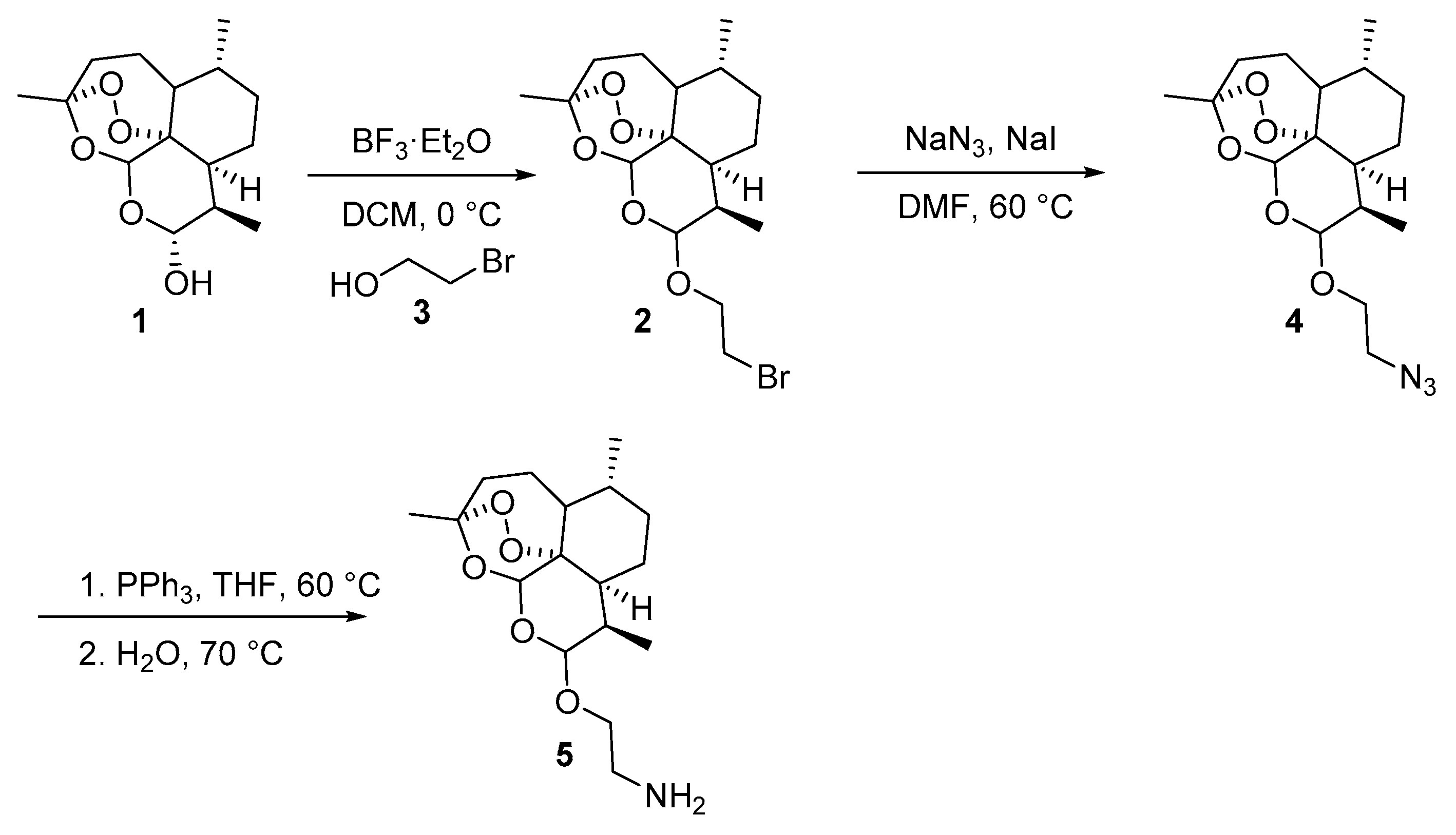
3.3. Characterization of the Products
1-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-Trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)ethyl)-4,5-dihydro-1H-naphtho[1,2-d][1,2,3]triazole (8a): 5 (60 mg, 0.183 mmol), α-tetralone (26.8 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 24 h. The product was purified by flash column chromatography (first DCM, followed by EtOAc/heptane = 3:2) to afford 8a (42 mg, 48%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 7.94 (d, J = 7.4 Hz, 1H), 7.30 (td, J = 7.4, 1.8 Hz, 1H), 7.24–7.17 (m, 2H), 5.38 (s, 1H), 4.62 (d, J = 8.0 Hz, 1H), 4.60–4.54 (m, 1H), 4.53–4.44 (m, 1H), 4.23 (dt, J = 10.6, 4.3 Hz, 1H), 3.91 (ddd, J = 10.7, 8.6, 4.0 Hz, 1H), 3.09–2.91 (m, 4H), 1.87–1.81 (m, 1H), 1.71–1.62 (m, 3H), 1.60–1.49 (m, 3H), 1.47 (s, 3H), 1.44–1.35 (m, 1H), 1.29 (dt, J = 14.4, 3.2 Hz, 1H), 1.22–1.09 (m, 2H), 1.06–0.95 (m, 1H), 0.88 (dd, J = 6.3, 3.6 Hz, 6H). 13C-NMR (101 MHz, CDCl3) δ 143.34, 134.03, 133.69, 129.04, 128.19, 127.37, 127.35, 122.16, 107.27, 101.43, 97.25, 82.73, 68.54, 48.50, 45.24, 44.11, 39.98, 35.27, 34.55, 34.52, 32.81, 28.81, 23.87, 22.17, 19.41, 19.34, 18.76.
3-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-Trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)ethyl)-4,5-dihydro-3H-naphtho[1,2-d][1,2,3]triazole (8b): 5 (60 mg, 0.183 mmol), β-tetralone (26.8 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 16 h. The product was purified by flash column chromatography (first DCM, then EtOAc/heptane = 3:2) to afford 8b (62 mg, 70%) as an off-white solid: m.p. 104–105 °C; 1H-NMR (400 MHz, CDCl3) δ 7.94 (d, J = 7.4 Hz, 1H), 7.30 (td, J = 7.4, 1.8 Hz, 1H), 7.24–7.17 (m, 2H), 5.38 (s, 1H), 4.62 (d, J = 8.0 Hz, 1H), 4.60–4.54 (m, 1H), 4.53–4.44 (m, 1H), 4.23 (dt, J = 10.6, 4.3 Hz, 1H), 3.91 (ddd, J = 10.7, 8.6, 4.0 Hz, 1H), 3.09–2.91 (m, 4H), 1.87–1.81 (m, 1H), 1.71–1.62 (m, 3H), 1.60–1.49 (m, 3H), 1.47 (s, 3H), 1.44–1.35 (m, 1H), 1.29 (dt, J = 14.4, 3.2 Hz, 1H), 1.22–1.09 (m, 2H), 1.06–0.95 (m, 1H), 0.88 (dd, J = 6.3, 3.6 Hz, 6H). 13C-NMR (101 MHz, CDCl3) δ 143.34, 134.03, 133.69, 129.04, 128.19, 127.37, 127.35, 122.16, 107.27, 101.43, 97.25, 82.73, 68.54, 48.50, 45.24, 44.11, 39.98, 35.27, 34.55, 34.52, 32.81, 28.81, 23.87, 22.17, 19.41, 19.34, 18.76.
7,8-Dimethoxy-3-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)ethyl)-4,5-dihydro-3H-naphtho[1,2-d][1,2,3]triazole (8c): 5 (60 mg, 0.183 mmol), 6,7-dimethoxy-3,4-dihydronaphthalen-2(1H)-one (37.8 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 16 h. The product was purified by flash column chromatography (first DCM, followed by EtOAc/heptane = 3:2) to give 8c (67 mg, 67%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 7.49 (s, 1H), 6.76 (s, 1H), 5.38 (s, 1H), 4.62 (d, J = 7.9 Hz, 1H), 4.59–4.53 (m, 1H), 4.51–4.44 (m, 1H), 4.26–4.18 (m, 1H), 3.95 (s, 3H), 3.89 (s, 4H), 3.05–2.84 (m, 4H), 1.88–1.79 (m, 1H), 1.71–1.62 (m, 3H), 1.60–1.50 (m, 3H), 1.47 (s, 3H), 1.44–1.35 (m, 1H), 1.33–1.23 (m, 2H), 1.21–1.10 (m, 1H), 1.08–0.95 (m, 1H), 0.91–0.85 (m, 6H). 13C-NMR (101 MHz, CDCl3) δ 148.40, 148.20, 143.46, 133.09, 125.92, 121.85, 111.89, 107.25, 105.67, 101.41, 97.24, 82.71, 68.50, 56.26, 56.18, 48.52, 45.23, 44.12, 39.97, 35.26, 34.55, 34.51, 32.80, 28.45, 23.85, 22.16, 19.62, 19.32, 18.74.
8-Nitro-3-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)ethyl)-4,5-dihydro-3H-naphtho[1,2-d][1,2,3]triazole (8d): 5 (60 mg, 0.183 mmol), 7-nitro-3,4-dihydronaphthalen-2(1H)-one (35 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 16 h. Flash column chromatography (first DCM, followed by EtOAc/heptane = 3:2) afforded 8d (70.4 mg, 73%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 8.78 (d, J = 2.2 Hz, 1H), 8.12 (dd, J = 8.3, 2.3 Hz, 1H), 7.48 (d, J = 8.4 Hz, 1H), 5.38 (s, 1H), 4.91–4.87 (m, 2H), 4.61 (d, J = 7.9 Hz, 1H), 4.45–4.39 (m, 1H), 4.15–4.01 (m, 1H), 3.15–3.02 (m, 4H), 1.86–1.79 (m, 1H), 1.69–1.61 (m, 3H), 1.55–1.48 (m, 3H), 1.44 (s, 3H), 1.36–1.32 (m, 1H), 1.32–1.27 (m, 2H), 1.21–1.11 (m, 1H), 1.02–0.92 (m, 1H), 0.88–0.76 (m, 6H). 13C-NMR (101 MHz, CDCl3) δ 147.43, 145.81, 144.66, 131.02, 129.79, 126.95, 123.16, 118.31, 107.19, 101.28, 97.24, 82.76, 77.16, 68.37, 50.36, 45.28, 44.08, 39.56, 35.21, 34.58, 34.51, 32.61, 30.78, 29.85, 23.85, 22.17, 20.38, 19.18, 18.80.
8-Methoxy-3-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)ethyl)-4,5-dihydro-3H-naphtho[1,2-d][1,2,3]triazole (8e): 5 (60 mg, 0.183 mmol), 7-methoxy-3,4-dihydronaphthalen-2(1H)-one (32.3 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 16 h. The product was purified by flash column chromatography (first DCM, followed by EtOAc/heptane = 3:2) to afford 8e (62 mg, 66%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 7.51 (d, J = 2.7 Hz, 1H), 7.13 (d, J = 8.3 Hz, 1H), 6.75 (dd, J = 8.3, 2.7 Hz, 1H), 5.37 (s, 1H), 4.62 (d, J = 8.0 Hz, 1H), 4.60–4.54 (m, 1H), 4.53–4.45 (m, 1H), 4.27–4.19 (m, 1H), 3.95–3.88 (m, 1H), 3.86 (s, 3H), 3.04–2.88 (m, 4H), 1.86–1.80 (m, 1H), 1.71–1.62 (m, 3H), 1.61–1.49 (m, 3H), 1.47 (s, 3H), 1.43–1.35 (m, 1H), 1.34–1.27 (m, 2H), 1.22–1.15 (m, 1H), 1.06–0.95 (m, 1H), 0.90–0.86 (m, 6H). 13C-NMR (101 MHz, CDCl3) δ 159.10, 143.39, 134.39, 129.97, 129.15, 125.68, 113.95, 107.26, 106.62, 101.41, 97.23, 82.71, 68.49, 55.62, 48.52, 45.22, 44.10, 39.95, 35.25, 34.54, 34.50, 32.79, 27.96, 23.85, 22.15, 19.63, 19.32, 18.74.
5-Phenyl-1-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)ethyl)-1H-1,2,3-triazole (8f): 5 (60 mg, 0.183 mmol), acetophenone (22 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 24 h. The product was purified by flash column chromatography (at first DCM followed by EtOAc/heptane = 3:2) to give 8f (41 mg, 49%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 7.69 (s, 1H), 7.53–7.44 (m, 5H), 5.35 (s, 1H), 4.64–4.46 (m, 3H), 4.32–4.21 (m, 1H), 4.12–4.00 (m, 1H), 1.88–1.79 (m, 1H), 1.70–1.62 (m, 3H), 1.61–1.49 (m, 3H), 1.47 (s, 3H), 1.42–1.33 (m, 1H), 1.31–1.22 (m, 2H), 1.21–1.12 (m, 1H), 1.06–0.94 (m, 1H), 0.89–0.81 (m, 6H). 13C-NMR (101 MHz, CDCl3) δ 138.90, 132.92, 129.45, 129.26, 129.10, 127.25, 107.23, 101.60, 97.23, 82.71, 68.08, 48.04, 45.27, 44.12, 39.70, 35.27, 34.57, 34.53, 32.79, 23.86, 22.17, 19.19, 18.76.
5-(3,4-Dibromophenyl)-1-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)ethyl)-1H-1,2,3-triazole (8g): 5 (60 mg, 0.183 mmol), 1-(3,4-dibromophenyl)ethan-1-one (51 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 36 h. Flash column chromatography purification (DCM first, followed by EtOAc/heptane = 3:2) afforded 8g (42 mg, 37%) as an off-white solid: m.p. 105–106 °C; 1H-NMR (400 MHz, CDCl3) δ 7.83 (d, J = 2.0 Hz, 1H), 7.73–7.69 (m, 2H), 7.32 (dd, J = 8.3, 2.1 Hz, 1H), 5.31 (s, 1H), 4.60–4.52 (m, 3H), 4.28–4.22 (m, 1H), 4.07–4.00 (m, 1H), 1.86–1.80 (m, 1H), 1.71–1.64 (m, 2H), 1.62–1.56 (m, 3H), 1.54–1.49 (m, 1H), 1.46 (s, 3H), 1.32–1.22 (m, 3H), 1.19–1.10 (m, 2H), 1.04–0.95 (m, 1H), 0.88 (d, J = 5.7 Hz, 3H), 0.84 (d, J = 7.1 Hz, 3H). 13C-NMR (151 MHz, CDCl3) δ 134.32, 134.22, 133.20, 129.19, 128.04, 126.35, 125.68, 107.26, 101.58, 97.21, 82.71, 68.42, 48.52, 45.24, 44.09, 39.71, 35.27, 34.56, 34.53, 32.81, 23.87, 22.17, 19.22, 18.78.
5-(Naphthalen-1-yl)-1-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)ethyl)-1H-1,2,3-triazole (8h): 5 (60 mg, 0.183 mmol), 1-(naphthalen-1-yl)ethan-1-one (31.2 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 36 h. The product was purified by flash column chromatography (first DCM, then EtOAc/heptane = 3:2) to afford 8h (49 mg, 53%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.2 Hz, 1H), 7.92 (d, J = 7.7 Hz, 1H), 7.79 (s, 1H), 7.58–7.46 (m, 5H), 5.24 (s, 1H), 4.51 (d, J = 8.0 Hz, 1H), 4.42–4.34 (m, 2H), 4.13–4.04 (m, 1H), 3.92–3.83 (m, 1H), 1.84–1.78 (m, 1H), 1.72–1.58 (m, 3H), 1.57–1.45 (m, 3H), 1.41 (s, 3H), 1.31–1.23 (m, 1H), 1.19–1.09 (m, 3H), 1.01–0.93 (m, 1H), 0.89–0.80 (m, 6H). 13C-NMR (101 MHz, CDCl3) δ 136.53, 134.52, 133.76, 132.25, 130.29, 129.05, 128.66, 127.34, 126.66, 125.27, 125.00, 124.59, 107.13, 101.35, 97.16, 82.65, 67.70, 48.24, 45.26, 44.05, 39.52, 35.23, 34.56, 34.52, 32.70, 23.83, 22.15, 19.25, 18.76.
2-Methoxy-4-(1-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)ethyl)-1H-1,2,3-triazol-5-yl)phenol (8i): 5 (60 mg, 0.183 mmol), 1-(4-hydroxy-3-methoxyphenyl)ethan-1-one (30.5 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 36 h. The product was purified by flash column chromatography (first DCM, followed by EtOAc/heptane = 3:2) to give 8i (35 mg, 38%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 7.64 (s, 1H), 7.01 (s, 3H), 5.86 (s, 1H), 5.34 (s, 1H), 4.59 (d, J = 7.9 Hz, 1H), 4.57–4.44 (m, 2H), 4.31–4.21 (m, 1H), 4.11–4.02 (m, 1H), 3.93 (s, 3H), 1.88–1.80 (m, 1H), 1.70–1.58 (m, 3H), 1.57–1.50 (m, 3H), 1.47 (s, 3H), 1.40–1.31 (m, 1H), 1.30–1.20 (m, 2H), 1.18–1.10 (m, 1H), 1.06–0.96 (m, 1H), 0.89–0.81 (m, 6H). 13C-NMR (101 MHz, CDCl3) δ 146.91, 138.98, 132.62, 122.83, 118.92, 115.07, 111.85, 107.27, 101.57, 97.25, 82.69, 68.20, 56.29, 47.85, 45.25, 44.10, 39.83, 35.27, 34.56, 34.53, 32.81, 29.84, 23.84, 22.17, 19.19, 18.77.
5-Benzyl-1-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)ethyl)-4,5,6,7-tetrahydro-1H-[1,2,3]triazolo[4,5-c]pyridine (8j): 5 (60 mg, 0.183 mmol), 1-benzylpiperidin-4-one (34.7 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 16 h. The product was purified by flash column chromatography (first DCM, next EtOAc/heptane = 3:2) to afford 8j (61 mg, 63%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 7.38–7.28 (m, 5H), 5.37 (s, 1H), 4.60 (d, J = 7.9 Hz, 1H), 4.52–4,46 (m, 1H), 4.42–4.34 (m, 1H), 4.23–4.17 (m, 1H), 3.90–3.82 (m, 1H), 3.75 (s, 2H), 3.72–3.64 (m, 2H), 2.85–2.68 (m, 4H), 1.85–1.81 (m, 1H), 1.70–1.53 (m, 6H), 1.47 (s, 3H), 1.39–1.36 (m, 1H), 1.34–1.26 (m, 1H), 1.95–1.12 (m, 2H), 1.08–0.99 (m, 1H), 0.92–0.83 (m, 6H). 13C-NMR (101 MHz, CDCl3) δ 142.03, 138.18, 131.76, 129.08, 128.52, 127.46, 107.26, 101.43, 97.23, 82.71, 68.54, 61.64, 49.92, 49.39, 48.23, 45.25, 44.14, 39.93, 35.26, 34.55, 34.52, 32.81, 29.82, 23.86, 22.16, 20.99, 19.28, 18.76.
(1S,3aS,3bR,5aS,10aS,10bS,12aS)-10a,12a-Dimethyl-7-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyl-decahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)oxy)ethyl)-1,2,3,3a,3b,4,5,5a,6,7,10,10a, 10b,11,12,12a-hexadecahydrocyclopenta[7,8]phenanthro[2,3-d][1,2,3]triazol-1-ol (8k): 5 (60 mg, 0.183 mmol), (5S,8R,9S,10S,13S,14S,17S)-17-hydroxy-10,13-dimethylhexadecahydro-3H-cyclopenta[a]phenanthren-3-one (53.2 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 16 h. Flash column chromatography (first DCM, followed by EtOAc/heptane = 3:2) yielded 8k (72 mg, 63%) as an off-white solid: m.p. 96–97 °C; 1H-NMR (400 MHz, CDCl3) δ 5.39 (s, 1H), 4.61 (d, J = 7.9 Hz, 1H), 4.48 (dt, J = 14.2, 4.2 Hz, 1H), 4.40–4.31 (m, 1H), 4.20 (dt, J = 8.9, 4.4 Hz, 1H), 3.91–3.85 (m, 1H), 3.66 (t, J = 8.5 Hz, 1H), 2.85 (d, J = 15.3 Hz, 1H), 2.55 (dd, J = 16.2, 5.0 Hz, 1H), 2.30 (t, J = 13.6 Hz, 2H), 2.11–2.03 (m, 1H), 1.89 - 1.82 (m, 2H), 1.75–1.51 (m, 10H), 1.48–1.47 (m, 3H), 1.45–1.38 (m, 3H), 1.32–1.25 (m, 3H), 1.20–1.12 (m, 3H), 1.05–0.96 (m, 2H), 0.91–0.87 (m, 6H), 0.76 (s, 3H), 0.72 (s, 3H). 13C-NMR (151 MHz, CDCl3) δ 142.96, 131.85, 107.28, 101.25, 97.27, 82.74, 81.99, 68.40, 53.86, 50.95, 48.03, 45.26, 44.14, 43.00, 42.39, 39.98, 36.96, 36.78, 36.31, 35.75, 35.32, 34.56, 34.54, 32.84, 31.34, 30.60, 29.05, 25.01, 23.87, 23.58, 22.18, 20.93, 19.39, 18.78, 11.73, 11.17.
(1R,3aS,5aR,5bR,7aR,12aR,12bR,14aR,14bR)-5a,5b,8,8,12a-Pentamethyl-1-(prop-1-en-2-yl)-9-(2-(((3R,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)-oxy)ethyl)-2,3,4,5,5a,5b,6,7,7a,8,9,12,12a,12b,13,14,14a,14b-octadecahydrocyclopenta[7,8]chryseno[2,3-d][1,2,3]triazole-3a(1H)-carboxylic acid (8l): 5 (60 mg, 0.183 mmol), (1R,3aS,5aR,5bR,7aR,11aR, 11bR,13aR,13bR)-5a,5b,8,8,11a-pentamethyl-9-oxo-1-(prop-1-en-2-yl)icosahydro-3aH-cyclopenta[a]chrysene-3a-carboxylic acid (83 mg, 0.183 mmol), 4-nitrophenyl azide (36.1 mg, 0.220 mmol). Reaction time was 16 h. The product was purified by flash column chromatography (first DCM, followed by EtOAc/heptane = 3:2) to afford 8l (68 mg, 47%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 5.39 (s, 1H), 4.76 (s, 1H), 4.69–4.63 (m, 2H), 4.59–4.51 (m, 1H), 4.28–4.23 (m, 1H), 4.19–4.11 (m, 1H), 3.06–3.00 (m, 1H), 2.91 (d, J = 15.3 Hz, 1H), 2.31–2.23 (m, 2H), 2.12 (d, J = 15.3 Hz, 1H), 2.03–1.96 (m, 2H), 1.86–1.76 (m, 3H), 1.71 (s, 3H), 1.69–1.63 (m, 3H), 1.60–1.50 (m, 8H), 1.48 (s, 5H), 1.45–1.39 (m, 2H), 1.37 (d, J = 2.5 Hz, 1H), 1.32 (d, J = 7.6 Hz, 4H), 1.29–1.22 (m, 9H), 1.18–1.10 (m, 3H), 1.00 (d, J = 4.6 Hz, 6H), 0.90 - 0.87 (m, 6H), 0.78 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 180.60, 150.37, 141.10, 138.35, 110.01, 107.26, 101.71, 97.31, 82.75, 68.55, 56.48, 54.86, 49.47, 49.31, 49.21, 47.00, 45.30, 44.10, 42.61, 40.75, 39.80, 39.09, 38.59, 38.42, 37.18, 35.29, 34.60, 34.56, 33.76, 33.54, 32.82, 32.22, 30.71, 29.94, 29.84, 28.82, 25.65, 23.90, 22.19, 21.57, 19.55, 19.33, 19.09, 18.80, 16.17, 15.80, 14.83.
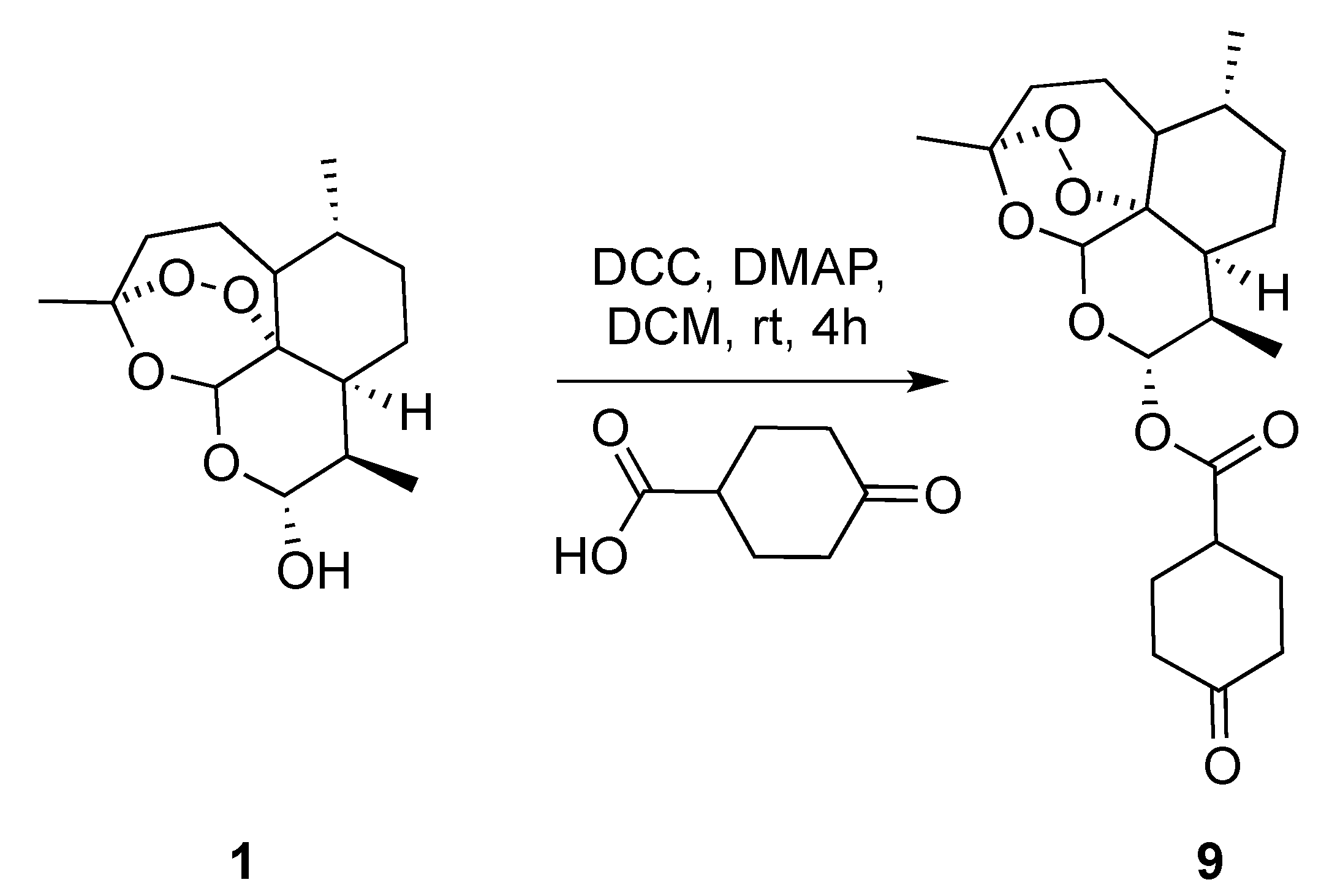
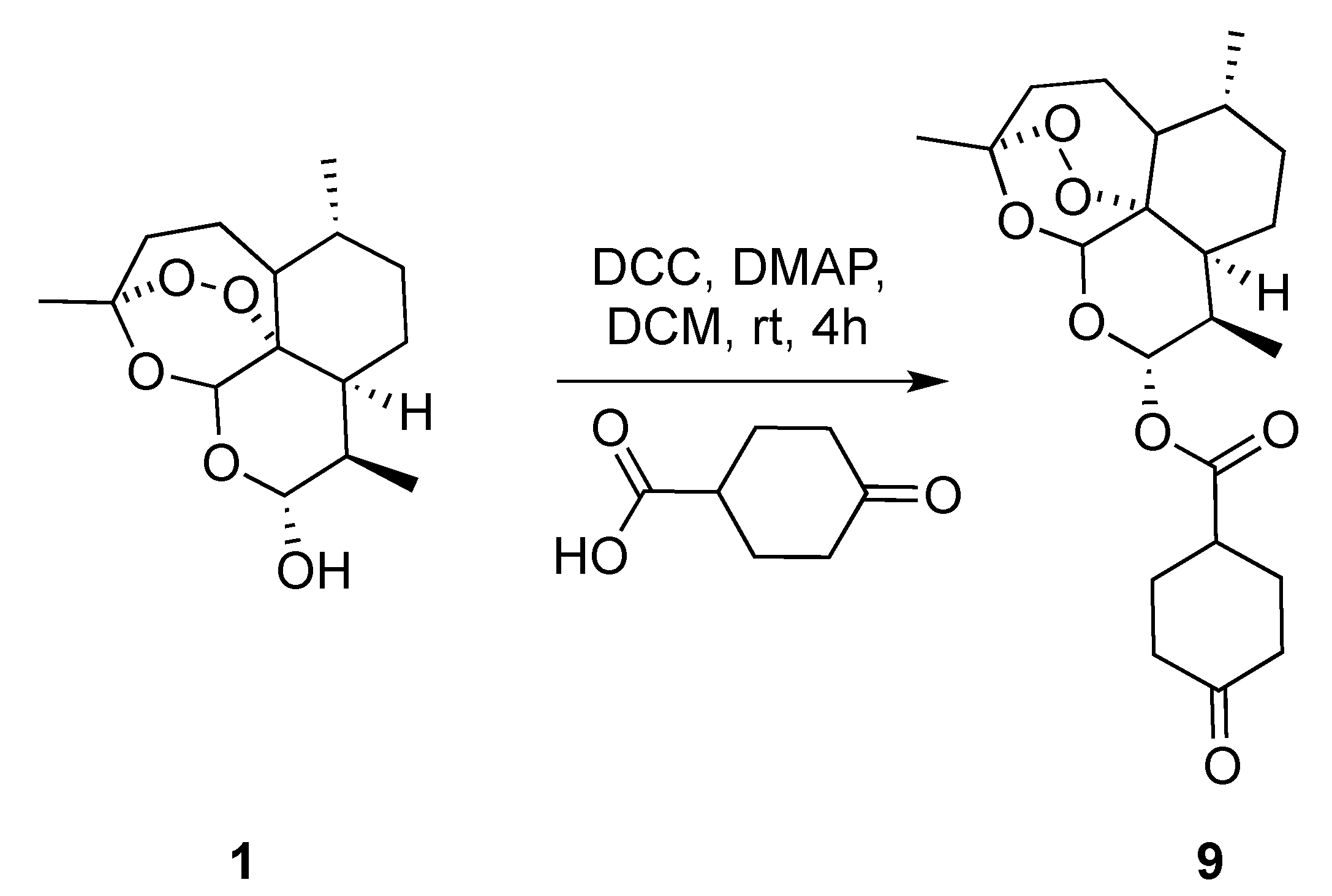
(3R,6R,8aS,9R,12R,12aR)-3,6,9-Trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl 4-oxocyclohexane-1-carboxylate (9): dihydroartemisinin (1 gm, 3.52 mmol), dicyclohexyl-methanediimine (0.726 gm, 3.52 mmol), 4-oxocyclohexane-1-carboxylic acid (0.600 gm, 4.22 mmol), N,N-dimethylpyridin-4-amine (0.064 gm,0.528 mmol.). Reaction time was 18 h. The product was purified by flash column chromatography (DCM/MeOH = 99/1) to give 9 (1.2 gm, 84%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 5.82 (d, J = 9.8 Hz, 1H), 5.45 (s, 1H), 2.91–2.75 (m, 1H), 2.60 (ddd, J = 9.9, 7.2, 4.6 Hz, 1H), 2.62–2.58 (m, 1H), 2.55–2.45 (m, 1H), 2.39–2.34 (m, 2H), 2.33–2.27 (m, 1H), 2.25–2.20 (m, 1H), 2.14–2.00 (m, 3H), 1.95–1.87 (m, 1H), 1.81–1.75 (m, 2H), 1.67–1.56 (m, 2H), 1.52–1.46 (m, 1H), 1.45–1.41 (m, 3H), 1.39–1.25 (m, 3H), 1.11–1.00 (m, 1H), 0.97 (d, J = 6.0 Hz, 3H), 0.86 (d, J = 7.1 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 210.13, 173.04, 104.63, 92.27, 91.67, 80.25, 77.48, 76.84, 51.70, 45.39, 40.70, 39.87, 39.66, 37.41, 36.33, 34.21, 31.94, 28.78, 28.05, 26.06, 24.71, 22.13, 20.34, 12.30.
(3R,6R,8aS,9R,10S,12R,12aR)-3,6,9-Trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl 1-(4-methylbenzyl)-4,5,6,7-tetrahydro-1H-benzo[d][1,2,3]triazole-5-carboxylate (11a): 9 (60 mg, 0.147 mmol), p-tolylmethanamine (18 mg, 0.147 mmol), 4-nitrophenyl azide (28.9 mg, 0.176 mmol). Reaction time was 16 h. Flash column chromatography (first DCM, followed by EtOAc/heptane = 3:2) afforded purified 11a (53 mg, 67%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 7.14 (d, J = 7.8 Hz, 2H), 7.08 (d, J = 7.8 Hz, 2H), 5.83–5.69 (m, 1H), 5.48–5.29 (m, 3H), 3.20–3.05 (m, 1H), 3.03–2.91 (m, 1H), 2.86–2.76 (m, 1H), 2.68–2.50 (m, 2H), 2.44–2.36 (m, H), 2.33 (s, 3H), 2.29–2.12 (m, 1H), 2.07–1.99 (m, 1H), 1.95–1.85 (m, 2H), 1.79–1.69 (m, 2H), 1.66–1.57 (m, 2H), 1.54–1.45 (m, 1H), 1.42 (d, J = 1.6 Hz, 3H), 1.38–1.25 (m, 3H), 0.96 (d, J = 5.8 Hz, 3H), 0.80 (d, J = 7.1 Hz, 3H). 13C-NMR (151 MHz, CDCl3) δ 173.24, 172.94, 141.10, 141.06, 136.27, 136.21, 132.13, 127.09, 127.02, 122.90, 122.73, 122.40, 122.24, 119.72, 119.63, 118.14, 118.05, 111.69, 111.62, 111.23, 104.76, 104.72, 92.59, 92.53, 91.82, 80.38, 80.22, 51.74, 51.70, 48.99, 48.80, 45.35, 39.53, 39.38, 37.40, 36.38, 34.18, 32.02, 31.90, 26.77, 26.68, 26.03, 25.92, 25.02, 24.80, 24.69, 24.35, 23.84, 22.15, 20.34, 18.23, 18.04, 12.32, 12.22.
(3R,6R,8aS,9R,10S,12R,12aR)-3,6,9-Trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl 1-(3,4,5-trimethoxybenzyl)-4,5,6,7-tetrahydro-1H-benzo[d][1,2,3]triazole-5-carboxylate (11b): 9 (60 mg, 0.147 mmol), (3,4,5-trimethoxyphenyl)methanamine (29 mg, 0.147 mmol), 4-nitrophenyl azide (28.9 mg, 0.176 mmol). Reaction time was 16 h. The product was purified by flash column chromatography (DCM at first, followed by EtOAc/heptane = 3:2) to afford 11b (55 mg, 61%) as an off-white solid: mp 80–81 °C; 1H-NMR (400 MHz, CDCl3) δ 6.40 (d, J = 6.8 Hz, 2H), 5.82–5.74 (m, 1H), 5.43 (t, J = 4.6 Hz, 1H), 5.39–5.28 (m, 2H), 3.83 (d, J = 1.0 Hz, 3H), 3.81 (d, J = 1.5 Hz, 6H), 3.20–2.94 (m, 2H), 2.88–2.81 (m, 1H), 2.74–2.54 (m, 2H), 2.52–2.42 (m, 1H), 2.42–2.33 (m, 1H), 2.31–2.20 (m, 1H), 2.02–1.86 (m, 2H), 1.79–1.68 (m, 2H), 1.66–1.59 (m, 1H), 1.42 (d, J = 3.5 Hz, 3H), 1.32–1.23 (m, 4H), 0.97 (d, J = 5.8 Hz, 3H), 0.90–0.84 (m, 2H), 0.81 (d, J = 7.1 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 173.06, 172.85, 153.77, 142.64, 142.33, 138.12, 131.47, 131.12, 130.33, 130.30, 104.73, 104.66, 104.61, 104.57, 92.30, 91.65, 80.22, 80.20, 60.97, 56.34, 52.31, 52.28, 51.67, 45.37, 45.33, 39.82, 39.47, 37.39, 36.31, 34.18, 31.92, 31.87, 29.81, 26.03, 25.47, 24.99, 24.68, 24.20, 22.09, 20.31, 19.29, 18.87, 12.24, 12.16.
(3R,6R,8aS,9R,10S,12R,12aR)-3,6,9-Trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl 1-(benzo[d][1,3]dioxol-5-ylmethyl)-4,5,6,7-tetrahydro-1H-benzo[d][1,2,3]triazole-5-carboxylate (11c): 9 (60 mg, 0.147 mmol), benzo[d][1,3]dioxol-5-ylmethanamine (22.2 mg, 0.147 mmol), 4-nitrophenyl azide (28.9 mg, 0.176 mmol). Reaction time was 16 h. The product was purified by flash column chromatography (first DCM, followed by EtOAc/heptane = 3:2) to give 11c (51 mg, 61%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 6.76 (dd, J = 6.9, 1.7 Hz, 1H), 6.68 (dt, J = 3.7, 1.6 Hz, 2H), 5.96 (s, 2H), 5.83–5.73 (m, 1H), 5.43 (d, J = 3.0 Hz, 1H), 5.39–5.25 (m, 2H), 3.18–3.06 (m, 1H), 3.04–2.92 (m, 1H), 2.87–2.78 (m, 1H), 2.69–2.54 (m, 2H), 2.47–2.33 (m, 2H), 2.28–2.19 (m, 1H), 2.06–1.86 (m, 3H), 1.79–1.69 (m, 2H), 1.66–1.58 (m, 1H), 1.51–1.45 (m, 1H), 1.42 (d, J = 1.9 Hz, 3H), 1.34–1.24 (m, 2H), 1.07–0.98 (m, 1H), 0.96 (d, J = 5.9 Hz, 3H), 0.91–0.84 (m, 1H), 0.83–0.78 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ 173.09, 172.92, 148.40, 147.87, 142.33, 131.34, 128.43, 121.33, 108.54, 108.21, 108.19, 104.59, 104.55, 101.45, 92.29, 91.64, 80.22, 80.19, 51.98, 51.67, 45.38, 45.33, 39.47, 37.38, 37.36, 36.31, 34.18, 31.92, 31.86, 26.04, 25.50, 24.99, 24.67, 24.23, 22.08, 20.31, 19.36, 18.86, 12.15.
(3R,6R,8aS,9R,10S,12R,12aR)-3,6,9-Trimethyldecahydro-12H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl 1-(2-(1H-indol-3-yl)ethyl)-4,5,6,7-tetrahydro-1H-benzo[d][1,2,3]triazole-5-carboxylate (11d): 9 (60 mg, 0.147 mmol), 2-(1H-indol-3-yl)ethan-1-amine (25.5 mg, 0.147 mmol), 4-nitrophenyl azide (28.9 mg, 0.176 mmol). Reaction time was 16 h. The product was purified by flash column chromatography (first DCM, followed by EtOAc/heptane = 3:2) to afford 11d (39 mg, 46%) as an off-white semisolid: 1H-NMR (400 MHz, CDCl3) δ 8.42 (d, J = 7.1 Hz, 1H), 7.43–7.33 (m, 2H), 7.22–7.16 (m, 1H), 7.11–7.06 (m, 1H), 6.70 (dd, J = 51.1, 2.3 Hz, 1H), 5.73 (dd, J = 9.9, 6.4 Hz, 1H), 5.47 (d, J = 13.4 Hz, 1H), 4.53–4.31 (m, 2H), 3.37–3.24 (m, 2H), 3.06–2.87 (m, 2H), 2.70–2.55 (m, 2H), 2.42–2.32 (m, 1H), 2.03 (ddd, J = 14.5, 7.3, 4.2 Hz, 1H), 1.93–1.85 (m, 3H), 1.82–1.74 (m, 2H), 1.72–1.61 (m, 4H), 1.54–1.41 (m, 1H), 1.41–1.34 (m, 4H), 1.34–1.25 (m, 2H), 0.97 (d, J = 5.6 Hz, 3H), 0.83 (d, J = 7.1 Hz, 3H). 13C-NMR (151 MHz, CDCl3) δ 173.24, 172.94, 141.10, 141.06, 136.27, 136.21, 132.13, 127.09, 127.02, 122.90, 122.73, 122.40, 122.24, 119.72, 119.63, 118.14, 118.05, 111.69, 111.62, 111.23, 104.76, 104.72, 92.59, 92.53, 91.82, 80.38, 80.22, 51.74, 51.70, 48.99, 48.80, 45.35, 39.53, 39.38, 37.40, 36.38, 34.18, 32.02, 31.90, 26.77, 26.68, 26.03, 25.92, 25.02, 24.80, 24.69, 24.35, 23.84, 22.15, 20.34, 18.23, 18.04, 12.32, 12.22.


 ,
, 

{kind=link}
{kind=link}
{kind=link}

