
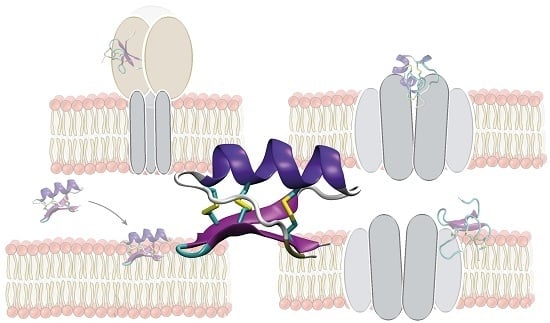
Molecular Simulations of Disulfide-Rich Venom Peptides with Ion Channels and Membranes


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
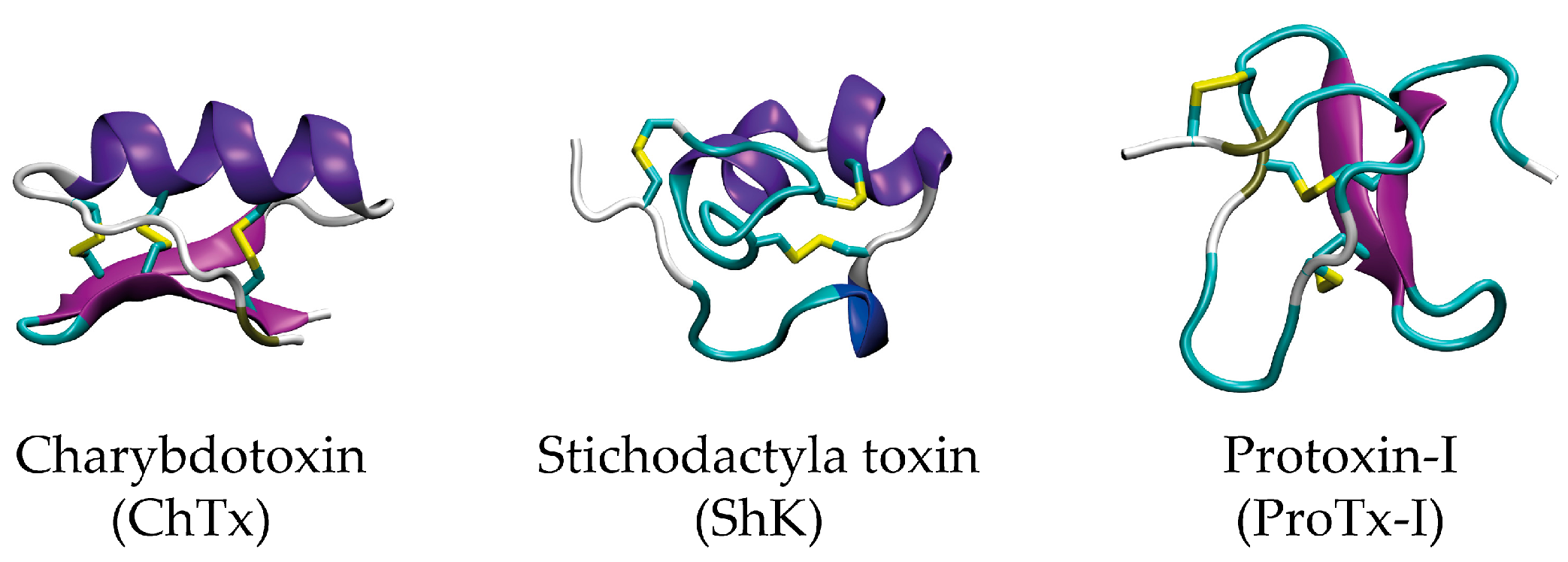
1. Introduction
2. Simulation Studies of Peptide-Channel Complexes
2.1. Predicting the Structure of Peptide-Channel Complexes without Experimental Data
2.2. Predicting the Structure of Peptide-Channel Complexes Guided by Experimental Data
2.3. Predicting Binding Free Energy for Peptide-Channel Complexes
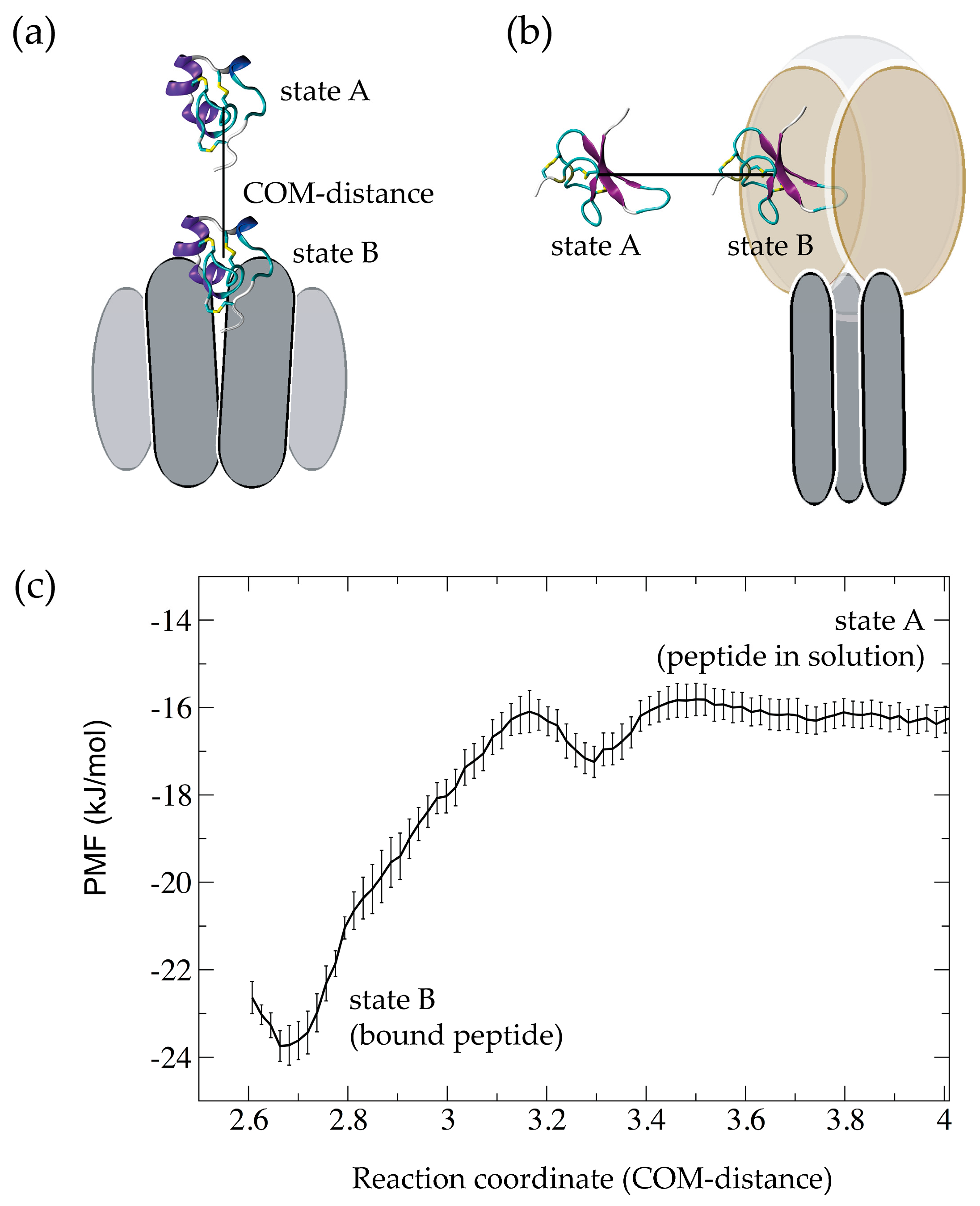
2.3.1. Predicting Binding Free Energies from the Potential of Mean Force
2.3.2. Predicting the Effect of Mutations on the Binding Affinity
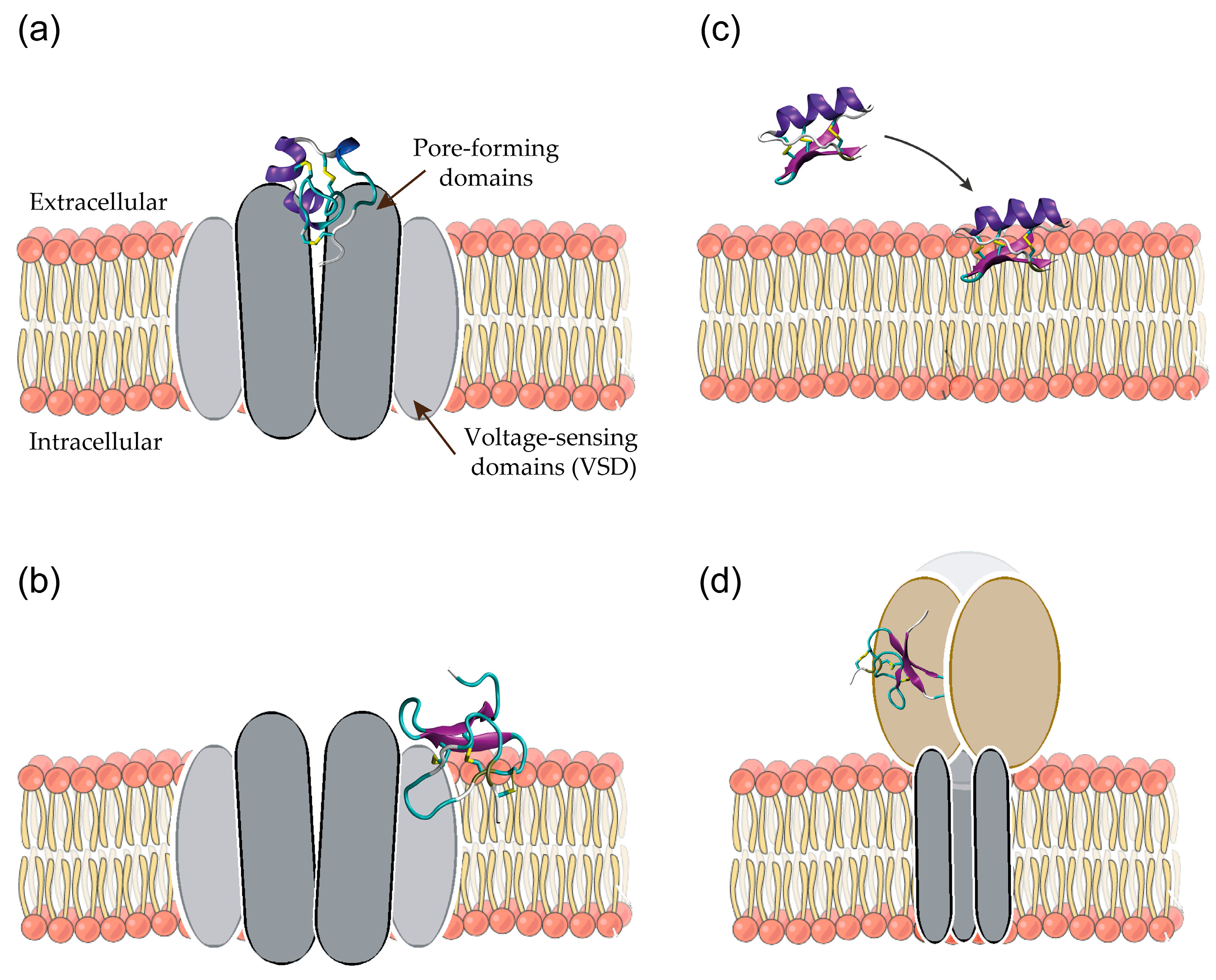
3. Simulations of Peptide-Membrane Interactions
4. Summary
Acknowledgments
Conflicts of Interest
References
- Lavergne, V.; Alewood, P.F.; Mobli, M.; King, G.F. The structural universe of disulfide-rich venom peptides. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; The Royal Society of Chemistry: Cambridge, UK, 2015; pp. 37–79. [Google Scholar]
- De la Vega, R.C.R.; Corzo, G.; Possani, L.D. Scorpion venoms as a platform for drug development. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; The Royal Society of Chemistry: Cambridge, UK, 2015; pp. 204–220. [Google Scholar]
- Dutertre, S.; Lewis, R.J. Use of venom peptides to probe ion channel structure and function. J. Biol. Chem. 2010, 285, 13315–13320. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: Pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Lau, C.H.Y.; Herzig, V.; Ikonomopoulou, M.P.; Rash, L.D.; King, G.F. Therapeutic applications of spider-venom peptides. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; The Royal Society of Chemistry: Cambridge, UK, 2015; pp. 221–244. [Google Scholar]
- Teichert, R.W.; Olivera, B.M.; McIntosh, J.M.; Bulaj, G.; Horvath, M.P. The molecular diversity of conoidean venom peptides and their targets: From basic research to therapeutic applications. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; The Royal Society of Chemistry: Cambridge, UK, 2015; pp. 163–203. [Google Scholar]
- Baron, A.; Diochot, S.; Salinas, M.; Deval, E.; Noël, J.; Lingueglia, E. Venom toxins in the exploration of molecular, physiological and pathophysiological functions of acid-sensing ion channels. Toxicon 2013, 75, 187–204. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- McCleary, R.J.R.; Kang, T.S.; Kini, R.M. Reptile venoms as a platform for drug development. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; The Royal Society of Chemistry: Cambridge, UK, 2015; pp. 129–162. [Google Scholar]
- Henriques, S.T.; Deplazes, E.; Lawrence, N.; Cheneval, O.; Inserra, M.; Thongyoo, P.; Mark, A.E.; Vetter, I.; Craik, D.J.; Schroeder, C. Interaction of tarantula venom peptide ProTx-II with lipid membranes is a prerequisite for its inhibition of human voltage-gated sodium channel NaV1.7. J. Biol. Chem. 2016, 291, 17049–17065. [Google Scholar] [CrossRef] [PubMed]
- Phillips, L.R.; Milescu, M.; Li-Smerin, Y.; Mindell, J.A.; Kim, J.I.; Swartz, K.J. Voltage-sensor activation with a tarantula toxin as cargo. Nature 2005, 436, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.-H. Computational studies of venom peptides targeting potassium channels. Toxins 2015, 7, 5194–5211. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Chen, R.; Chung, S.H. Computational methods of studying the binding of toxins from venomous animals to biological ion channels: Theory and applications. Physiol. Rev. 2013, 93, 767–802. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F.; Bakowies, D.; Baron, R.; Chandrasekhar, I.; Christen, M.; Daura, X.; Gee, P.; Geerke, D.P.; Glattli, A.; Hunenberger, P.H.; et al. Biomolecular modeling: Goals, problems, perspectives. Angew. Chem. Int. Ed. Engl. 2006, 45, 4064–4092. [Google Scholar] [CrossRef] [PubMed]
- Novoseletsky, V.; Volyntseva, A.; Shaitan, K.; Kirpichnikov, M.; Feofanov, A. Modeling of the binding of peptide blockers to voltage-gated potassium channels: Approaches and evidence. Acta Nat. 2016, 8, 35–46. [Google Scholar]
- Rashid, M.H.; Mahdavi, S.; Kuyucak, S. Computational studies of marine toxins targeting ion channels. Mar. Drugs 2013, 11, 848–869. [Google Scholar] [CrossRef] [PubMed]
- Deplazes, E.; Davies, J.; Bonvin, A.M.J.J.; King, G.F.; Mark, A.E. Combination of ambiguous and unambiguous data in the restraint-driven docking of flexible peptides with HADDOCK: The binding of the spider toxin PcTx1 to the acid sensing ion channel (ASIC) 1a. J. Chem. Inf. Model. 2016, 56, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Pietra, F. Docking and MD simulations of the interaction of the tarantula peptide psalmotoxin-1 with ASIC1a channels using a homology model. J. Chem. Inf. Model. 2009, 49, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Qadri, Y.J.; Berdiev, B.K.; Song, Y.; Lippton, H.L.; Fuller, C.M.; Benos, D.J. Psalmotoxin-1 docking to human acid-sensing ion channel-1. J. Biol. Chem. 2009, 284, 17625–17633. [Google Scholar] [CrossRef] [PubMed]
- Saez, N.J.; Deplazes, E.; Cristofori-Armstrong, B.; Chassagnon, I.R.; Lin, X.; Mobli, M.; Mark, A.E.; Rash, L.D.; King, G.F. Molecular dynamics and functional studies define a hot spot of crystal contacts essential for PcTx1 inhibition of acid-sensing ion channel 1a. Br. J. Pharmacol. 2015, 172, 4985–4995. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Systematic validation of protein force fields against experimental data. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Martín-García, F.; Papaleo, E.; Gomez-Puertas, P.; Boomsma, W.; Lindorff-Larsen, K. Comparing molecular dynamics force fields in the essential subspace. PLoS ONE 2015, 10. [Google Scholar]
- Abrams, C.; Bussi, G. Enhanced sampling in molecular dynamics using metadynamics, replica-exchange, and temperature-acceleration. Entropy 2014, 16, 163. [Google Scholar] [CrossRef]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. BBA-Gen. Subjects 2015, 1850, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Sawle, L.; Ghosh, K. Convergence of molecular dynamics simulation of protein native states: Feasibility vs self-consistency dilemma. J. Chem. Theory Comput. 2016, 12, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Soares, T.A.; Straatsma, T.P. Assessment of the convergence of molecular dynamics simulations of lipopolysaccharide membranes. Mol. Simul. 2008, 34, 295–307. [Google Scholar] [CrossRef]
- Moal, I.H.; Torchala, M.; Bates, P.A.; Fernández-Recio, J. The scoring of poses in protein-protein docking: Current capabilities and future directions. BMC Bioinformatics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Sandor, V.; Kozakov, D. Sampling and scoring: A marriage made in heaven. Proteins 2013, 81, 1874–1884. [Google Scholar]
- Gordon, D.; Chen, R.; Ho, J.; Coote, M.L.; Chung, S.-H. Rigid body brownian dynamics as a tool for studying ion channel blockers. J. Phys. Chem. B 2012, 116, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Chung, S.-H. Extension of brownian dynamics for studying blockers of ion channels. J. Phys. Chem. B 2012, 116, 14285–14294. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-C.; Kuyucak, S. Mechanism and energetics of charybdotoxin unbinding from a potassium channel from molecular dynamics simulations. Biophys. J. 2009, 96, 2577–2588. [Google Scholar] [CrossRef] [PubMed]
- Cestele, S.; Yarov-Yarovoy, V.; Qu, Y.; Sampieri, F.; Scheuer, T.; Catterall, W.A. Structure and function of the voltage sensor of sodium channels probed by a beta-scorpion toxin. J. Biol. Chem. 2006, 281, 21332–21344. [Google Scholar] [CrossRef] [PubMed]
- Khabiri, M.; Nikouee, A.; Cwiklik, L.; Grissmer, S.; Ettrich, R. Charybdotoxin unbinding from the mKv1.3 potassium channel: A combined computational and experimental study. J. Phys. Chem. B 2011, 115, 11490–11500. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wu, B.F.; Feng, Y.J.; Tao, J.; Ji, Y.H. Mapping the interaction anatomy of BmP02 on Kv1.3 channel. Sci. Rep. 2016, 6, 29431. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Mashiach, E.; Nussinov, R.; Wolfson, H.J. Principles of flexible protein–protein docking. Proteins: Struct. Funct. Bioinf. 2008, 73, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.J. High-resolution protein–protein docking. Curr. Opin. Struct. Biol. 2006, 16, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.P.G.L.M.; Bonvin, A.M.J.J. Integrative computational modeling of protein interactions. FEBS J. 2014, 281, 1988–2003. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.R.; Sternberg, M.J.E. Prediction of protein–protein interactions by docking methods. Curr. Opin. Struct. Biol. 2002, 12, 28–35. [Google Scholar] [CrossRef]
- Xue, L.C.; Dobbs, D.; Bonvin, A.M.; Honavar, V. Computational prediction of protein interfaces: A review of data driven methods. FEBS Lett. 2015, 583, 3516–3526. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.X.; Qin, S. Interaction-site prediction for protein complexes: A critical assessment. Bioinformatics 2007, 23, 2203–2209. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.-H. Binding modes of μ-conotoxin to the bacterial sodium channel (NaVAb). Biophys. J. 2012, 102, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Wee, C.L.; Gavaghan, D.; Sansom, M.S.P. Interactions between a voltage sensor and a toxin via multiscale simulations. Biophys. J. 2010, 98, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.H. Engineering a potent and specific blocker of voltage-gated potassium channel Kv1.3, a target for autoimmune diseases. Biochemistry 2012, 51, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, N.; di Luccio, E.; Sampieri, F.; De Waard, M.; Sabatier, J.M. Molecular modeling and docking simulations of scorpion toxins and related analogs on human SKCa2 and SKCa3 channels. Peptides 2005, 26, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Robinson, A.; Gordon, D.; Chung, S.H. Modeling the binding of three toxins to the voltage-gated potassium channel (Kv1.3). Biophys. J. 2011, 101, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, S.; Kuyucak, S. Why the drosophila shaker K+ channel is not a good model for ligand binding to voltage-gated Kv1 channels. Biochemistry 2013, 52, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.W.; Harunur Rashid, M.; Tajhya, R.B.; Beeton, C.; Kuyucak, S.; Norton, R.S. A C-terminally amidated analogue of ShK is a potent and selective blocker of the voltage-gated potassium channel Kv1.3. FEBS Lett. 2012, 586, 3996–4001. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Kuyucak, S. Affinity and selectivity of ShK toxin for the Kv1 potassium channels from free energy simulations. J. Phys. Chem. B 2012, 116, 4812–4822. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yarov-Yarovoy, V.; Kahn, R.; Gordon, D.; Gurevitz, M.; Scheuer, T.; Catterall, W.A. Mapping the receptor site for α-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. USA 2011, 108, 15426–15431. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cao, Z.; Yi, H.; Jiang, D.; Mao, X.; Liu, H.; Li, W. Simulation of the interaction between scytx and small conductance calcium-activated potassium channel by docking and MM-PBSA. Biophys. J. 2004, 87, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.R.; Zhao, H.Y.; Zheng, Z.X.; Wang, Y.; Niu, Y.Y.; Wang, H.; Xu, J.; Lu, Y.; Chen, H.Z. Structural determinants for the interactions between muscarinic toxin 7 and muscarinic acetylcholine receptors. J. Mol. Recognit. 2015, 28, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Qiu, S.; Wu, Y.; Li, W.; Wang, B. Differential molecular information of maurotoxin peptide recognizing ikca and Kv1.2 channels explored by computational simulation. BMC Struct. Biol. 2011, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Palma, P.N.; Krippahl, L.; Wampler, J.E.; Moura, J.J. Bigger: A new (soft) docking algorithm for predicting protein interactions. Proteins 2000, 39, 372–384. [Google Scholar] [CrossRef]
- De Vries, S.J.; van Dijk, A.D.; Krzeminski, M.; van Dijk, M.; Thureau, A.; Hsu, V.; Wassenaar, T.; Bonvin, A.M. HADDOCK versus HADDOCK: New features and performance of HADDOCK 2.0 on the CAPRI targets. Proteins 2007, 69, 726–733. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK web server for data-driven biomolecular docking. Nature Protocols 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.J.; Moughon, S.; Wang, C.; Schueler-Furman, O.; Kuhlman, B.; Rohl, C.A.; Baker, D. Protein–protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [Google Scholar] [CrossRef]
- Rashid, M.H.; Kuyucak, S. Free energy simulations of binding of HsTx1 toxin to Kv1 potassium channels: The basis of Kv1.3/Kv1.1 selectivity. J. Phys. Chem. B 2014, 118, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Kuyucak, S. Developing a comparative docking protocol for the prediction of peptide selectivity profiles: Investigation of potassium channel toxins. Toxins 2012, 4, 110–138. [Google Scholar] [CrossRef] [PubMed]
- Saez, N.J.; Mobli, M.; Bieri, M.; Chassagnon, I.R.; Malde, A.K.; Gamsjaeger, R.; Mark, A.E.; Gooley, P.R.; Rash, L.D.; King, G.F. A dynamic pharmacophore drives the interaction between psalmotoxin-1 and the putative drug target acid-sensing ion channel 1a. Mol. Pharmacol 2011, 80, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.-H. Structural basis of the selective block of Kv1.2 by maurotoxin from computer simulations. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.-H. Binding modes of two scorpion toxins to the voltage-gated potassium channel Kv1.3 revealed from molecular dynamics. Toxins 2014, 6, 2149–2161. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.H. Molecular dynamics simulations of scorpion toxin recognition by the Ca2+-activated potassium channel KCa3.1. Biophys. J. 2013, 105, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Robinson, A.; Chung, S.H. Mechanism of mu-conotoxin PIIIA binding to the voltage-gated Na+ channel NaV1.4. PLoS ONE 2014, 9. [Google Scholar]
- Choudhary, G.; Aliste, M.P.; Tieleman, D.P.; French, R.J.; Dudley, J.S.C. Docking of μ-conotoxin GIIIA in the sodium channel outer vestibule. Channels 2007, 1, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.A.L.; Roux, B. Modeling the structure of agitoxin in complex with the Shaker K+ channel: A computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys J. 2002, 83, 2595–2609. [Google Scholar] [CrossRef]
- Li, D.; Chen, R.; Chung, S.H. Molecular dynamics of the honey bee toxin tertiapin binding to Kir3.2. Biophys. Chem. 2016, 219, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez de la Vega, R.C.; Possani, L.D. Current views on scorpion toxins specific for K+-channels. Toxicon 2004, 43, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Chipot, C. Frontiers in free-energy calculations of biological systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 71–89. [Google Scholar] [CrossRef]
- Hansen, N.; van Gunsteren, W.F. Practical aspects of free-energy calculations: A review. J. Chem. Theory Comput. 2014, 10, 2632–2647. [Google Scholar] [CrossRef] [PubMed]
- Trzesniak, D.; Kunz, A.-P.E.; van Gunsteren, W.F. A comparison of methods to compute the potential of mean force. ChemPhysChem 2007, 8, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F.; Daura, X.; Mark, A.E. Computation of free energy. Helv. Chim. Acta 2002, 85, 3113–3129. [Google Scholar] [CrossRef]
- Gumbart, J.C.; Roux, B.; Chipot, C. Standard binding free energies from computer simulations: What is the best strategy? J. Chem. Theory Comput. 2013, 9, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecher, T.; Labahn, A. Towards accurate free energy calculations in ligand protein-binding studies. Curr. Med. Chem 2010, 17, 767–785. [Google Scholar] [CrossRef] [PubMed]
- Baştuğ, T.; Chen, P.-C.; Patra, S.M.; Kuyucak, S. Potential of mean force calculations of ligand binding to ion channels from jarzynski’s equality and umbrella sampling. J. Chem. Phys. 2008, 128, 155104. [Google Scholar] [CrossRef] [PubMed]
- Baştuğ, T.; Kuyucak, S. Application of jarzynski’s equality in simple versus complex systems. Chem. Phys. Lett. 2007, 436, 383–387. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar]
- Kästner, J.; Thiel, W. Bridging the gap between thermodynamic integration and umbrella sampling provides a novel analysis method: “Umbrella integration”. J. Chem. Phys. 2005, 123, 144104. [Google Scholar] [CrossRef] [PubMed]
- Kästner, J.; Thiel, W. Analysis of the statistical error in umbrella sampling simulations by umbrella integration. J. Chem. Phys. 2006, 124, 234106. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Souaille, M.; Roux, B. Extension to the weighted histogram analysis method: Combining umbrella sampling with free energy calculations. Comput. Phys. Commun. 2001, 135, 40–57. [Google Scholar] [CrossRef]
- Dellago, C.; Hummer, G. Computing equilibrium free energies using non-equilibrium molecular dynamics. Entropy 2014, 16, 41. [Google Scholar] [CrossRef]
- Park, S.; Khalili-Araghi, F.; Tajkhorshid, E.; Schulten, K. Free energy calculation from steered molecular dynamics simulations using jarzynski’s equality. J. Chem. Phys. 2003, 119, 3559–3566. [Google Scholar] [CrossRef]
- Park, S.; Schulten, K. Calculating potentials of mean force from steered molecular dynamics simulations. J. Chem. Phys. 2004, 120, 5946–5961. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics poisson−boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods: I. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem 2010, 31, 797–810. [Google Scholar] [CrossRef] [PubMed]
- General, I.J. A note on the standard state’s binding free energy. J. Chem. Theory Comput. 2010, 6, 2520–2524. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Given, J.A.; Bush, B.L.; McCammon, J.A. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys. J. 1997, 72, 1047–1069. [Google Scholar] [CrossRef]
- Woo, H.-J. Calculation of absolute protein–ligand binding constants with the molecular dynamics free energy perturbation method. In Molecular Modeling of Proteins; Kukol, A., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 109–120. [Google Scholar]
- Chen, P.-C.; Kuyucak, S. Accurate determination of the binding free energy for kcsa-charybdotoxin complex from the potential of mean force calculations with restraints. Biophys J. 2011, 100, 2466–2474. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.-H. Conserved functional surface of antimammalian scorpion β-toxins. J. Phys. Chem. B 2012, 116, 4796–4800. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Heinzelmann, G.; Huq, R.; Tajhya, R.B.; Chang, S.C.; Chhabra, S.; Pennington, M.W.; Beeton, C.; Norton, R.S.; Kuyucak, S. A potent and selective peptide blocker of the Kv1.3 channel: Prediction from free-energy simulations and experimental confirmation. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Hummer, G.; Szabo, A. Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc. Natl. Acad. Sci. USA 2001, 98, 3658–3661. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Heinzelmann, G.; Kuyucak, S. Calculation of free energy changes due to mutations from alchemical free energy simulations. J. Theor. Comput. Chem. 2015, 14, 1550023. [Google Scholar] [CrossRef]
- Han, S.; Yi, H.; Yin, S.-J.; Chen, Z.-Y.; Liu, H.; Cao, Z.-J.; Wu, Y.-L.; Li, W.-X. Structural basis of a potent peptide inhibitor designed for Kv1.3 channel, a therapeutic target of autoimmune disease. J. Biol. Chem. 2008, 283, 19058–19065. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wu, Y. Molecular mechanism of the sea anemone toxin ShK recognizing the Kv1.3 channel explored by docking and molecular dynamic simulations. J. Chem. Inf. Model. 2007, 47, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.; Gilles, N.; Karbat, I.; Ilan, N.; Gordon, D.; Gurevitz, M. Direct evidence that receptor site-4 of sodium channel gating modifiers is not dipped in the phospholipid bilayer of neuronal membranes. J. Biol. Chem. 2006, 281, 20673–20679. [Google Scholar] [CrossRef] [PubMed]
- Deplazes, E.; Henriques, S.T.; Smith, J.J.; King, G.F.; Mark, A.E.; Craik, D.J.; Schroeder, C. Membrane-binding properties of gating-modifier and pore-blocking toxins: Membrane interaction is not a prerequisite for modification of channel gating. BBA Biomembr. 2016, 1858, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Posokhov, Y.O.; Gottlieb, P.A.; Morales, M.J.; Sachs, F.; Ladokhin, A.S. Is lipid bilayer binding a common property of inhibitor cysteine knot ion-channel blockers? Biophys. J. 2007, 93, L20–L22. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Luo, X.; Kuang, F.; Deng, M.; Wang, M.; Zeng, X.; Liang, S. Synthesis and characterization of huwentoxin-IV, a neurotoxin inhibiting central neuronal sodium channels. Toxicon 2008, 51, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.H.Y.; King, G.F.; Mobli, M. Molecular basis of the interaction between gating modifier spider toxins and the voltage sensor of voltage-gated ion channels. Sci. Rep. 2016, 6, 34333. [Google Scholar] [CrossRef] [PubMed]
- Bemporad, D.; Sands, Z.A.; Wee, C.L.; Grottesi, A.; Sansom, M.S. Vstx1, a modifier of Kv channel gating, localizes to the interfacial region of lipid bilayers. Biochemistry 2006, 45, 11844–11855. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Nishizawa, K. Molecular dynamics simulations of a stretch-activated channel inhibitor GsMTx4 with lipid membranes: Two binding modes and effects of lipid structure. Biophys. J. 2007, 92, 4233–4243. [Google Scholar] [CrossRef] [PubMed]
- Wee, C.L.; Bemporad, D.; Sands, Z.A.; Gavaghan, D.; Sansom, M.S. SGTx1, a Kv channel gating-modifier toxin, binds to the interfacial region of lipid bilayers. Biophys. J. 2007, 92, L07–L09. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Nishizawa, K. Interaction between K+ channel gate modifier hanatoxin and lipid bilayer membranes analyzed by molecular dynamics simulation. Eur. Biophys. J. 2006, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.-H. Effect of gating modifier toxins on membrane thickness: Implications for toxin effect on gramicidin and mechanosensitive channels. Toxins 2013, 5, 456–471. [Google Scholar] [CrossRef] [PubMed]
- Wee, C.L.; Gavaghan, D.; Sansom, M.S.P. Lipid bilayer deformation and the free energy of interaction of a Kv channel gating-modifier toxin. Biophys. J. 2008, 95, 3816–3826. [Google Scholar] [CrossRef] [PubMed]
- Wee, C.L.; Ulmschneider, M.B.; Sansom, M.S.P. Membrane/toxin interaction energetics via serial multiscale molecular dynamics simulations. J. Chem. Theory Comput. 2010, 6, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Agwa, A.J.; Lawrence, N.; Deplazes, E.; Cheneval, O.; Chen, R.M.; Craik, D.J.; Schroeder, C.I.; Henriques, S.T. Spider peptide toxin HwTx-IV engineered to bind to lipid membranes has an increased inhibitory potency at human voltage-gated sodium channel hNaV1.7. BBA Biomembr. 2017, 1859, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; Wood, D.L.A.; Newell, F.; Chaumeil, P.-A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. Arachnoserver 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res. 2010, 39, D653–D657. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deplazes, E. Molecular Simulations of Disulfide-Rich Venom Peptides with Ion Channels and Membranes. Molecules 2017, 22, 362. https://doi.org/10.3390/molecules22030362
Deplazes E. Molecular Simulations of Disulfide-Rich Venom Peptides with Ion Channels and Membranes. Molecules. 2017; 22(3):362. https://doi.org/10.3390/molecules22030362
Chicago/Turabian StyleDeplazes, Evelyne. 2017. "Molecular Simulations of Disulfide-Rich Venom Peptides with Ion Channels and Membranes" Molecules 22, no. 3: 362. https://doi.org/10.3390/molecules22030362