
High-Resolution α-Glucosidase Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of Antidiabetic Compounds in Eremanthus crotonoides (Asteraceae)

 , and
, and 
Abstract
:
1. Introduction
2. Results and Discussion
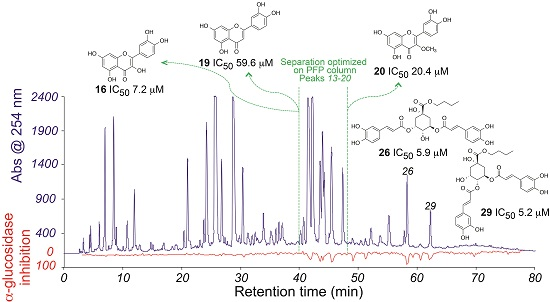
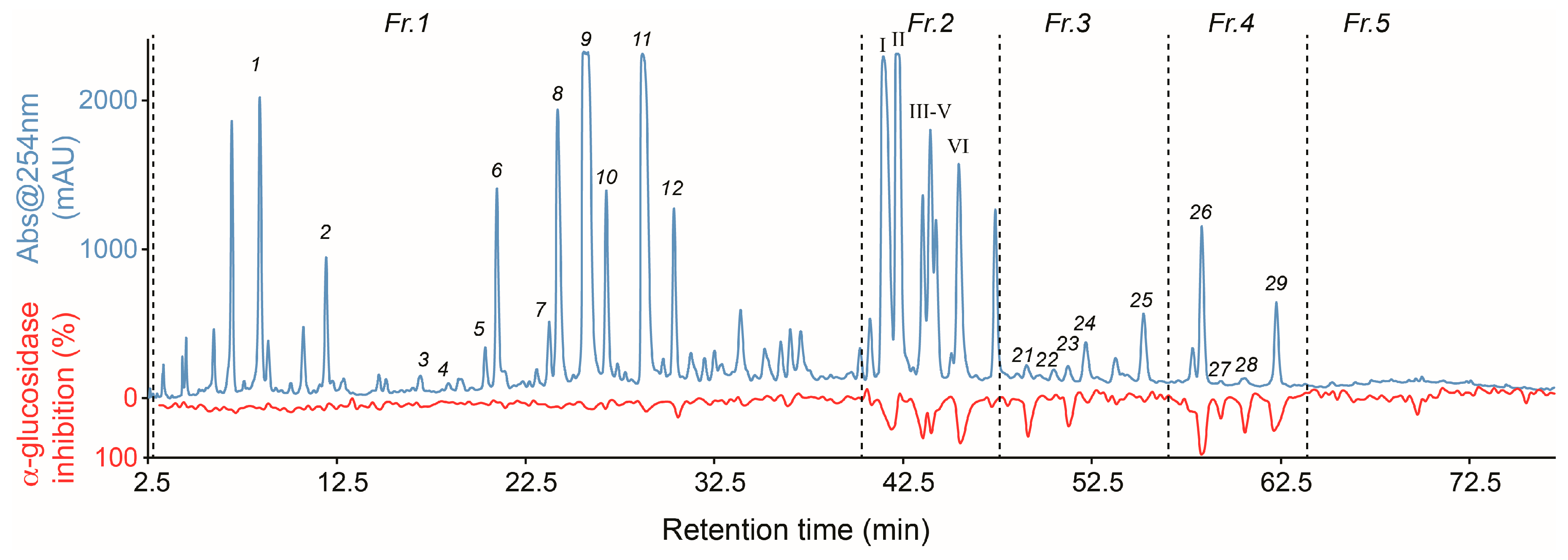
2.1. High-Resolution α-Glucosidase Inhibition Profiling of Extract
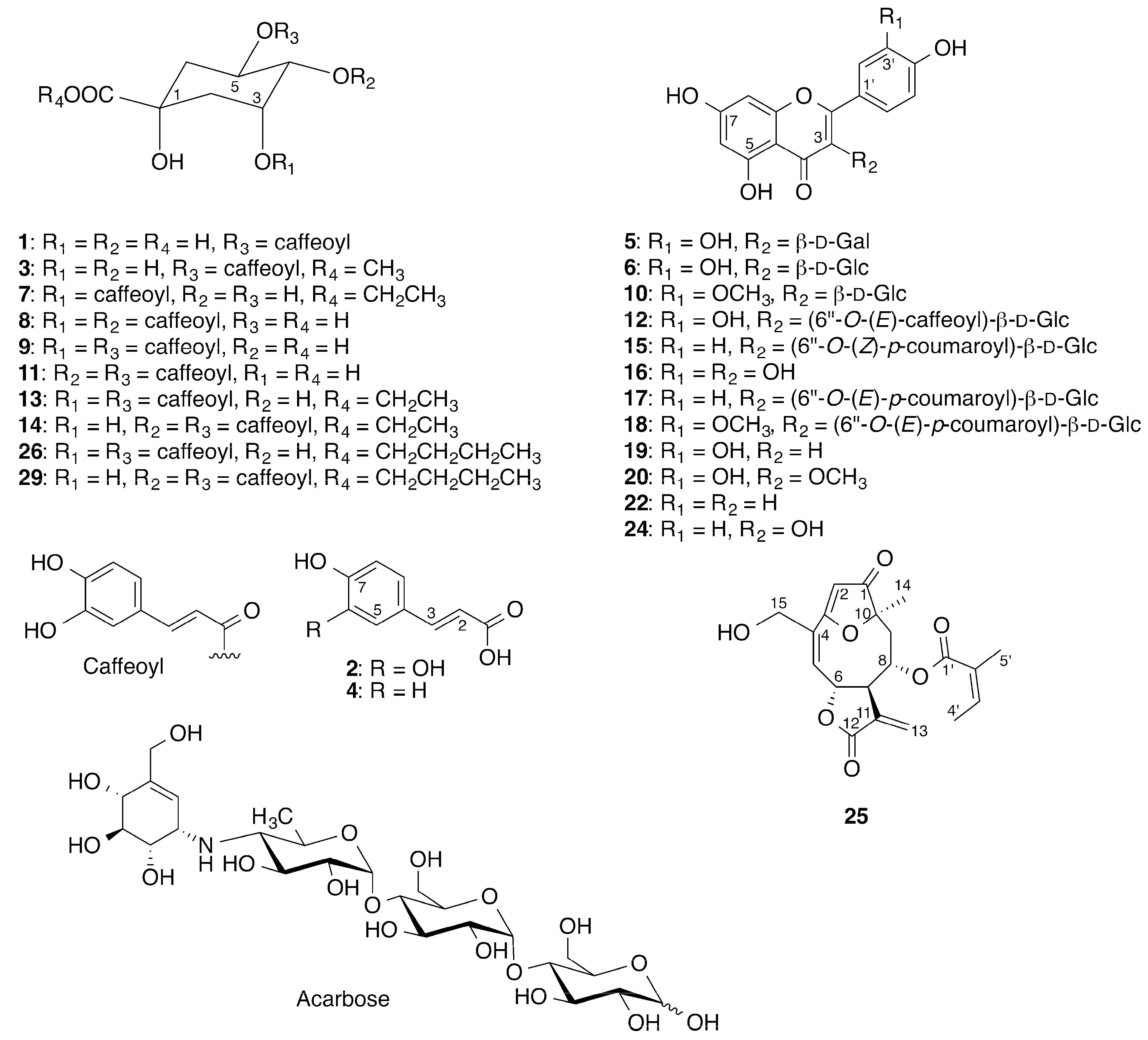
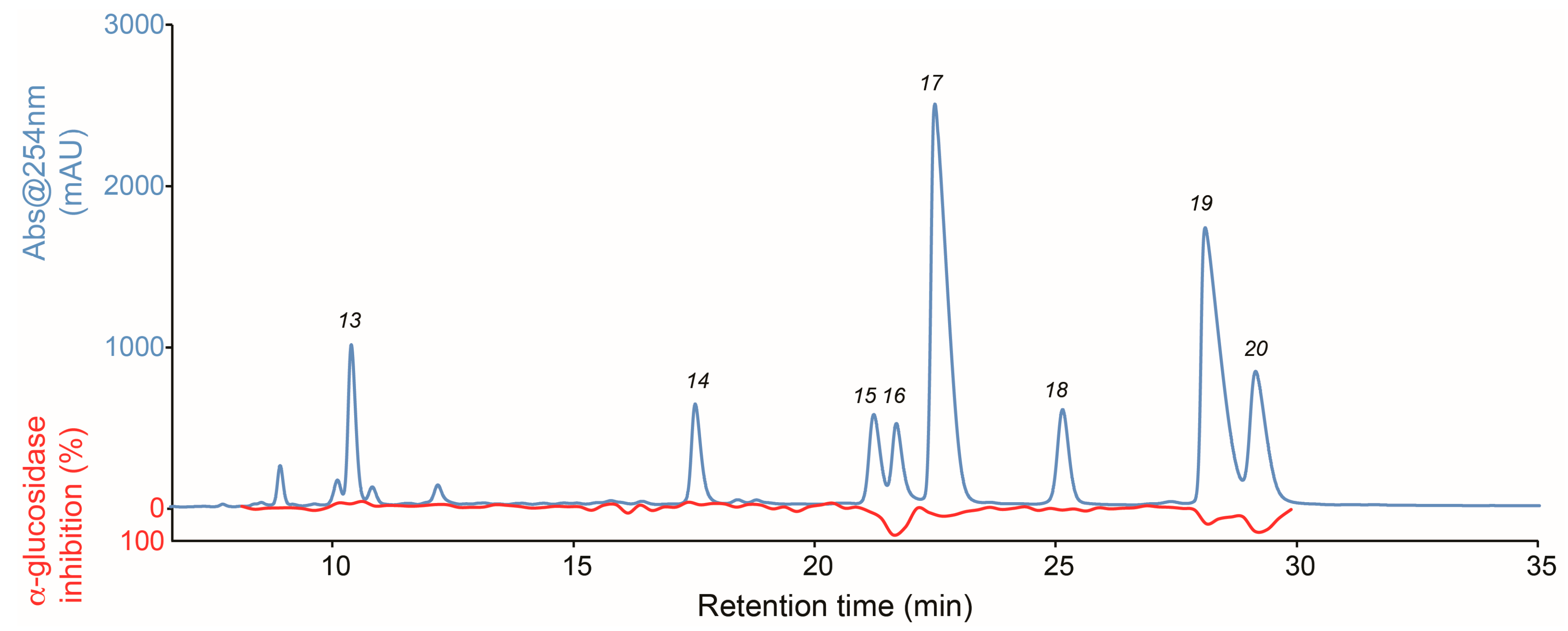
2.2. Identification of Inhibitory Constituents
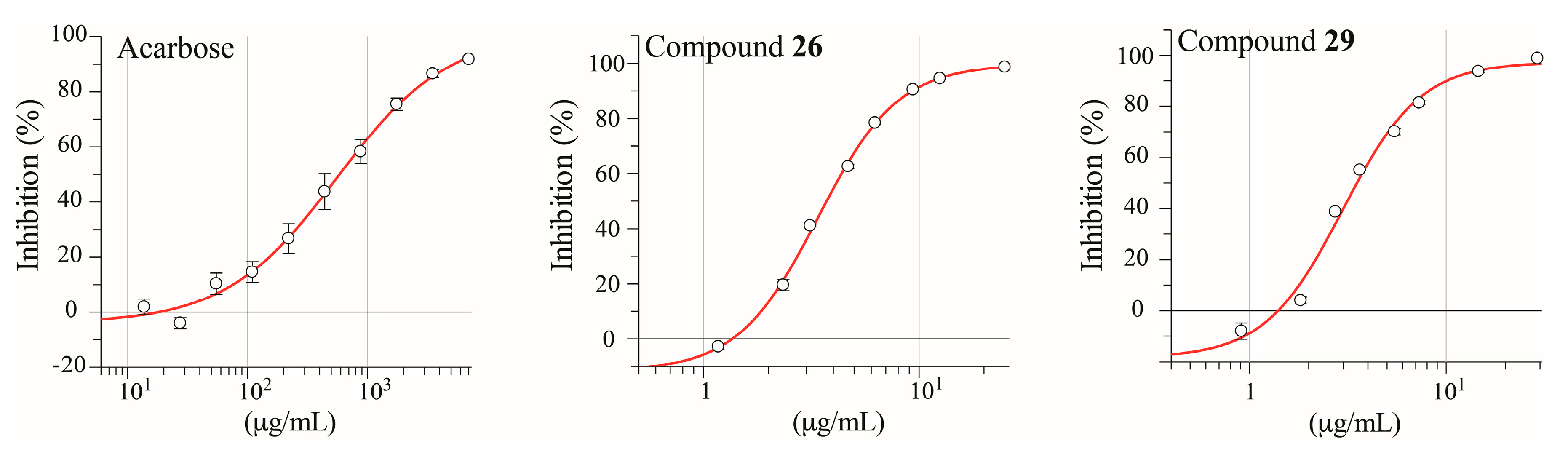
2.3. Isolation and Pharmacological Evaluation of the α-Glucosidase Inhibitors
3. Materials and Methods
3.1. Reagents
3.2. Plant Material and Sample Preparation
3.3. High-Resolution α-Glucosidase Biochromatogram
3.4. HPLC-HRMS-SPE-NMR Analysis
3.5. NMR Experiments
3.6. Isolation and Evaluation of the Bioactive Compounds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scully, T. Diabetes in numbers. Nature 2012, 485, S2–S3. [Google Scholar] [CrossRef] [PubMed]
- Flores-Bocanegra, L.; Pérez-Vásquez, A.; Torres-Piedra, M.; Bye, R.; Linares, E.; Mata, R. α-Glucosidase inhibitors from Vauquelinia corymbosa. Molecules 2015, 20, 15330–15342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, X.; Brown, J.; Vistisen, D.; Sicree, R.; Shaw, J. Global healthcare expenditure on diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Dai, H.; Pan, S.; Wang, H.; Hu, Y.; Ma, W. Isolation and characterization of an α-Glucosidase inhibitor from Musa spp. (Baxijiao) flowers. Molecules 2014, 19, 10563–10573. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. Postprandial hyperglycemia and diabetes complications: Is it time to treat? Diabetes 2005, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Ikegami, H.; Inoue, K.; Kawabata, Y.; Ogihara, T. Effect of two α-glucosidase inhibitors, voglibose and acarbose, on postprandial hyperglycemia correlates with subjective abdominal symptoms. Metabolism 2005, 54, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Krentz, A.J.; Bailey, C.J. Oral antidiabetic agents: Current role in type 2 diabetes mellitus. Drugs 2005, 65, 385–411. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.; Stærk, D.; Hansen, S.H.; Jaroszewski, J.W. HPLC-SPE-NMR hyphenation in natural products research: Optimization of analysis of Croton membranaceus extract. Magn. Reson. Chem. 2005, 43, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.S.; Lauridsen, M.B.; Dragsted, L.O.; Nielsen, J.; Staerk, D. Development of a bioassay-coupled HPLC-SPE-ttNMR platform for identification of α-glucosidase inhibitors in apple peel (Malus × domestica borkh.). Food Chem. 2012, 135, 1692–1699. [Google Scholar] [CrossRef] [PubMed]
- Wubshet, S.G.; Schmidt, J.S.; Wiese, S.; Staerk, D. High-resolution screening combined with HPLC-HRMS-SPE-NMR for identification of potential health-promoting constituents in sea aster and searocket—New Nordic food ingredients. J. Agric. Food Chem. 2013, 61, 8616–8623. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.S.; Nyberg, N.T.; Staerk, D. Assessment of constituents in Allium by multivariate data analysis, high-resolution α-glucosidase inhibition assay and HPLC-SPE-NMR. Food Chem. 2014, 161, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Kongstad, K.T.; Özdemir, C.; Barzak, A.; Wubshet, S.G.; Staerk, D. Combined use of high-resolution α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR for investigation of antidiabetic principles in crude plant extracts. J. Agric. Food Chem. 2015, 63, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Wubshet, S.G.; Moresco, H.H.; Tahtah, Y.; Brighente, I.M.C.; Staerk, D. High-resolution bioactivity profiling combined with HPLC–HRMS–SPE–NMR: α-Glucosidase inhibitors and acetylated ellagic acid rhamnosides from Myrcia palustris DC. (Myrtaceae). Phytochemistry 2015, 116, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kongstad, K.T.; Wiese, S.; Jäger, A.K.; Staerk, D. Edible seaweed as future functional food: Identification of α-glucosidase inhibitors by combined use of high-resolution α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR. Food Chem. 2016, 203, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kongstad, K.T.; Qinglei, S.; Nyberg, N.T.; Jäger, A.K.; Staerk, D. Dual high-resolution α-glucosidase and radical scavenging profiling combined with HPLC-HRMS-SPE-NMR for identification of minor and major constituents directly from the crude extract of Pueraria lobata. J. Nat. Prod. 2015, 78, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Tahtah, Y.; Kongstad, K.T.; Wubshet, S.G.; Nyberg, N.T.; Jønsson, L.H.; Jäger, A.K.; Qinglei, S.; Staerk, D. Triple aldose reductase/α-glucosidase/radical scavenging high-resolution profiling combined with high-performance liquid chromatography-High-resolution mass spectrometry-solid-phase extraction-nuclear magnetic resonance spectroscopy for identification of antidiabetic constituents in crude extract of Radix Scutellariae. J. Chromatogr. A 2015, 1408, 125–132. [Google Scholar] [PubMed]
- Wubshet, S.G.; Tahtah, Y.; Heskes, A.M.; Kongstad, K.T.; Pateraki, I.; Hamberger, B.; Møller, B.M.; Staerk, D. Identification of PTP1B and α-glucosidase inhibitory serrulatanes from Eremophila spp. by combined use of dual high-resolution PTP1B and α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR. J. Nat. Prod. 2016, in press. [Google Scholar]
- Okutan, L.; Kongstad, K.T.; Jäger, A.K.; Staerk, D. High-resolution α-amylase assay combined with high-performance liquid chromatography-solid-phase extraction-nuclear magnetic resonance spectroscopy for expedited identification of α-amylase inhibitors: Proof of concept and α-amylase inhibitor in cinnamon. J. Agric. Food Chem. 2014, 62, 11465–11471. [Google Scholar] [CrossRef] [PubMed]
- Kongstad, K.T.; Wubshet, S.G.; Johannesen, A.; Kjellerup, L.; Winther, A.-M.L.; Jäger, A.K.; Staerk, D. High-resolution screening combined with HPLC-HRMS-SPE-NMR for identification of fungal plasma membrane H+-ATPase inhibitors from plants. J. Agric. Food Chem. 2014, 62, 5595–5602. [Google Scholar] [CrossRef] [PubMed]
- Wiese, S.; Wubshet, S.G.; Nielsen, J.; Staerk, D. Coupling HPLC-SPE-NMR with a microplate-based high-resolution antioxidant assay for efficient analysis of antioxidants in food—Validation and proof-of-concept study with caper buds. Food Chem. 2013, 141, 4010–4018. [Google Scholar] [CrossRef] [PubMed]
- Grosso, C.; Jäger, A.K.; Staerk, D. Coupling of a high-resolution monoamine oxidase-A inhibitor assay and HPLC-SPE-NMR for advanced bioactivity-profiling of plant extracts. Phytochem. Anal. 2013, 24, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Sidhy, M.C.; Sharma, T. A database of antidiabetic plant species of Family Asteraceae, Euphorbiaceae, Fabaceae, Lamiaceae and Moraceae. Int. J. Herbal Med. 2013, 2, 187–199, 2321–2187. [Google Scholar]
- Lobo, J.F.R.; Castro, E.S.; Gouvea, D.R.; Fernandes, C.P.; Almeida, F.B.; Amorim, L.M.F.; Burth, P.; Rocha, L.; Santos, M.G.; Harmerski, L.; et al. Antiproliferative activity of Eremanthus crotonoides extracts and centratherin demonstrated in brain tumor cell lines. Rev. Bras. Farmacogn. 2012, 22, 1295–1300. [Google Scholar] [CrossRef]
- Bohlmann, F.; Zdero, C.; King, R.M.; Robinson, H. Sesquiterpene lactones from Eremanthus species. Phytochemistry 1980, 19, 2663–2668. [Google Scholar] [CrossRef]
- Vichnewski, W.; Takahashi, A.M.; Nasi, A.M.T.; Rodrigues, D.C.; Gonçalves, G.; Dias, D.A.; Lopes, J.N.C.; Goedken, V.L.; Gutiérrez, A.B.; Herz, W. Sesquiterpene lactones and other constituents from Eremanthus seidelii, E. goyazensis and Vanillosmopsis erythropappa. Phytochemistry 1989, 28, 1441–1151. [Google Scholar] [CrossRef]
- Gouvea, D.R.; Gobbo-Neto, L.; Sakamoto, H.T.; Lopes, N.P.; Lopes, J.L.C.; Meloni, F.; Amaral, J.G. Seasonal variation of the major secondary metabolites present in the extract of Eremanthus mattogrossensis Less (Asteracea: Vernonieae) leaves. Quim. Nova 2012, 35, 2139–2145. [Google Scholar] [CrossRef]
- Silvério, M.S.; Del-Vechio-Vieira, G.; Pinto, M.A.O.; Alves, M.S.; Souza, O.V. Chemical composition and biological activities of essential oils of Eremanthus erythropappus (DC) McLeisch (Asteraceae). Molecules 2013, 18, 9785–9796. [Google Scholar] [CrossRef] [PubMed]
- Bohlmann, F.; Singh, P.; Zdero, C.; Ruhe, A.; King, R.M.; Robinson, H. Furanoheliangolides from two Eremanthus species and from Chresta sphaerocephala. Phytochemistry 1982, 21, 1669–1673. [Google Scholar] [CrossRef]
- Lee, E.J.; Kim, J.S.; Kim, H.P.; Lee, J.H.; Kang, S.S. Phenolic constituents from the flower buds of Lonicera japonica and their 5-lipoxygenase inhibitory activities. Food Chem. 2010, 120, 134–139. [Google Scholar] [CrossRef]
- Fuchs, C.; Spiteller, G. Rapid and easy identification of isomers of coumaroyl- and caffeoyl-d-quinic acid by gas chromatography mass spectrometry. J. Mass. Spectrom. 1996, 31, 602–608. [Google Scholar] [CrossRef]
- Chen, J.; Mangelinckx, S.; Ma, L.; Wang, Z.; Li, W.; de Kimpe, N. Caffeoylquinic acid derivatives isolated from the aerial parts of Gynura divaricata and their yeast alpha-glucosidase and PTP1B inhibitory activity. Fitoterapia 2014, 99, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.S.; Kim, J.A.; Qian, Z.J.; Kim, Y.A.; Lee, J.I.; Kim, S.K.; Nam, T.J.; Seo, Y. Protective effect of isorhamnetin 3-O-β-d-glucopyranoside from Salicornia herbacea against oxidation-induced cell damage. Food Chem. Toxicol. 2009, 47, 1914–1920. [Google Scholar] [CrossRef] [PubMed]
- Calzada, F.; Cedillo-Rivera, R.; Mata, R. Antiprotozoal activity of the constituents of Conyza filaginoides. J. Nat. Prod. 2001, 64, 671–673. [Google Scholar] [CrossRef] [PubMed]
- Rashed, K.; Sahuc, M.E.; Deloison, G.; Calland, N.; Brodin, P.; Rouille, Y.; Seron, K. Potent antiviral activity of Solanum rantonnetii and the isolated compounds against hepatitis C virus in vitro. J. Funct. Foods 2014, 11, 185–191. [Google Scholar] [CrossRef]
- Soares, A.C.F.; Silva, A.N.; Matos, P.M.; da Silva, E.H.; Heleno, V.C.G.; Lopes, N.P.; Lopes, J.L.C.; Sass, D.C. Complete 1H and 13C NMR structural assignments for a group of four goyazensolide-type furanoheliangolides. Quim. Nova 2012, 35, 2205–2207. [Google Scholar] [CrossRef]
- Wei, X.Y.; Huang, H.J.; Wu, P.; Cao, H.L.; Ye, W.H. Phenolic constituents from Mikania micrantha. Biochem. Syst. Ecol. 2004, 32, 1091–1096. [Google Scholar] [CrossRef]
- Yi, B.; Hu, L.; Mei, W.; Zhou, K.; Wang, H.; Luo, Y.; Wei, X.; Dai, H. Antioxidant phenolic compounds of cassava (Manihot esculenta) from Hainan. Molecules 2011, 16, 10157–10167. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Shrestha, S.; Seo, W.D.; Lee, M.H.; Jeong, T.S.; Cho, J.H.; Song, Y.C.; Kang, H.W.; Rho, Y.D.; Baek, N.I. Structural and quantitative analysis of antioxidant and low-density lipoprotein-antioxidant flavonoids from the grains of sugary rice. J. Med. Food 2012, 15, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hamburger, M.; Gueho, J.; Hostettmann, K. Cyclohexanecarboxylic-acid derivatives from Psiadia trinervia. Helv. Chim. Acta 1992, 75, 269–275. [Google Scholar] [CrossRef]
- Timmers, M.; Urban, S. On-line (HPLC-NMR) and off-line phytochemical profiling of the australian plant, Lasiopetalum macrophyllum. Nat. Prod. Commun. 2012, 7, 551–560. [Google Scholar] [PubMed]
- Jou, S.J.; Chen, C.H.; Guh, J.H.; Lee, C.N.; Lee, S.S. Flavonol glycosides and cytotoxic triterpenoids from Alphitonia philippinensis. J. Chin. Chem. Soc. 2004, 51, 827–834. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, G.; Pan, J.; Wang, Y. α-Glucosidase inhibition by luteolin: Kinetics, interaction and molecular docking. Int. J. Biol. Macromol. 2014, 64, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Zhang, W.; Feng, F.; Zhang, Y.; Kang, W. α-Glucosidase inhibitors isolated from medicinal plants. Food Sci. Hum. Wellness 2014, 3, 136–174. [Google Scholar] [CrossRef]
- Enomoto, S.; Okada, Y.; Guvenc, A.; Erdurak, C.S.; Coskun, M.; Okuyama, T. Inhibitory effect of traditional Turkish folk medicines on aldose reductase (AR) and hematological activity, and on AR inhibitory activity of quercetin-3-O-methyl ether isolated from Cistus laurifolius L. Biol. Pharm. Bull. 2004, 27, 1140–1143. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Kim, M.H.; Choi, Y.Y.; Lee, H.; Hong, J.; Yang, W.M. Effects of Lonicera japonica Thunb. on type 2 diabetes via PPAR-γ activation in rats. Phytother. Res. 2015, 29, 1616–1621. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.Y.; Mei, S.X.; Jiang, B.; Zhou, H.; Sun, H.D. Constituents from Lonicera japonica. Fitoterapia 2000, 71, 713–715. [Google Scholar] [CrossRef]
- Borah, S.; Das, A.K.; Saikia, D.; Borah, J. A note on the use of ethnomedicine in treatment of diabetes by Mishing communities in Assam, India. Ethnobot. Leaflets 2009, 13, 1348–1352. [Google Scholar]
- Sample Availability: Not available.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | IC50 (μM) a |
|---|---|
| Acarbose b | 859.79 ± 0.09 |
| 16 | 7.19 ± 0.06 |
| 17 | ND c,d |
| 19 | 59.64 ± 1.32 |
| 20 | 20.36 ± 1.30 |
| 26 | 5.93 ± 0.12 |
| 29 | 5.20 ± 0.30 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, E.L.e.; Lobo, J.F.R.; Vinther, J.M.; Borges, R.M.; Staerk, D. High-Resolution α-Glucosidase Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of Antidiabetic Compounds in Eremanthus crotonoides (Asteraceae). Molecules 2016, 21, 782. https://doi.org/10.3390/molecules21060782
Silva ELe, Lobo JFR, Vinther JM, Borges RM, Staerk D. High-Resolution α-Glucosidase Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of Antidiabetic Compounds in Eremanthus crotonoides (Asteraceae). Molecules. 2016; 21(6):782. https://doi.org/10.3390/molecules21060782
Chicago/Turabian StyleSilva, Eder Lana e, Jonathas Felipe Revoredo Lobo, Joachim Møllesøe Vinther, Ricardo Moreira Borges, and Dan Staerk. 2016. "High-Resolution α-Glucosidase Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of Antidiabetic Compounds in Eremanthus crotonoides (Asteraceae)" Molecules 21, no. 6: 782. https://doi.org/10.3390/molecules21060782
APA StyleSilva, E. L. e., Lobo, J. F. R., Vinther, J. M., Borges, R. M., & Staerk, D. (2016). High-Resolution α-Glucosidase Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of Antidiabetic Compounds in Eremanthus crotonoides (Asteraceae). Molecules, 21(6), 782. https://doi.org/10.3390/molecules21060782