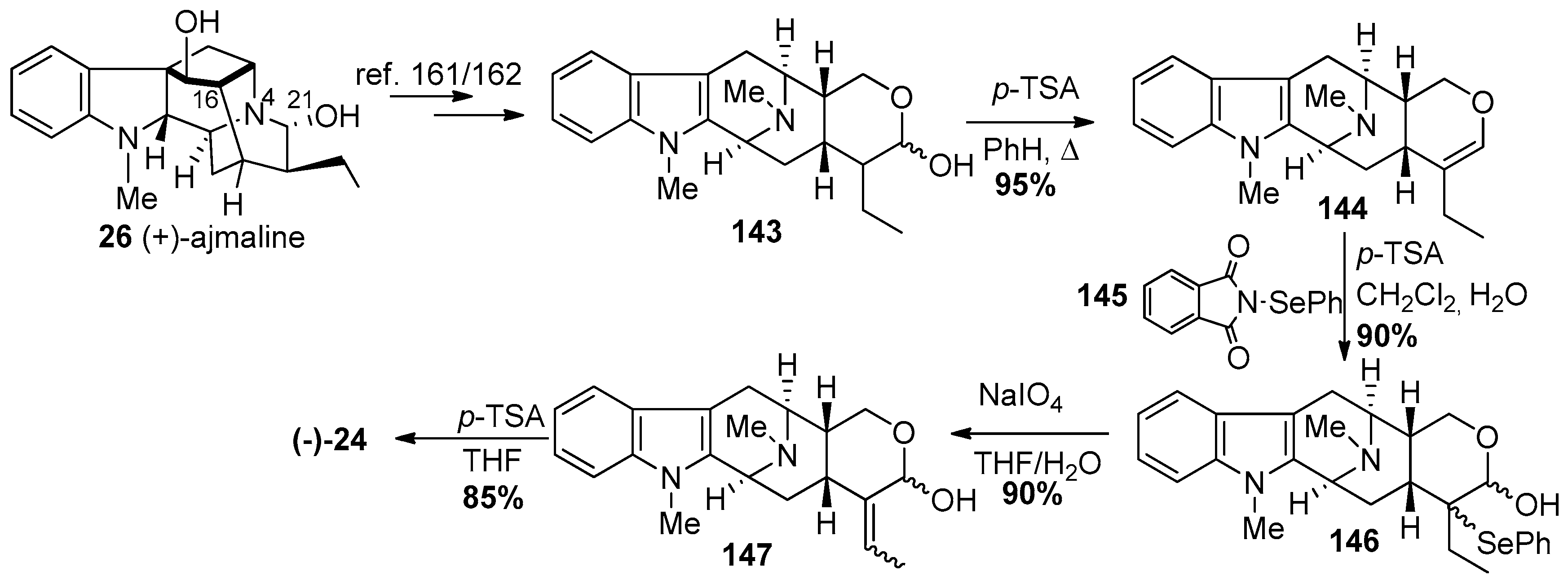
4.6. (−)-Macrocarpamine
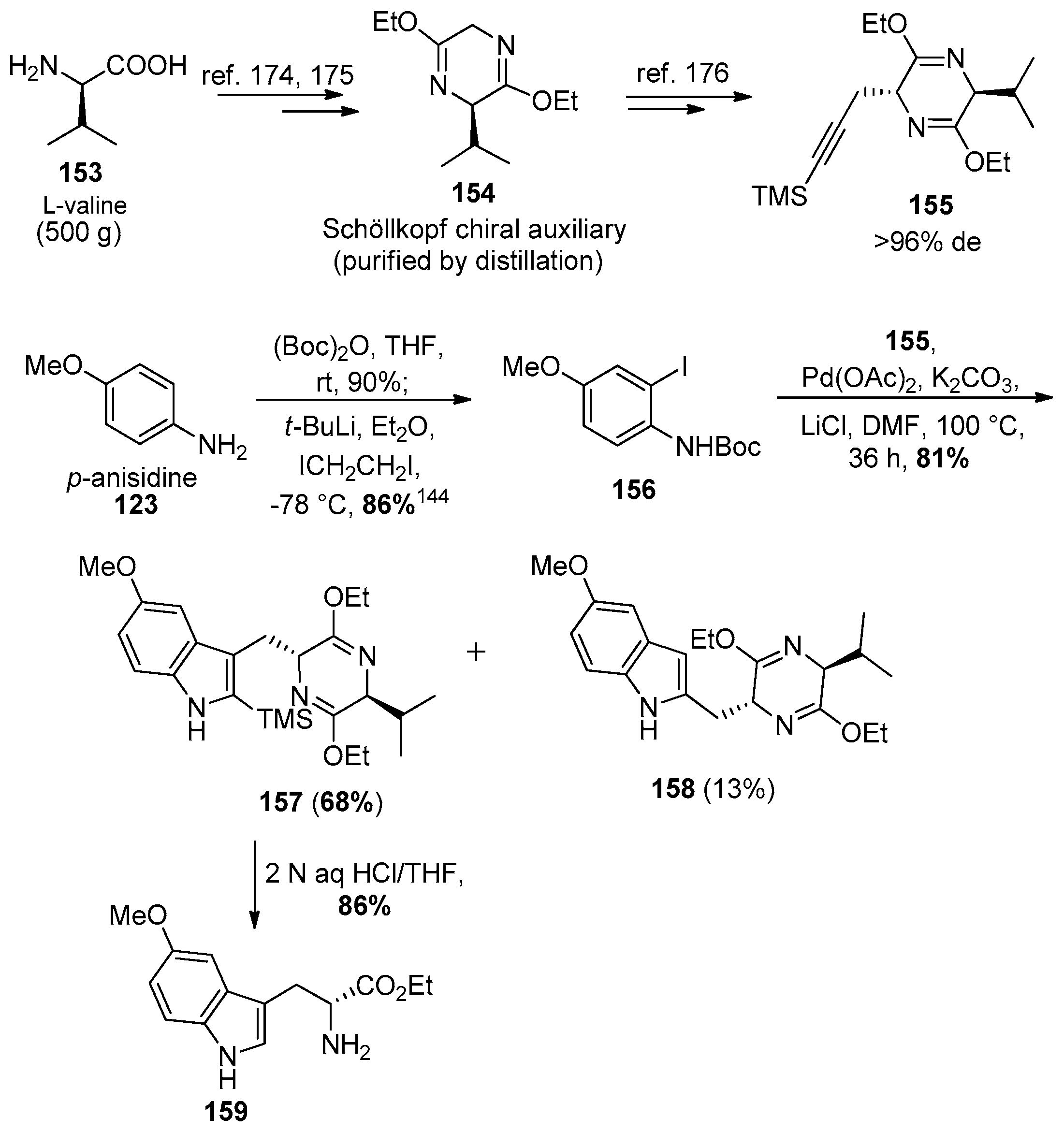
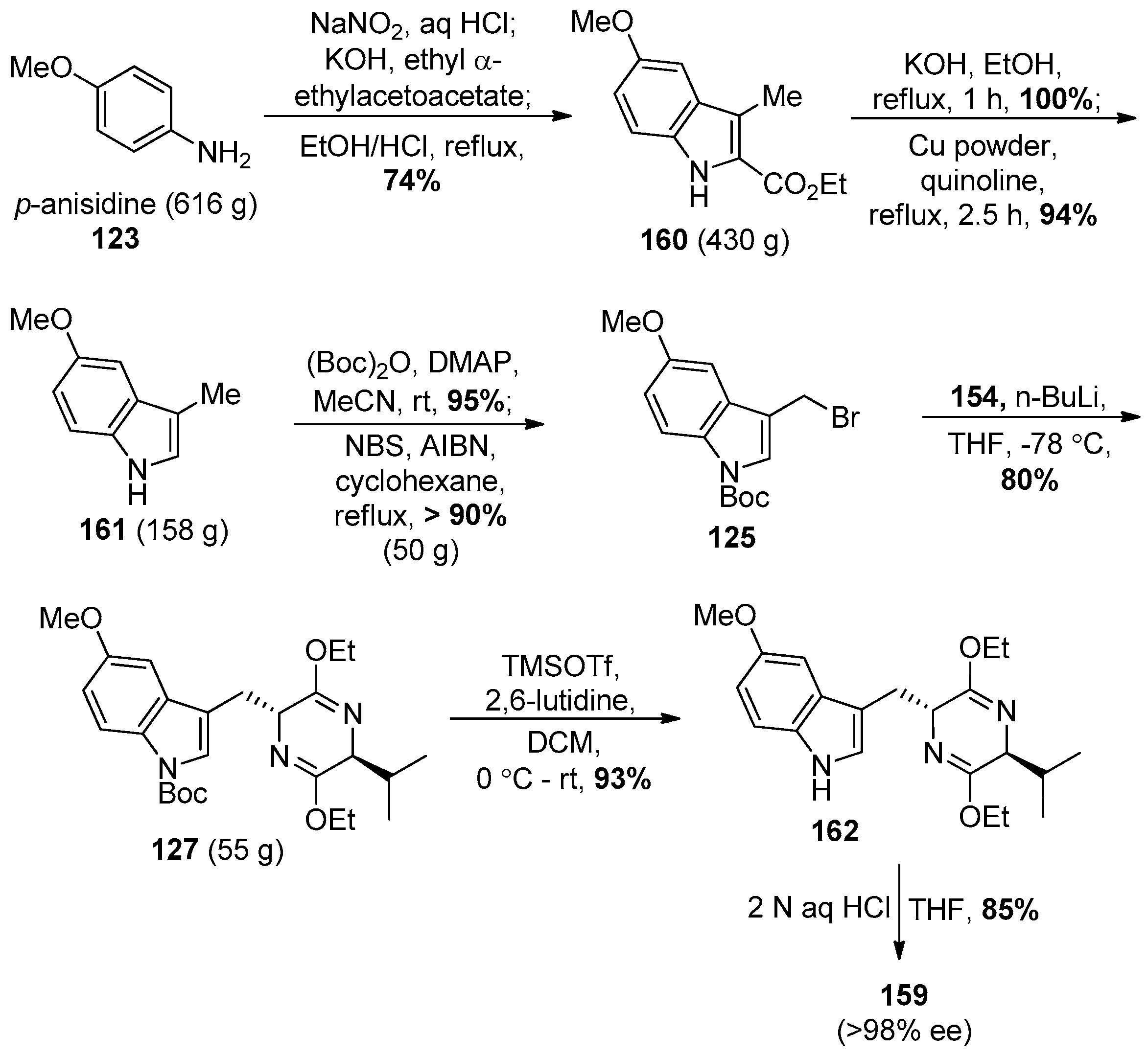
4.7. (+)-Dispegatrine
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- O’Neil, M.J. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals; RSC Publishing: London, UK, 2013. [Google Scholar]
- Henry, T.A. The Plant Alkaloids, 4th ed.; J & A Churchill Ltd.: London, UK, 1949. [Google Scholar]
- Fabricant, D.S.; Farnsworth, N.R. The value of plants used in traditional medicine for drug discovery. Environ. Health Perspect. 2001, 109 (Suppl. S1), 69–75. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, N.R.; Akerele, O.; Bingel, A.S.; Soejarto, D.D.; Guo, Z. Medicinal plants in therapy. Bull. World Health Organ. 1985, 63, 965–981. [Google Scholar] [CrossRef]
- Wang, J.-F.; Wei, D.-Q.; Chou, K.-C. Drug candidates from traditional Chinese medicines. Curr. Top. Med. Chem. 2008, 8, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S. Natural products to drugs: Natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. [Google Scholar] [CrossRef] [PubMed]
- Cordell, G.A.; Quinn-Beattie, M.L.; Farnsworth, N.R. The potential of alkaloids in drug discovery. Phytother. Res. 2001, 15, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem. Int. Ed. 2003, 42, 3996–4028. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.B.; Tiwari, V.K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem. 2011, 46, 4769–4807. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Biodiversity: A continuing source of novel drug leads. Pure Appl. Chem. 2005, 77, 7–24. [Google Scholar] [CrossRef]
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov. Today 2003, 8, 536–544. [Google Scholar] [CrossRef]
- Butler, M.S. The role of natural product chemistry in drug discovery. J. Nat. Prod. 2004, 67, 2141–2153. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S. Natural products to drugs: Natural product derived compounds in clinical trials. Nat. Prod. Rep. 2005, 22, 162–195. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Cordell, G.A.; Saxton, J.E. Bisindole Alkaloids. In The Alkaloids: Chemistry and Physiology; Rodrigo, G.A., Ed.; Academic Press: New York, NY, USA, 1981; Volume 20, pp. 3–295. [Google Scholar]
- Cordell, G.A. The bisindole alkaloids. In Chemistry of Heterocyclic Compounds: Indoles, Part Four, the Monoterpenoid Indole Alkaloids; Saxton, J.E., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1983; Volume 25, pp. 539–728. [Google Scholar]
- Kam, T.-S.; Choo, Y.-M. Bisindole alkaloids. In The Alkaloids: Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: San Diego, CA, USA, 2006; Volume 63, pp. 181–337. [Google Scholar]
- Kitajima, M.; Takayama, H. Chapter Four-Monoterpenoid Bisindole Alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: San Diego, CA, USA, 2016; Volume 76, pp. 259–310. [Google Scholar]
- Ban, Y.; Murakami, Y.; Iwasawa, Y.; Tsuchiya, M.; Takano, N. Indole alkaloids in medicine. Med. Res. Rev. 1988, 8, 231–308. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; Allen, D.; Cai, Y.; Phillipson, J.; Said, I.; Kirby, G.; Warhurst, D. In vitro antiamoebic and antiplasmodial activities of alkaloids isolated from Alstonia angustifolia roots. Phytother. Res. 1992, 6, 121–124. [Google Scholar] [CrossRef]
- Keawprdub, N.; Houghton, P.; Eno-Amooquaye, E.; Burke, P. Activity of extracts and alkaloids of Thai Alstonia species against human lung cancer cell lines. Planta Med. 1997, 63, 97–101. [Google Scholar] [CrossRef]
- Johnson, I.S.; Wright, H.F.; Svoboda, G.H.; Vlantis, J. Antitumor principles derived from Vinca rosea Linn I. Vincaleukoblastine and leurosine. Cancer Res. 1960, 20, 1016–1022. [Google Scholar] [PubMed]
- Noble, R.; Beer, C.; Cutts, J. Role of chance observations in chemotherapy: Vinca rosea. Ann. N. Y. Acad. Sci. 1958, 76, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, G.H.; Johnson, I.S.; Gorman, M.; Neuss, N. Current status of research on the alkaloids of Vinca rosea Linn. (Catharanthus roseus G. Don). J. Pharm. Sci. 1962, 51, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Neuss, N.; Gorman, M.; Svoboda, G.; Maciak, G.; Beer, C. Vinca alkaloids. III. 1. Characterization of leurosine and vincaleukoblastine, new alkaloids from Vinca rosea LINN. J. Am. Chem. Soc. 1959, 81, 4754–4755. [Google Scholar] [CrossRef]
- Johnson, I. Historical background of Vinca alkaloid research and areas of future interest. Cancer Chemother. Rep. 1968, 52, 455–461. [Google Scholar] [PubMed]
- Neuss, N.; Neuss, M.N. Therapeutic use of bisindole alkaloids from catharanthus. Alkaloids Chem. Pharmacol. 1990, 37, 229–240. [Google Scholar]
- Sieber, S.; Mead, J.; Adamson, R. Pharmacology of antitumor agents from higher plants. Cancer Treat. Rep. 1976, 60, 1127. [Google Scholar] [PubMed]
- Carney, D.W.; Lukesh, J.C.; Brody, D.M.; Brütsch, M.M.; Boger, D.L. Ultrapotent vinblastines in which added molecular complexity further disrupts the target tubulin dimer–dimer interface. Proc. Natl. Acad. Sci. USA 2016, 113, 9691–9698. [Google Scholar] [CrossRef] [PubMed]
- Kutney, J.P. Synthetic Vinblastine and Vincristine Derivatives. U.S. Patent 4144237 A, 13 March 1979. [Google Scholar]
- Fahy, J.; Duflos, A.; Ribet, J.-P.; Jacquesy, J.-C.; Berrier, C.; Jouannetaud, M.-P.; Zunino, F. Vinca alkaloids in superacidic media: A method for creating a new family of antitumor derivatives. J. Am. Chem. Soc. 1997, 119, 8576–8577. [Google Scholar] [CrossRef]
- Keglevich, P.; Hazai, L.; Kalaus, G.; Szántay, C. Modifications on the basic skeletons of vinblastine and vincristine. Molecules 2012, 17, 5893–5914. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Giannakakou, P. Targeting microtubules for cancer chemotherapy. Curr. Med. Chem. Anticancer Agents 2005, 5, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Va, P.; Campbell, E.L.; Robertson, W.M.; Boger, D.L. Total synthesis and evaluation of a key series of C5-substituted vinblastine derivatives. J. Am. Chem. Soc. 2010, 132, 8489–8495. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Colby, D.A.; Seto, S.; Va, P.; Tam, A.; Kakei, H.; Rayl, T.J.; Hwang, I.; Boger, D.L. Total synthesis of vinblastine, vincristine, related natural products, and key structural analogues. J. Am. Chem. Soc. 2009, 131, 4904–4916. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Van Linn, M.; Cook, J. The asymmetric Pictet-Spengler reaction. Curr. Org. Synth. 2010, 7, 189–223. [Google Scholar] [CrossRef]
- Munoz, V.; Moretti, C.; Sauvain, M.; Caron, C.; Porzel, A.; Massiot, G.; Richard, B.; Le Men-Olivier, L. Isolation of bis-indole alkaloids with antileishmanial and antibacterial activities from Perschiera van heurkii (Syn. Tabernaemontana van heurkii). Planta Med. 1994, 60, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Pant, N.; Jain, D.; Bhakuni, R. Antimalarial agents from plant sources. Curr. Sci. 2003, 85, 1314–1329. [Google Scholar]
- Keawpradub, N.; Eno-Amooquaye, E.; Burke, P.; Houghton, P. Cytotoxic activity of indole alkaloids from Alstonia macrophylla. Planta Med. 1999, 65, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.W.; Allen, D.; Phillipson, J.D.; Kirby, G.C.; Warhurst, D.C.; Massiot, G.; Le Men-Olivier, L. Alstonia species: Are they effective in malaria treatment? J. Ethnopharmacol. 1993, 40, 41–45. [Google Scholar] [CrossRef]
- Wright, C.W.; Phillipson, J.D. Natural products and the development of selective antiprotozoal drugs. Phytother. Res. 1990, 4, 127–139. [Google Scholar] [CrossRef]
- Kam, T.-S.; Choo, Y.-M.; Komiyama, K. Unusual spirocyclic macroline alkaloids, nitrogenous derivatives, and a cytotoxic bisindole from Alstonia. Tetrahedron 2004, 60, 3957–3966. [Google Scholar] [CrossRef]
- Feng, Y.; Gao, H.; Zeng, G. Effect of higenamine on alpha-adrenoceptors. Acta pharmacol. Sin. 1986, 7, 208–211. [Google Scholar]
- Talapatra, S.; Aditya Chaudhury, N. Macralstonine, an alkaloid of the trunk bark of Alstonia macrophylla Wall. Sci. Cult. 1958, 24, 243. [Google Scholar]
- Isidro, N.; Manalo, G.D. J. Phillipine Pharm. Assoc. 1967, 53, 8.
- Keawpradub, N.; Kirby, G.; Steele, J.; Houghton, P. Antiplasmodial activity of extracts and alkaloids of three Alstonia species from Thailand. Planta Med. 1999, 65, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Kingston, D.G. Plant anticancer agents VI: Isolation of voacangine, voacamine, and epivoacorine from Tabernaemontana arborea sap. J. Pharm. Sci. 1978, 67, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.-S.; Tan, S.-J.; Ng, S.-W.; Komiyama, K. Bipleiophylline, an unprecedented cytotoxic bisindole alkaloid constituted from the bridging of two indole moieties by an aromatic spacer unit. Org. Lett. 2008, 10, 3749–3752. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, A.; Lounasmaa, M. The sarpagine-ajmaline group of indole alkaloids. In Progress in the Chemistry of Organic Natural Products; Ingham, J.L., Ed.; Springer: Wien, Austria, 1983; Volume 43, pp. 267–346. [Google Scholar]
- Brugada, J.; Brugada, P. What to do in patients with no structural heart disease and sudden arrhythmic death? Am. J. Cardiol. 1996, 78, 69–75. [Google Scholar] [CrossRef]
- Creasey, W.A. Pharmacology, biochemistry, and clinical applications of the monoterpenoid alkaloids. In Chemistry of Heterocyclic Compounds: Indoles, Part Four, The Monoterpenoid Indole Alkaloids; Saxton, J.E., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1983; Volume 25, pp. 783–829. [Google Scholar]
- Lounasmaa, M.; Hanhinen, P.; Westersund, M.; Halonen, N. The Sarpagine Group of Indole Alkaloids. In The Alkaloids: Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: San Diego, CA, USA, 1999; Volume 52, pp. 103–195. [Google Scholar]
- Namjoshi, O.A.; Cook, J.M. Chapter Two-Sarpagine and Related Alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: San Diego, CA, USA, 2016; Volume 76, pp. 63–169. [Google Scholar]
- Le Men, J.; Taylor, W.I. A uniform numbering system for indole alkaloids. Experientia 1965, 21, 508–510. [Google Scholar] [CrossRef] [PubMed]
- Hamaker, L.K.; Cook, J.M. Chapter Two: The Synthesis of Macroline Related Sarpagine Alkaloids. In Alkaloids: Chemical and Biological Perspectives; Pelletier, S.W., Ed.; Elsevier Science: New York, NY, USA, 1995; Volume 9, pp. 23–84. [Google Scholar]
- Esmond, R.W.; Le Quesne, P.W. Biomimetic synthesis of macroline. J. Am. Chem. Soc. 1980, 102, 7116–7117. [Google Scholar] [CrossRef]
- Woodward, R. Neuere entwicklungen in der chemie der naturstoffe. Angew. Chem. 1956, 68, 13–20. [Google Scholar] [CrossRef]
- Bartlett, M.; Lambert, B.; Werblood, H.; Taylor, W. Rauwolfia alkaloids. XLIII. 1 A facile ring closure of deoxyajmalal-A to deoxyajmaline. J. Am. Chem. Soc. 1963, 85, 475–477. [Google Scholar] [CrossRef]
- Van Tamelen, E.; Oliver, L. The biogenetic-type total synthesis of ajmaline. Bioorg. Chem. 1976, 5, 309–326. [Google Scholar] [CrossRef]
- Koskinen, A.; Lounasmaa, M. Biogenesis of the Ajmaline Type Alkaloids. Planta Med. 1982, 45, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Pfitzner, A.; Stöckigt, J. Biogenetic link between sarpagine and ajmaline type alkaloids. Tetrahedron Lett. 1983, 24, 5197–5200. [Google Scholar] [CrossRef]
- Mattern-Dogru, E.; Ma, X.; Hartmann, J.; Decker, H.; Stöckigt, J. Potential active-site residues in polyneuridine aldehyde esterase, a central enzyme of indole alkaloid biosynthesis, by modelling and site-directed mutagenesis. Eur. J. Biochem. 2002, 269, 2889–2896. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, M.; Woll, J.; Giritch, A.; Genady, E.; Ma, X.; Stöckigt, J. Functional expression of an ajmaline pathway-specific esterase from Rauvolfia in a novel plant-virus expression system. Planta 2005, 222, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, N. Synthesis of 12-methyl-1,2,3,4,6,7,12,12b-octahydro-2, 6-methanoindolo[2,3-a]quinolizine. Chem. Pharm. Bull. 1965, 13, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, K.; Sato, Y. On the synthesis of ajmaline. Tetrahedron Lett. 1969, 10, 905–906. [Google Scholar] [CrossRef]
- Cloudsdale, I.S.; Kluge, A.F.; McClure, N.L. Synthetic studies in the ajmaline series. J. Org. Chem. 1982, 47, 919–928. [Google Scholar] [CrossRef]
- Soerens, D.; Sandrin, J.; Ungemach, F.; Mokry, P.; Wu, G.; Yamanaka, E.; Hutchins, L.; DiPierro, M.; Cook, J. Study of the Pictet-Spengler reaction in aprotic media: Synthesis of the. beta.-galactosidase inhibitor, pyridindolol. J. Org. Chem. 1979, 44, 535–545. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Cook, J. Pictet-Spengler reactions in aprotic media: Nb-benzyl promoted retention of optical activity in the synthesis of an indolo substituted azabicyclo[3.3.1]nonane, a key template for the synthesis of macroline alkaloids. Heterocycles 1988, 27, 2795–2802. [Google Scholar] [CrossRef]
- Li, J.; Wang, T.; Yu, P.; Peterson, A.; Weber, R.; Soerens, D.; Grubisha, D.; Bennett, D.; Cook, J. General approach for the synthesis of ajmaline/sarpagine indole alkaloids: Enantiospecific total synthesis of (+)-ajmaline, alkaloid g, and norsuaveoline via the asymmetric pictet-spengler reaction. J. Am. Chem. Soc. 1999, 121, 6998–7010. [Google Scholar] [CrossRef]
- Li, J. Enantiospecific Total Synthesis of (+)-ajmaline and Alkaloid G as Well as Studies Directed toward the Total Synthesis of 19-hydroxy-Nb-methylraumacline via the Asymmetric Pictet-Spengler Reaction. Ph.D. Thesis, University of Wisconsin-Milwaukee, Milwaukee, WI, USA, 1999. [Google Scholar]
- Elderfield, R.C.; Gilman, R.E. Alkaloids of Alstonia muelleriana. Phytochemistry 1972, 11, 339–343. [Google Scholar] [CrossRef]
- Burke, D.E.; Cook, G.A.; Cook, J.M.; Haller, K.G.; Lazar, H.A.; Le Quesne, P.W. Further alkaloids of Alstonia muelleriana. Phytochemistry 1973, 12, 1467–1474. [Google Scholar] [CrossRef]
- Cook, J.M.; Le Quesne, P. Structure of alstonisidine, a novel dimeric indole alkaloid from Alstonia muelleriana. J. Org. Chem. 1971, 36, 582–586. [Google Scholar] [CrossRef]
- Burke, D.E.; Cook, J.M.; Le Quesne, P. Biomimetic synthesis and structure of the bisindole alkaloid alstonisidine. J. Chem. Soc. Chem. Commun. 1972, 697. [Google Scholar] [CrossRef]
- Hoard, L.G. The Crystal Structures of Altstonisidine, C42H48N4O4, and Anhydrous Cholesterol, C27H46O. Ph.D. Thesis, University of Michigan, Ann Arbor, MI, USA, 1977. [Google Scholar]
- Cook, J.M.; Le Quesne, P.; Elderfield, R. Alstonerine, a new indole alkaloid from Alstonia muelleriana. J. Chem. Soc. Chem. Commun. 1969, 1306–1307. [Google Scholar] [CrossRef]
- Kishi, T.; Hesse, M.; Gemenden, C.; Taylor, W.; Schmid, H. Alstophyllin, ein neues Indolalkaloid aus Alstonia macrophylla WALL. Helv. Chim. Acta 1965, 48, 1349–1362. [Google Scholar] [CrossRef] [PubMed]
- Aynilian, G.; Farnsworth, N. Alkaloids of Vinca species. 3. Isolation and characterization of indole alkaloids from Vinca libanotica. Lloydia 1974, 37, 299. [Google Scholar] [PubMed]
- Salim, A.A.; Garson, M.J.; Craik, D.J. New indole alkaloids from the roots of Ochrosia a cuminata. J. Nat. Prod. 2004, 67, 1719–1721. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.; Fong, H.; Farnsworth, N.; Svoboda, G. Biological and phytochemical evaluation of plants XI: Isolation of aspidospermine, quebrachidine, rhazinilam, (—)-pyrifolidine, and akuammidine from Aspidosperma quebracho-blanco (apocynaceae). J. Pharm. Sci. 1973, 62, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Gorman, M.; Burlingame, A.; Biemann, K. Application of mass spectrometry to structure problems. The structure of quebrachidine. Tetrahedron Lett. 1963, 4, 39–46. [Google Scholar] [CrossRef]
- Burke, D.E.; Cook, J.M.; Le Quesne, P. Biomimetic synthesis of the bisindole alkaloids villalstonine and alstonisidine. J. Am. Chem. Soc. 1973, 95, 546–552. [Google Scholar] [CrossRef]
- Hesse, M.; Bodmer, F.; Gemenden, C.; Joshi, B.; Taylor, W.; Schmid, H. Die struktur des Alstonia-alkaloides villalstonin. Helv. Chim. Acta 1966, 49, 1173–1182. [Google Scholar] [CrossRef]
- Bi, Y.; Cook, J.M. General approach for the synthesis of macroline/sarpagine alkaloids. The total synthesis of (+)-macroline. Tetrahedron Lett. 1993, 34, 4501–4504. [Google Scholar] [CrossRef]
- Bi, Y.; Zhang, L.-H.; Hamaker, L.K.; Cook, J.M. Enantiospecific Synthesis of (−)-Alstonerine and (+)-Macroline as Well as a Partial Synthesis of (+)-Villalstonine. J. Am. Chem. Soc. 1994, 116, 9027–9041. [Google Scholar] [CrossRef]
- Liao, X.; Zhou, H.; Yu, J.; Cook, J.M. An improved total synthesis of (+)-macroline and alstonerine as well as the formal total synthesis of (−)-talcarpine and (−)-anhydromacrosalhine-methine. J. Org. Chem. 2006, 71, 8884–8890. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, T.; Liu, X.; Deschamps, J.; Flippen-Anderson, J.; Liao, X.; Cook, J.M. General approach for the synthesis of sarpagine indole alkaloids. Enantiospecific total synthesis of (+)-vellosimine, (+)-normacusine B, (−)-alkaloid Q3, (−)-panarine, (+)-Na-methylvellosimine, and (+)-Na-methyl-16-epi-pericyclivine. J. Org. Chem. 2003, 68, 7565–7581. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, C.; Liao, X.; Cook, J.M. Enantiospecific total synthesis of the enantiomer of the indole alkaloid intermediate macroline. Tetrahedron Lett. 2002, 43, 7373–7377. [Google Scholar] [CrossRef]
- Yu, J.; Wearing, X.Z.; Cook, J.M. A general strategy for the synthesis of vincamajine-related indole alkaloids: Stereocontrolled total synthesis of (+)-dehydrovoachalotine, (−)-vincamajinine, and (−)-11-methoxy-17-epivincamajine as well as the related quebrachidine diol, vincamajine diol, and vincarinol. J. Org. Chem. 2005, 70, 3963–3979. [Google Scholar] [PubMed]
- Neukomm, G.; Kletzhäundler, E.; Hesse, M. Die absolute konfiguration von macrolin, einem abbauprodukt des alkaloides villalstonin 179. Mitteilung über organische naturstoffe. Helv. Chim. Acta 1981, 64, 90–96. [Google Scholar] [CrossRef]
- Gorman, M.; Sweeny, J. Perivine. Tetrahedron Lett. 1964, 5, 3105–3111. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Cook, J.M. Pictet-Spengler reactions in aprotic media. Stereospecific conversion of optically active cis-1, 3-disubstituted 1,2,3,4-tetrahydro-β-carbolines into their corresponding trans diastereomers. Heterocycles 1988, 27, 1357–1363. [Google Scholar] [CrossRef]
- Yu, P.; Wang, T.; Yu, F.; Cook, J.M. General approach for the synthesis of macroline/sarpagine related indole alkaloids via the asymmetric pictet-spengler reaction: The enantiospecific synthesis of the Na-H, azabicyclo[3.3.1]nonone template. Tetrahedron Lett. 1997, 38, 6819–6822. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Bi, Y.-Z.; Yu, F.-X.; Menzia, G.; Cook, J.M. Stereospecificity in the Pictet-Spengler reaction. Enantiospecific synthesis of (6S, 10S)-5#75)-5-methyl-9-oxo-12-benzyl-6,7,8,9,10,11-hexahydro-6,10-imino-5H-cyclooct[b]indole, a template for preparation of macroline/sarpagine alkaloids. Heterocycles 1992, 34, 517–547. [Google Scholar]
- Trudell, M.L.; Cook, J.M. Total synthesis of (±)-suaveoline. J. Am. Chem. Soc. 1989, 111, 7504–7507. [Google Scholar] [CrossRef]
- Bornack, W.K.; Bhagwat, S.S.; Ponton, J.; Helquist, P. Preparation of lactone systems. Total synthesis of (±)-quadrone. J. Am. Chem. Soc. 1981, 103, 4647–4648. [Google Scholar] [CrossRef]
- Amer, M.A.; Court, W.E. Alkaloids of Rauwolfia nitida root bark. Phytochemistry 1981, 20, 2569–2573. [Google Scholar] [CrossRef]
- Banerji, A.; Chakrabarty, M.; Mukherjee, B. Minor indole alkaloids of Alstonia macrophylla. Phytochemistry 1972, 11, 2605–2607. [Google Scholar] [CrossRef]
- Tran, Y.S.; Kwon, O. An application of the phosphine-catalyzed [4 + 2] annulation in indole alkaloid synthesis: Formal syntheses of (±)-alstonerine and (±)-macroline. Org. Lett. 2005, 7, 4289–4291. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, T.; Cheung, M.; Kan, T. N-carboalkoxy-2-nitrobenzenesulfonamides: A practical preparation of N-Boc-, N-Alloc-, and N-Cbz-protected primary amines. Synlett 1999, 1999, 1301–1303. [Google Scholar] [CrossRef]
- Eschweiler, W. Ersatz von an stickstoff gebundenen wasserstoffatomen durch die methylgruppe mit hülfe von formaldehyd. Ber. Dtsch. Chem. Ges. 1905, 38, 880–882. [Google Scholar] [CrossRef]
- Clarke, H.; Gillespie, H.; Weisshaus, S. The action of formaldehyde on amines and amino acids1. J. Am. Chem. Soc. 1933, 55, 4571–4587. [Google Scholar] [CrossRef]
- Achenbach, H.; Schaller, E. Alkaloide in Tabernaemontana-Arten, VII. Über einige Bisindolalkaloide aus Tabernaemontana accedens. Chem. Ber. 1976, 109, 3527–3536. [Google Scholar] [CrossRef]
- Azoug, M.; Loukaci, A.; Richard, B.; Nuzillard, J.-M.; Moreti, C.; Zèches-Hanrot, M.; Le Men-Olivier, L. Alkaloids from stem bark and leaves of Peschiera buchtieni. Phytochemistry 1995, 39, 1223–1228. [Google Scholar] [CrossRef]
- Yang, J.; Sarma, P.; Cook, J.M. Progress toward the enantiospecific total synthesis of accedinisine and N′-demethylaccedinisine. In Proceedings of the 235th American Chemical Society National Meeting, New Orleans, LA, USA, 6–10 April 2008; p. ORGN 648.
- Rallapalli, S.K.; Cook, J.M. Progress toward the total synthesis of the sarpagine related alkaloids amervolfine and ervincidine. In Proceedings of the 237th American Chemical Society National Meeting, Salt Lake City, UT, USA, 22–26 March 2009; p. ORGN 517.
- Edwankar, C.R.; Edwankar, R.V.; Namjoshi, O.A.; Rallapalli, S.K.; Yang, J.; Cook, J.M. Recent progress in the total synthesis of indole alkaloids. Curr. Opin. Drug Discov. Devel. 2009, 12, 752–771. [Google Scholar] [PubMed]
- Edwankar, C.R.; Edwankar, R.V.; Rallapalli, S.; Cook, J.M. General approach to the total synthesis of macroline-related sarpagine and ajmaline alkaloids. Nat. Prod. Commun. 2008, 3, 1839–1870. [Google Scholar]
- Yin, W.; Ma, J.; Rivas, F.M.; Cook, J.M. First enantiospecific total synthesis of the important biogenetic intermediates, (+)-polyneuridine and (+)-polyneuridine aldehyde, as well as 16-epi-vellosimine and macusine A. Org. Lett. 2007, 9, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cook, J.M. General approach for the synthesis of sarpagine/ajmaline indole alkaloids. stereospecific total synthesis of the sarpagine alkaloid (+)-vellosimine. Org. Lett. 2000, 2, 2057–2059. [Google Scholar] [CrossRef] [PubMed]
- Warmuth, R.; Munsch, T.E.; Stalker, R.A.; Li, B.; Beatty, A. Enantioselective synthesis of benzocyclic α, α-dialkyl-amino acids: New insight into the solvent dependent stereoselectivity of the TMSCN addition to phenylglycinol derived imines. Tetrahedron 2001, 57, 6383–6397. [Google Scholar] [CrossRef]
- Wender, P.A.; Schaus, J.M.; White, A.W. General methodology for cis-hydroisoquinoline synthesis: Synthesis of reserpine. J. Am. Chem. Soc. 1980, 102, 6157–6159. [Google Scholar] [CrossRef]
- Bonjoch, J.; Fernàndez, J.-C.; Valls, N. Total syntheses of (±)-deethylibophyllidine using a crisscross annulation: Ring cleavage of octahydroindolo[2,3-a]quinolizines followed by tandem cyclizations of octahydroazecino[5,4-b]indoles. J. Org. Chem. 1998, 63, 7338–7347. [Google Scholar] [CrossRef] [PubMed]
- Schill, G.; Löwer, H.; Priester, C.U.; Windhövel, U.F.; Fritz, H. Eine neue synthese von vinblastin-derivaten II: Synthesekonzept und modelluntersuchungen. Tetrahedron 1987, 43, 3729–3745. [Google Scholar] [CrossRef]
- Schill, G.; Priester, C.U.; Windhovel, U.F.; Fritz, H. Eine neue synthese von vinblastin-derivaten v. konzept und untersuchungen zur synthese von 20′-desethyl-20′-desoxy-c′-homovinblastin—octahydro-3H-azecino[5,4-b]inidol-derivate. Tetrahedron 1990, 46, 1211–1220. [Google Scholar] [CrossRef]
- Calverley, M.; Harley-Mason, J.; Quarrie, S.; Edwards, P. On the stereochemistry of the solvolytic c/d ring cleavage of the 1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizine system. Tetrahedron 1981, 37, 1547–1556. [Google Scholar] [CrossRef]
- Liu, C.T.; Sun, S.C.; Yu, Q.S. Synthesis and photooxidation of the condensation products of tryptamine and catechol derivatives. An approach to the synthesis of a probable precursor of koumine. J. Org. Chem. 1983, 48, 44–47. [Google Scholar] [CrossRef]
- Takayama, H.; Masubuchi, K.; Kitajima, M.; Aimi, N.; Sakai, S.-I. A biomimetic construction of humantenine skeleton. Tetrahedron 1989, 45, 1327–1336. [Google Scholar] [CrossRef]
- Takayama, H.; Tominaga, Y.; Kitajima, M.; Aimi, N.; Sakai, S.-I. First synthesis of the novel Gelsemium alkaloids, gelselegine, gelsenicine, and gelsedine using a biomimetic approach. J. Org. Chem. 1994, 59, 4381–4385. [Google Scholar] [CrossRef]
- Banks, B.; Calverley, M.; Edwards, P.; Harley-Mason, J. A new synthesis of indolo[2,3-α]quinolizidine derivatives: A formal total synthesis of (±)-geissoschizine. Tetrahedron Lett. 1981, 22, 1631–1634. [Google Scholar] [CrossRef]
- Weisbach, J.A.; Raffauf, R.F.; Ribeiro, O.; Macko, E.; Douglas, B. Problems in chemotaxonomy I. Alkaloids of Peschiera affinis. J. Pharm. Sci. 1963, 52, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Clivio, P.; Richard, B.; Deverre, J.-R.; Sevenet, T.; Zeches, M.; Le Men-Oliver, L. Alkaloids from leaves and root bark ofErvatamia hirta. Phytochemistry 1991, 30, 3785–3792. [Google Scholar] [CrossRef]
- Liu, X.; Wang, T.; Xu, Q.; Ma, C.; Cook, J.M. Enantiospecific total synthesis of the enantiomer of the indole alkaloid affinisine. Tetrahedron Lett. 2000, 41, 6299–6303. [Google Scholar] [CrossRef]
- Buchi, G.; Manning, R.; Monti, S. Voacamine and voacorine. J. Am. Chem. Soc. 1964, 86, 4631–4641. [Google Scholar] [CrossRef]
- Buchi, G.; Manning, R.; Monti, S. Voacamine. J. Am. Chem. Soc. 1963, 85, 1893–1894. [Google Scholar] [CrossRef]
- Sharp, T.M. 265. The alkaloids of Alstonia barks. Part II. A. macrophylla, wall., A. somersetensis, FM Bailey, A. verticillosa, F. Muell., A. villosa, blum. J. Chem. Soc. 1934, 1227–1232. [Google Scholar] [CrossRef]
- Kishi, T.; Hesse, M.; Vetter, W.; Gemenden, C.; Taylor, W.; Schmid, H. Macralstonin. Helv. Chim. Acta 1966, 49, 946–964. [Google Scholar] [CrossRef]
- Lim, S.-H.; Low, Y.-Y.; Tan, S.-J.; Lim, K.-H.; Thomas, N.F.; Kam, T.-S. Perhentidines A–C: Macroline-macroline bisindoles from Alstonia and the absolute configuration of perhentinine and macralstonine. J. Nat. Prod. 2012, 75, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Changwichit, K.; Khorana, N.; Suwanborirux, K.; Waranuch, N.; Limpeanchob, N.; Wisuitiprot, W.; Suphrom, N.; Ingkaninan, K. Bisindole alkaloids and secoiridoids from Alstonia macrophylla Wall. ex G. Don. Fitoterapia 2011, 82, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Ghedira, K.; Zeches-Hanrot, M.; Richard, B.; Massiot, G.; Le Men-Olivier, L.; Sevenet, T.; Goh, S. Alkaloids of Alstonia angustifolia. Phytochemistry 1988, 27, 3955–3962. [Google Scholar] [CrossRef]
- Cook, J.; Le Quesne, P. Macralstonine from Alstonia muelleriana. Phytochemistry 1971, 10, 437–439. [Google Scholar] [CrossRef]
- Hart, N.; Johns, S.; Lamberton, J. Tertiary alkaloids of Alstonia spectabilis and Alstonia glabriflora (Apocynaceae). Aust. J. Chem. 1972, 25, 2739–2741. [Google Scholar] [CrossRef]
- Schmid, H. 4. Internationales Symposium Biochemie und Physiologie der Alkaloide (1969); Akademie Verlag: Berlin, Germany, 1972; p. 348. [Google Scholar]
- Burke, D.E.; DeMarkey, C.A.; Le Quesne, P.; Cook, J.M. Biomimetic synthesis of the bis-indole alkaloid macralstonine. J. Chem. Soc. Chem. Commun. 1972, 1346–1347. [Google Scholar] [CrossRef]
- Liu, X. The Enantiospecific Stereospecific Total Synthesis of the Enantiomers of the Indole Alkaloids Na-methylvellosimine, Affinisine and Macroline as Well as the Total Synthesis of the Indole Alkaloids Trinervine, Alstophylline and the Antimalarial Bisindole Macralstonine. Ph.D. Thesis, University of Wisconsin-Milwaukee, Milwaukee, WI, USA, 2002. [Google Scholar]
- Allen, M.S.; Hamaker, L.K.; La Loggia, A.J.; Cook, J.M. Entry into 6-methoxy-D(+)-tryptophans. Stereospecific synthesis of 1-benzenesulfonyl-6-methoxy-D(+)-tryptophan ethyl ester. Synth. Commun. 1992, 22, 2077–2102. [Google Scholar] [CrossRef]
- Ma, C.; Liu, X.; Li, X.; Flippen-Anderson, J.; Yu, S.; Cook, J.M. Efficient asymmetric synthesis of biologically important tryptophan analogues via a palladium-mediated heteroannulation reaction. J. Org. Chem. 2001, 66, 4525–4542. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Deschamp, J.R.; Cook, J.M. Regiospecific, enantiospecific total synthesis of the alkoxy-substituted indole bases, 16-epi-Na-methylgardneral, 11-methoxyaffinisine, and 11-methoxymacroline as well as the indole alkaloids alstophylline and macralstonine. Org. Lett. 2002, 4, 3339–3342. [Google Scholar] [CrossRef] [PubMed]
- Garnick, R.L.; Le Quesne, P.W. Biomimetic transformations among monomeric macroline-related indole alkaloids. J. Am. Chem. Soc. 1978, 100, 4213–4219. [Google Scholar] [CrossRef]
- Liao, X.; Zhou, H.; Wearing, X.Z.; Ma, J.; Cook, J.M. The first regiospecific, enantiospecific total synthesis of 6-oxoalstophylline and an improved total synthesis of alstonerine and alstophylline as well as the bisindole alkaloid macralstonine. Org. Lett. 2005, 7, 3501–3504. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.; Baran, P.S.; Zhong, Y.-L. Selective oxidation at carbon adjacent to aromatic systems with IBX. J. Am. Chem. Soc. 2001, 123, 3183–3185. [Google Scholar] [PubMed]
- Tsuji, J.; Nagashima, H.; Hori, K. A new preparative method for 1,3-dicarbonyl compounds by the regioselective oxidation of alpha, beta-unsaturated carbonyl compounds, catalyzed by PdCl2 using hydroperoxides as the reoxidant of Pd0. Chem. Lett. 1980, 257–260. [Google Scholar] [CrossRef]
- Zhao, S.; Liao, X.; Cook, J.M. Enantiospecific, stereospecific total synthesis of (+)-majvinine, (+)-10-methoxyaffinisine, and (+)-Na-methylsarpagine as well as the total synthesis of the alstonia bisindole macralstonidine. Org. Lett. 2002, 4, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Liao, X.; Wang, T.; Flippen-Anderson, J.; Cook, J.M. The enantiospecific, stereospecific total synthesis of the ring-A oxygenated sarpagine indole alkaloids (+)-majvinine, (+)-10-methoxyaffinisine, and (+)-Na-methylsarpagine, as well as the total synthesis of the Alstonia bisindole alkaloid macralstonidine. J. Org. Chem. 2003, 68, 6279–6295. [Google Scholar] [CrossRef] [PubMed]
- Heath-Brown, B.; Philpott, P. Studies in the indole series. Part III. The Japp–Klingemann reaction. J. Chem. Soc. 1965, 7185–7193. [Google Scholar] [CrossRef]
- Liao, X. The First Total Synthesis of the Indole Alkaloids, Macralstonidine, 6-oxoalstophylline, 10-methoxyvellosimine, Lochnerine, Sarpagine and an Improved Total Synthesis of Macralstonine and Macroline, as Well as the Formal Total Synthesis of Dispegatrine. Ph.D. Thesis, University of Wisconsin-Milwaukee, Milwaukee, WI, USA, 2007. [Google Scholar]
- Hesse, M.; Hürzeler, H.; Gemenden, C.; Joshi, B.; Taylor, W.; Schmid, H. Die Struktur des Alstonia-Alkaloides Villalstonin Vorläufige Mitteilung. Helv. Chim. Acta 1965, 48, 689–704. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Terrazas, C.; Acuña, U.M.; Ninh, T.N.; Chai, H.; de Blanco, E.J.C.; Soejarto, D.D.; Satoskar, A.R.; Kinghorn, A.D. Bioactive indole alkaloids isolated from Alstonia angustifolia. Phytochem. Lett. 2014, 10, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Nordman, C.; Kumra, S. The structure of villalstonine1. J. Am. Chem. Soc. 1965, 87, 2059–2060. [Google Scholar] [CrossRef]
- Kump, W.; Schmid, H. Über die alkaloide von Pleiocarpa mutica BENTH. Helv. Chim. Acta 1961, 44, 1503–1516. [Google Scholar] [CrossRef]
- Kam, T.-S.; Subramaniam, G.; Chen, W. Alkaloids from Kopsia dasyrachis. Phytochemistry 1999, 51, 159–169. [Google Scholar] [CrossRef]
- Bartlett, M.; Sklar, R.; Smith, A.; Taylor, W. The alkaloids of Hunteria eburnea Pichon. III. 1 The tertiary bases. J. Org. Chem. 1963, 28, 2197–2199. [Google Scholar] [CrossRef]
- Hesse, M.; Philipsborn, W.; Schumann, D.; Spiteller, G.; Spiteller-Friedmann, M.; Taylor, W.; Schmid, H.; Karrer, P. Die strukturen von C-fluorocurin, C-mavacurin und pleiocarpamin. 57. Mitteilung über Curare-alkaloide. Helv. Chim. Acta 1964, 47, 878–911. [Google Scholar] [CrossRef]
- Mayerl, F.; Hesse, M. Macrocarpamin, ein neues bisindolalkaloid aus Alstonia macrophylla WALL. 167. Mittelung über organische naturstoffe. Helv. Chim. Acta 1978, 61, 337–351. [Google Scholar] [CrossRef]
- Said, I.M.; Din, L.B.; Yusoff, N.I.; Wright, C.W.; Cai, Y.; Phillipson, J.D. A new alkaloid from the roots of Alstonia angustifolia. J. Nat. Prod. 1992, 55, 1323–1324. [Google Scholar] [CrossRef]
- Khan, Z.M.; Hesse, M.; Schmid, H. Die Struktur des quartären Alkaloides Macrosalhin. Helv. Chim. Acta 1967, 50, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Gan, T.; Cook, J.M. General approach for the synthesis of macroline/sarpagine related indole alkaloids via the asymmetric Pictet-Spengler reaction: The enantiospecific synthesis of (−)-anhydromacrosalhine-methine. Tetrahedron Lett. 1996, 37, 5033–5036. [Google Scholar] [CrossRef]
- Gan, T.; Cook, J.M. Partial synthesis of the antiamoebic bisindole alkaloid (−)-macrocarpamine. Tetrahedron Lett. 1996, 37, 5037–5038. [Google Scholar] [CrossRef]
- Gan, T.; Cook, J.M. Enantiospecific total synthesis of (−)-anhydromacrosalhine-methine and partial synthesis of the antiamoebic bisindole alkaloid (−)-macrocarpamine. J. Org. Chem. 1998, 63, 1478–1483. [Google Scholar] [CrossRef]
- Takayama, H.; Phisalaphong, C.; Kitajima, M.; Aimi, N.; Sakai, S.-I. An efficient synthetic pathway to the macroline-type indole alkaloids, talcarpine and alstonerine from ajmaline. Tetrahedron 1991, 47, 1383–1392. [Google Scholar] [CrossRef]
- Wearing, X.Z. Enantiospecific Stereospecific Total Synthesis of the Oxindole Alkaloid (+)-alstonisine and Stereocontrolled Total Synthesis of (−)-11-methoxy-17-epivincamajine as Well as Studies Directed toward the Total Synthesis of Nb-demethylalstophylline Oxindole. Ph.D. Thesis, Univeristy of Wisconsin-Milwaukee, Milwaukee, WI, USA, 2004. [Google Scholar]
- Zhang, L.; Cook, J. General approach to the synthesis of macroline-related alkaloids. Stereospecific total synthesis of (−)-alstonerine. J. Am. Chem. Soc. 1990, 112, 4088–4090. [Google Scholar] [CrossRef]
- Lin, M.; Yang, B.-Q.; Yu, D.-Q. Studies on the quaternary alkaloids of Rauvolfia verticillata (lour.) Baill var. Hainanensis Tsiang. Acta Pharmacol. Sin. 1986, 21, 114–118. [Google Scholar]
- Lin, M.; Yu, D.-Q.; Liu, X.; Fu, F.; Zheng, Q.; He, C.; Bao, G.; Xu, C. Chemical studies on the quaternary alkaloids of Rauvolfia verticillata (Lour.) Baill. F. ruberocarpa HT Chang. mss. Acta Pharmacol. Sin. 1985, 20, 198–202. [Google Scholar]
- Arbain, D.; Dachriyanus, F.; Sargent, M.V.; Skelton, B.W.; White, A.H. Unusual indole alkaloids from Ophiorrhiza blumeana Korth. J. Chem. Soc. Perkin Trans. 1998, 1, 2537–2540. [Google Scholar] [CrossRef]
- Orazi, O.O.; Corral, R.A.; Stoichevich, M.E. Studies on plants: XI. Alkaloids of Aspidosperma spegazzinii. Can. J. Chem. 1966, 44, 1523–1529. [Google Scholar] [CrossRef]
- Madinaveitia, A.; Valencia, E.; Bermejo, J.; Gonzalez, A. Indole alkaloids from Rauwolfia sprucei. Biochem. Syst. Ecol. 1995, 23, 877. [Google Scholar] [CrossRef]
- Edwankar, C.R.; Edwankar, R.V.; Deschamps, J.R.; Cook, J.M. Nature-inspired stereospecific total synthesis of P-(+)-dispegatrine and four other monomeric sarpagine indole alkaloids. Angew. Chem. Int. Ed. 2012, 51, 11762–11765. [Google Scholar] [CrossRef] [PubMed]
- Edwankar, C.R.; Edwankar, R.V.; Namjoshi, O.A.; Liao, X.; Cook, J.M. Stereospecific approach to the synthesis of ring-A oxygenated sarpagine indole alkaloids. Total synthesis of the dimeric indole alkaloid P-(+)-dispegatrine and six other monomeric indole alkaloids. J. Org. Chem. 2013, 78, 6471–6487. [Google Scholar] [CrossRef] [PubMed]
- Tohma, H.; Morioka, H.; Takizawa, S.; Arisawa, M.; Kita, Y. Efficient oxidative biaryl coupling reaction of phenol ether derivatives using hypervalent iodine (III) reagents. Tetrahedron 2001, 57, 345–352. [Google Scholar] [CrossRef]
- Larock, R.C.; Yum, E.K. Synthesis of indoles via palladium-catalyzed heteroannulation of internal alkynes. J. Am. Chem. Soc. 1991, 113, 6689–6690. [Google Scholar] [CrossRef]
- Larock, R.; Yum, E.; Refvik, M. Synthesis of 2,3-disubstituted indoles via palladium-catalyzed annulation of internal alkynes. J. Org. Chem. 1998, 63, 7652–7662. [Google Scholar] [CrossRef]
- Schollkopf, U. Asymmetric syntheses of amino acids via metalated bis-lactim ethers of 2,5-diketopiperazines. Pure Appl. Chem. 1983, 55, 1799–1806. [Google Scholar] [CrossRef]
- Schöllkopf, U.; Groth, U.; Deng, C. Enantioselective syntheses of (R)-amino acids using l-valine as chiral agent. Angew. Chem. Int. Ed. 1981, 20, 798–799. [Google Scholar] [CrossRef]
- Ma, J.; Yin, W.; Zhou, H.; Cook, J.M. Total synthesis of the opioid agonistic indole alkaloid mitragynine and the first total syntheses of 9-methoxygeissoschizol and 9-methoxy-Nb-methylgeissoschizol. Org. Lett. 2007, 9, 3491–3494. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kojima, S.; Sakamoto, T. General and facile synthesis of indoles with oxygen-bearing substituents at the benzene moiety. J. Org. Chem. 1997, 62, 6507–6511. [Google Scholar] [CrossRef]
- Lizos, D.E.; Murphy, J.A. Concise synthesis of (±)-horsfiline and (±)-coerulescine by tandem cyclisation of iodoaryl alkenyl azides. Org. Biomol. Chem. 2003, 1, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Snieckus, V. Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics. Chem. Rev. 1990, 90, 879–933. [Google Scholar] [CrossRef]
- Zhou, H.; Liao, X.; Cook, J.M. Regiospecific, enantiospecific total synthesis of the 12-alkoxy-substituted indole alkaloids, (+)-12-methoxy-Na-methylvellosimine, (+)-12-methoxyaffinisine, and (−)-fuchsiaefoline. Org. Lett. 2004, 6, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Abramovitch, R.; Shapiro, D. 880. Tryptamines, carbolines, and related compounds. Part II. A convenient synthesis of tryptamines and β-carbolines. J. Chem. Soc. 1956, 4589–4592. [Google Scholar] [CrossRef]
- Robinson, B. The Fischer Indole Synthesis; John Wiley & Sons Ltd.: New York, NY, USA, 1982. [Google Scholar]
- Nelson, T.D.; Crouch, R.D. Cu, Ni, and Pd mediated homocoupling reactions in biaryl syntheses: The Ullmann reaction. Org. React. 2004. [Google Scholar] [CrossRef]
- Cox, E.D.; Cook, J.M. The Pictet-Spengler condensation: A new direction for an old reaction. Chem. Rev. 1995, 95, 1797–1842. [Google Scholar] [CrossRef]
- Klausen, R.S.; Jacobsen, E.N. Weak Brønsted acid–thiourea co-catalysis: Enantioselective, catalytic Protio-Pictet–Spengler reactions. Org. Lett. 2009, 11, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Stöckigt, J.; Antonchick, A.P.; Wu, F.; Waldmann, H. The Pictet–Spengler reaction in nature and in organic chemistry. Angew. Chem. Int. Ed. 2011, 50, 8538–8564. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Rallapalli, S.K.; Cook, J.M. The first enantiospecific total synthesis of the 3-oxygenated sarpagine indole alkaloids affinine and 16-epiaffinine, as well as vobasinediol and 16-epivobasinediol. Tetrahedron Lett. 2010, 51, 815–817. [Google Scholar] [CrossRef]
- Solé, D.; Urbaneja, X.; Bonjoch, J. Palladium-catalyzed intramolecular coupling of amino-tethered vinyl halides with ketones, esters, and nitriles using potassium phenoxide as the base. Adv. Synth. Catal. 2004, 346, 1646–1650. [Google Scholar] [CrossRef]
- Keller, P.A.; Yepuri, N.R.; Kelso, M.J.; Mariani, M.; Skelton, B.W.; White, A.H. Oxidative coupling of indoles using thallium (III) trifluoroacetate. Tetrahedron 2008, 64, 7787–7795. [Google Scholar] [CrossRef]
- McKillop, A.; Turrell, A.G.; Young, D.W.; Taylor, E.C. Thallium in organic synthesis. 58. Regiospecific intermolecular oxidative dehydrodimerization of aromatic compounds to biaryls using thallium (III) trifluoroacetate. J. Am. Chem. Soc. 1980, 102, 6504–6512. [Google Scholar] [CrossRef]
- Mckillop, A.; Taylor, E.C. Recent advances in organothallium chemistry. Adv. Organomet. Chem. 1973, 11, 147–206. [Google Scholar]
- Elson, I.H.; Kochi, J.K. Thallium (III) in one-electron oxidation of arenes by electron spin resonance. J. Am. Chem. Soc. 1973, 95, 5060–5062. [Google Scholar] [CrossRef]
- Lau, W.; Kochi, J. Kinetics and mechanism of aromatic thallation. Identification and proof of competing electrophilic and electron-transfer pathways. J. Am. Chem. Soc. 1984, 106, 7100–7112. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.T.; Tiruveedhula, V.V.N.P.B.; Cook, J.M. Synthesis of Bisindole Alkaloids from the Apocynaceae Which Contain a Macroline or Sarpagine Unit: A Review. Molecules 2016, 21, 1525. https://doi.org/10.3390/molecules21111525
Rahman MT, Tiruveedhula VVNPB, Cook JM. Synthesis of Bisindole Alkaloids from the Apocynaceae Which Contain a Macroline or Sarpagine Unit: A Review. Molecules. 2016; 21(11):1525. https://doi.org/10.3390/molecules21111525
Chicago/Turabian StyleRahman, Md Toufiqur, Veera V. N. Phani Babu Tiruveedhula, and James M. Cook. 2016. "Synthesis of Bisindole Alkaloids from the Apocynaceae Which Contain a Macroline or Sarpagine Unit: A Review" Molecules 21, no. 11: 1525. https://doi.org/10.3390/molecules21111525