
Quantum Chemical Calculations on CHOP Derivatives—Spanning the Chemical Space of Phosphinidenes, Phosphaketenes, Oxaphosphirenes, and COP− Isomers


Abstract
:1. Introduction
2. Results and Discussion
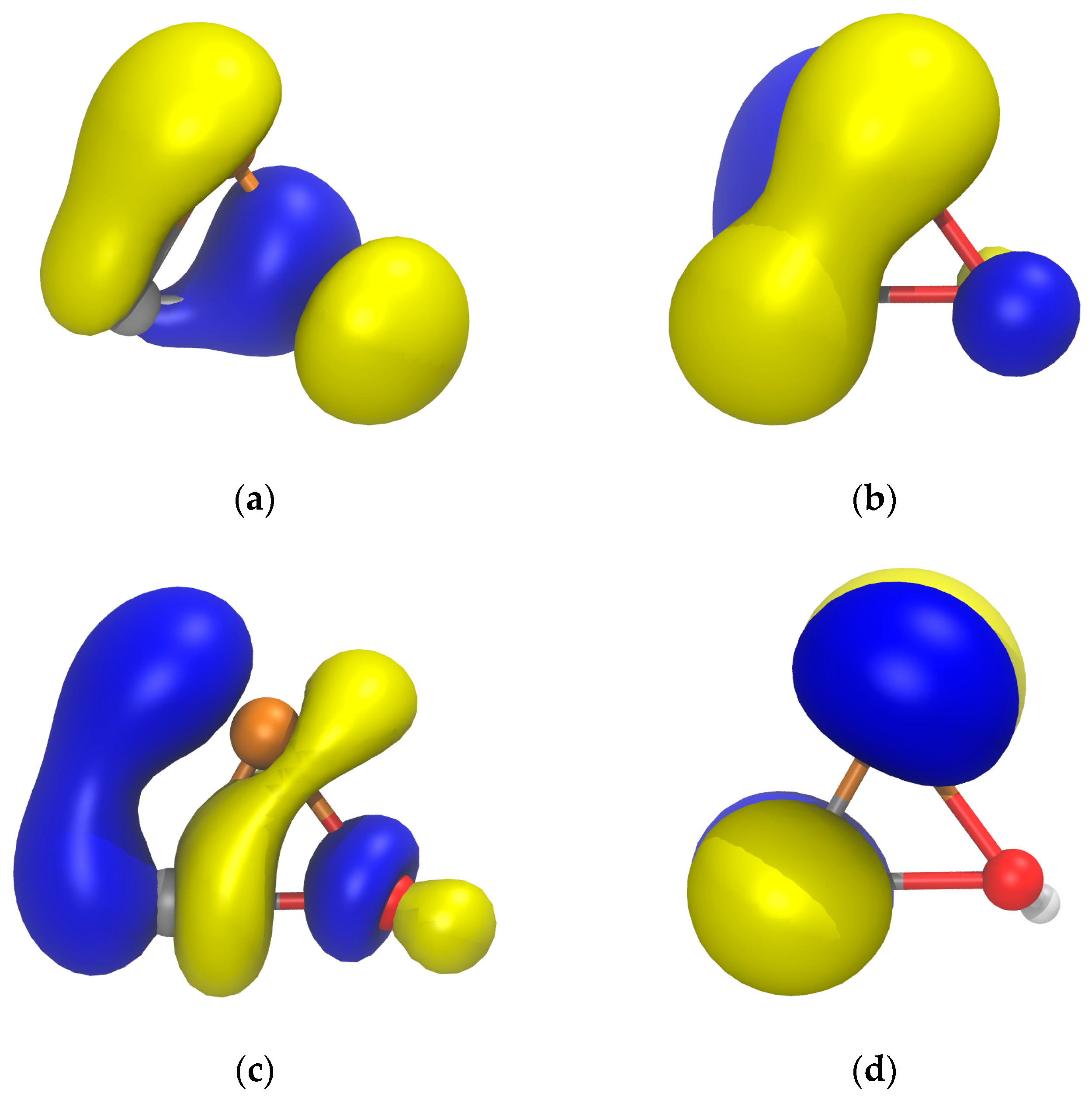
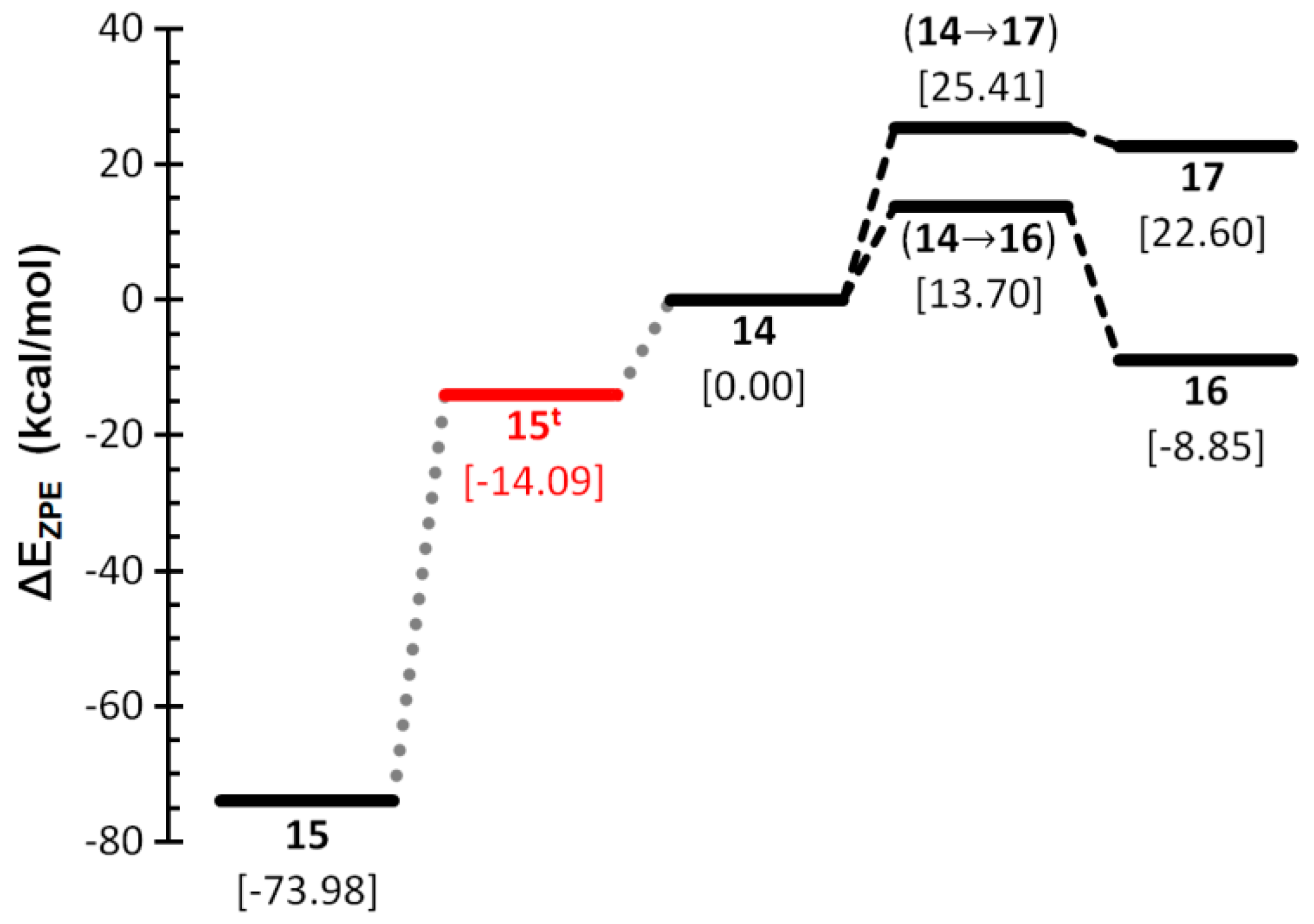
2.1. PES of the Parent Oxaphosphirene (1a)
2.2. Isomers of Substituted Oxaphosphirenes
2.3. Anionic COP− Isomers
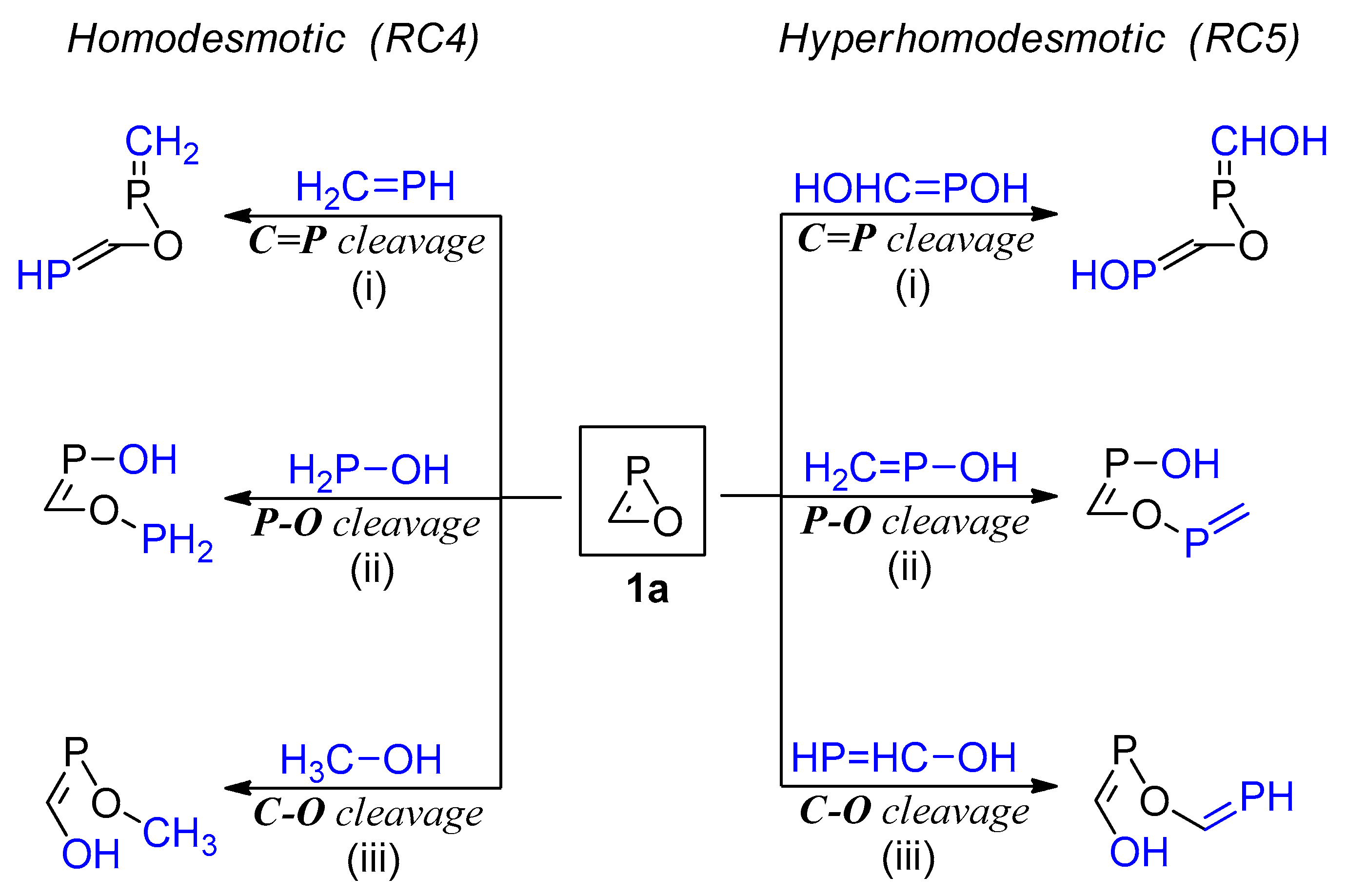
2.4. Ring Strain Energy
3. Materials and Methods
Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mathey, F.; Tran Huy, N.H.; Marinetti, A. Electrophilic terminal-phosphinidene complexes: Versatile phosphorus analogues of singlet carbenes. Helvet. Chim. Acta 2001, 84, 2938–2957. [Google Scholar] [CrossRef]
- Mathey, F. Phosphaorganic chemistry: Panorama and perspectives. Angew. Chem. Int. Ed. 2003, 42, 1578–1604. [Google Scholar] [CrossRef] [PubMed]
- Lammertsma, K. Phosphinidenes. Top. Curr. Chem. 2003, 229, 95–119. [Google Scholar] [CrossRef]
- Regitz, M.; Scherer, O.J. Multiple Bonds and Low Coordination in Phosphorus Chemistry; Thieme: Stuttgart, Germany, 1990; ISBN-10: 3137522013. [Google Scholar]
- Mathey, F.; Regitz, M. Phosphiranes, Phosphirenes, and Heavier Analogues. In Comprehensive Heterocyclic Chemistry II; Elsevier Science Ltd.: Amsterdam, The Netherlands, 1996; Volume 1A, pp. 277–304. [Google Scholar]
- Bauer, S.; Marinetti, A.; Ricard, L.; Mathey, F. Epoxidation of phosphorus-carbon double bond of phosphaalkene complexes: Crystal structure analysis of a stable oxaphosphirane complex. Angew. Chem. Int. Ed. 1990, 102, 1166–1167. [Google Scholar] [CrossRef]
- Streubel, R.; Kusenberg, A.; Jeske, J.; Jones, P.G. Thermisch-induzierte Ringspaltung eines 2H-Azaphosphiren-Wolframkomplexes. Angew. Chem. Int. Ed. Engl. 1994, 33, 2427–2428. [Google Scholar] [CrossRef]
- Dimur, C.; Pauzat, F.; Ellinger, Y.; Berthier, G. Looking for the PC bond in space: HPCO and HPCS as possible tracers. Spectrochim. Acta A 2001, 57, 859–873. [Google Scholar] [CrossRef]
- Fu, H.; Yu, H.; Chi, Y.; Li, Z.; Huang, X.; Sun, C. Theoretical study on the singlet potential energy surface of CHOP system. Chem. Phys. Lett. 2002, 361, 62–70. [Google Scholar] [CrossRef]
- Septelean, R.; Petrar, P.M.; Gabriela, N.; Escudié, J.; Silaghi-Dumitrescu, I. Theoretical study of structural patterns in CH2OP2 isomers. J. Molec. Model. 2010, 17, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R. Antiaromaticity. Acc. Chem. Res. 1973, 6, 393–398. [Google Scholar] [CrossRef]
- Appel, R.; Paulen, W. Das erste Phosphaketen, spektroskopischer Nachweis und Folgereaktionen. Tetrahedron Lett. 1983, 24, 2639–2642. [Google Scholar] [CrossRef]
- Appel, R.; Paulen, W. Das erste stabile Phosphaketen. Angew. Chem. 1983, 95, 807–808. [Google Scholar] [CrossRef]
- Puschmann, F.F.; Stein, D.; Heift, D.; Hendriksen, C.; Gál, Z.A.; Grützmacher, H.-F.; Grützmacher, H. Phosphination of Carbon Monoxide: A Simple Synthesis of Sodium Phosphaethynolate (NaOCP). Angew. Chem. Int. Ed. 2011, 50, 8420–8423. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P.; Zhao, Y. Ab initio study of bonding trends 6. The X≡Y and X=Y=Z species containing phosphorus. Mol. Phys. 1990, 70, 701–714. [Google Scholar] [CrossRef]
- Goicoechea, J.M.; Grützmacher, H. The Chemistry of the 2-Phosphaethynolate Anion. Angew. Chem. Int. Ed. 2018. [Google Scholar] [CrossRef] [PubMed]
- Weber, L. 2-Phospha- and 2-Arsaethynolates–Versatile Building Blocks in Modern Synthetic Chemistry. Eur. J. Inorg. Chem. 2018, 2175–2227. [Google Scholar] [CrossRef]
- Suter, R.; Mei, Y.; Baker, M.; Benkö, Z.; Li, Z.; Grützmacher, H. 2,4,6-Tri(hydroxy)-1,3,5-triphosphinine, P3C3(OH)3: The Phosphorus Analogue of Cyanuric Acid. Angew. Chem. Int. Ed. 2017, 56, 1356–1380. [Google Scholar] [CrossRef] [PubMed]
- Szkop, K.M.; Jupp, A.R.; Suter, R.; Grützmacher, H.; Stephan, D.W. Borane-Stabilized Isomeric Dimers of the Phosphaethynolate Anion. Angew. Chem. Int. Ed. 2017, 56, 14174–14177. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, X.; Benko, Z.; Liu, L.; Ruiz, D.A.; Peltier, J.L.; Bertrand, G.; Su, C.-Y.; Grützmacher, H. N-Heterocyclic Carbenes as Promoters for the Rearrangement of Phosphaketenes to Phosphaheteroallenes: A Case Study for OCP to OPC Constitutional Isomerism. Angew. Chem. Int. Ed. 2016, 56, 6018–6022. [Google Scholar] [CrossRef]
- Hinz, A.; Labbow, R.; Rennick, R.; Schulz, A.; Goicoechea, J.M. HPCO—A Phosphorus-Containing Analogue of Isocyanic Acid. Angew. Chem. Int. Ed. 2016, 56, 3911–3915. [Google Scholar] [CrossRef]
- Mielke, Z.; Andrews, L. Infrared detection of the PCO radical and HPCO molecule. Chem. Phys. Lett. 1991, 181, 355–360. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Hegarty, A.F.; McGinn, M.A.; Ruelle, P. Structure and properties of phosphaketene (H–P=C=O): Phosphorus versus oxygen protonation? J. Chem. Soc. Perkin 2 1985, 1991–1997. [Google Scholar] [CrossRef]
- Thorwirth, S.; Lattanzi, V.; McCarthy, M.C. Phosphorus and silicon analogs of isocyanic acid: Microwave detection of HPCO and HNSiO. J. Molec. Spectr. 2015, 310, 119–125. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Ruelle, P. Comparative SCF study of the nature of the carbon-phosphorus bond in phospha-alkynes, RCP, and of the boron–sulphur bond in sulphidoborons, RBS. J. Chem. Soc. Faraday 2 1984, 80, 1225–1234. [Google Scholar] [CrossRef]
- Ermolaeva, L.V.; Konovalov, A.I. Ab initio study of oxaphosphaalkyne P≡C–OH and its isomers. Russ. Chem. Bull. 1994, 43, 169–170. [Google Scholar] [CrossRef]
- Cheng, X.; Zhao, Y.; Li, L.; Tao, X. Analysis of vibrational spectra of phosphaalkynes R–C≡P (R=-BH2, -CH3, -NH2, -OH) and their isomers based on DFT methods. J. Mol. Struct. Teochem. 2004, 682, 137–143. [Google Scholar] [CrossRef]
- Lattelais, M.; Pauzat, F.; Pilmé, J.; Ellinger, Y. Electronic structure of simple phosphorus containing molecules [C,xH,O,P] candidate for astrobiology (x=1, 3, 5). Phys. Chem. Chem. Phys. 2008, 10, 2089–2097. [Google Scholar] [CrossRef]
- Borisov, E.V.; Mebel, A.M.; Knyazez, B.A.; Zabrodin, V.B.; Korkin, A.A. Comparative non-empirical study and isodesmic calculations of heats of formation of HNCO and HPCO isomers. Russ. Chem. Bull. 1992, 41, 1222–1226. [Google Scholar] [CrossRef]
- Frantzius, G.v.; Espinosa Ferao, A.; Streubel, R. Coordination of CO to low-valent phosphorus centres and other related P–C bonding situations. A theoretical case study. Chem. Sci. 2013, 4, 4309–4322. [Google Scholar] [CrossRef]
- Decius, J.C. Compliance matrix and molecular vibrations. J. Chem. Phys. 1963, 38, 241–248. [Google Scholar] [CrossRef]
- Jones, L.H.; Ryan, R.R. Interaction coordinates and compliance constants. J. Chem. Phys. 1970, 52, 2003–2004. [Google Scholar] [CrossRef]
- Baker, J. A critical assessment of the use of compliance constants as bond strength descriptors for weak interatomic interactions. J. Chem. Phys. 2006, 125, 014103. [Google Scholar] [CrossRef] [PubMed]
- Streubel, R.; Faßbender, J.; Schnakenburg, G.; Espinosa Ferao, A. Formation of transient and stable 1,3-dipole complexes with P,S,C and S,P,C ligand skeletons. Organometallics 2015, 34, 3103–3106. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the Pople-Santry-Segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Mayer, I. Charge, bond order and valence in the AB initio SCF theory. Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar] [CrossRef]
- Mayer, I. Bond order and valence: Relations to Mulliken’s population analysis. Int. J. Quant. Chem. 1984, 26, 151–154. [Google Scholar] [CrossRef]
- Mayer, I. Bond orders and valences in the SCF theory: A comment. Theor. Chim. Acta 1985, 67, 315–322. [Google Scholar] [CrossRef]
- Mayer, I. Modelling of Structure and Properties of Molecules; Maksic, Z.B., Ed.; John Wiley & Sons: New York, NY, USA, 1987. [Google Scholar]
- Bridgeman, A.J.; Cavigliasso, G.; Ireland, L.R.; Rothery, J. The Mayer bond order as a tool in inorganic chemistry. J. Chem. Soc. Dalton Trans. 2001, 2095–2108. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules; Matta, C.F., Boyd, R.J., Eds.; Wiley-VCH: New York, NY, USA, 2007; pp. 1–34. [Google Scholar]
- Espinosa Ferao, A.; Streubel, R. Thiaphosphiranes and their complexes: Systematic study on ring strain and ring cleavage reactions. Inorg. Chem. 2016, 55, 9611–9619. [Google Scholar] [CrossRef] [PubMed]
- Kyri, A.W.; Gleim, F.; García Alcaraz, A.; Schnakenburg, G.; Espinosa Ferao, A.; Streubel, R. ‘Low-coordinate’ 1,2-oxaphosphetanes—A new opportunity in coordination and main group chemistry. Chem Commun. 2018, 54, 7123–7126. [Google Scholar] [CrossRef] [PubMed]
- Espinosa Ferao, A. Kinetic energy density per electron as quick insight into ring strain energies. Tetrahedron Lett. 2016, 57, 5616–5619. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Ford, G.P. Relationship between olefinic π-complexes and three-membered rings. J. Am. Chem. Soc. 1979, 101, 783–791. [Google Scholar] [CrossRef]
- Stirling, C.J.M. Some quantitative effects of strain on reactivity. Pure Appl. Chem. 1984, 56, 1781–1796. [Google Scholar] [CrossRef]
- Krahe, O.; Neese, F.; Streubel, R. The Quest for Ring Opening of Oxaphosphirane Complexes: A Coupled-Cluster and Density Functional Study of CH3PO Isomers and Their Cr(CO)5 Complexes. Chem. Eur. J. 2009, 15, 2594–2601. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Estimating ring strain energies in small carbocycles by means of the Bader’s theory of ‘atoms-in-molecules’. Chem. Phys. Lett. 2012, 536, 165–169. [Google Scholar] [CrossRef]
- Espinosa, A.; Streubel, R. Computational studies on azaphosphiridines and the quest of how to effect ring-opening processes via selective bond activation. Chem. Eur. J. 2011, 17, 3166–3178. [Google Scholar] [CrossRef]
- Espinosa, A.; Gómez, C.; Streubel, R. Single electron transfer-mediated selective endo- and exocyclic bond cleavage processes in azaphosphiridine chromium(0) complexes: A computational study. Inorg. Chem. 2012, 51, 7250–7256. [Google Scholar] [CrossRef]
- Albrecht, C.; Schneider, E.; Engeser, M.; Schnakenburg, G.; Espinosa, A.; Streubel, R. Synthesis and DFT Calculations of Spirooxaphosphirane Complexes. Dalton Trans. 2013, 42, 8897–8906. [Google Scholar] [CrossRef]
- Espinosa, A.; de las Heras, É.; Streubel, R. Oxaphosphirane-borane complexes: Ring strain and migratory insertion reactions. Inorg. Chem. 2014, 53, 6132–6140. [Google Scholar] [CrossRef] [PubMed]
- Villalba Franco, J.M.; Schnakenburg, G.; Sasamori, T.; Espinosa Ferao, A.; Streubel, R. Stimuli-Responsive Frustrated Lewis-Pair-Type Reactivity of a Tungsten Iminoazaphosphiridine Complex. Chem. Eur. J. 2015, 21, 9650–9655. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Houk, K.N.; Schleyer, P.v.R.; Allen, W.D. A Hierarchy of Homodesmotic Reactions for Thermochemistry. J. Am. Chem. Soc. 2009, 131, 2547–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. Available online: https://cec.mpg.de/fileadmin/media/Forschung/ORCA/orca_manual_4_0_1.pdf (accessed on 11 November 2018). [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient approximate and parallel Hartree-Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Basis Set Exchange (BSE). Available online: https://bse.pnl.gov/bse/portal (accessed on 11 November 2018).
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comp. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef] [PubMed]
- Pople, J.A.; Head-Gordon, M.; Raghavachari, K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A. Efficient and accurate local approximations to coupled-electron pair approaches: An attempt to revive the pair natural orbital method. J. Chem. Phys. 2009, 130, 114108. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Hansen, A.; Wennmohs, F.; Grimme, S. Accurate theoretical chemistry with coupled pair models. Acc. Chem. Res. 2009, 42, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Wennmohs, F.; Neese, F. A comparative study of single reference correlation methods of the coupled-pair type. Chem. Phys. 2008, 343, 217–230. [Google Scholar] [CrossRef]
- Grimme, S. Improved second-order Møller–Plesset perturbation theory by separate scaling of parallel- and antiparallel-spin pair correlation energies. J. Chem. Phys. 2003, 118, 9095–9102. [Google Scholar] [CrossRef]
- Grimme, S.; Goerigk, L.; Fink, R.F. Spin-component-scaled electron correlation methods. WIREs Comput. Mol. Sci. 2012, 2, 886–906. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals—Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2011, 7, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys Chem Chem Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef]
- Espinosa Ferao, A. On the mechanism of trimethylphosphine-mediated reductive dimerization of ketones. Inorg. Chem. 2018, 57, 8058–8064. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Molec. Graphics. 1996, 14, 33–38. Available online: http://www.ks.uiuc.edu/Research/vmd/ (accessed on 11 November 2018). [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |















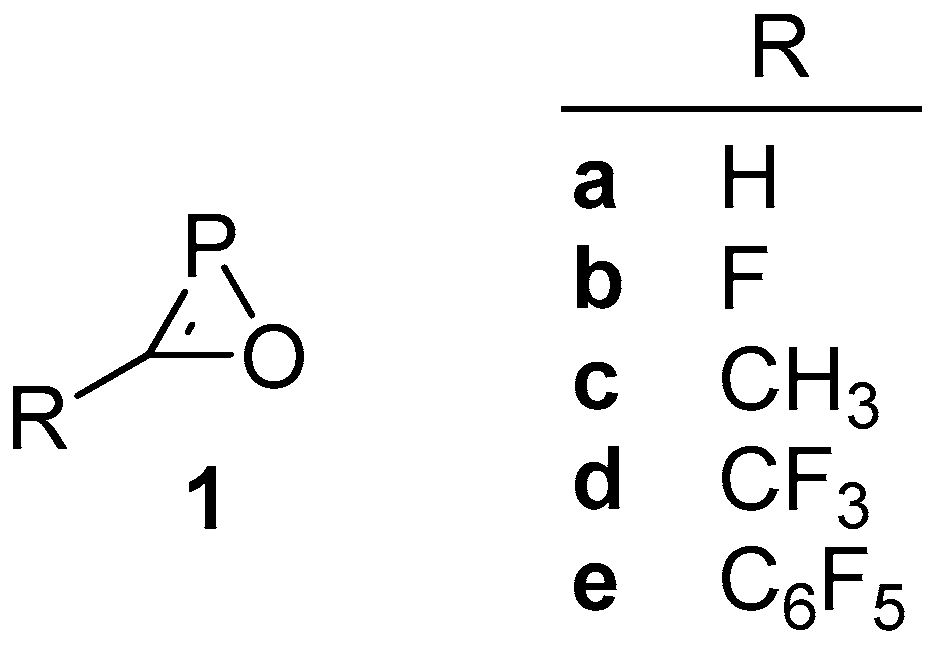{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | d (Å) | WBI | MBO | ρ (au) | G (au) | G/ρ (au) | Atom | qnat (e) | |
|---|---|---|---|---|---|---|---|---|---|
| 1a | P–C P–O C–O | 1.671 2.046 1.288 | 1.571 0.779 1.338 | 1.694 0.785 1.339 | 0.183 1 0.353 | 0.234 1 0.463 | 1.279 1 1.311 | P C O | 0.469 −0.137 −0.525 |
| 2a | P–C P–O C–O | 1.956 1.870 1.229 | 0.997 0.626 1.582 | 0.991 0.735 1.555 | 0.116 0.101 0.393 | 0.044 0.105 0.700 | 0.376 1.041 1.780 | P C O | 0.359 0.236 −0.537 |
| 3asp | P–C P–O | 1.677 1.649 | 2.107 0.818 | 1.984 1.036 | 0.202 0.162 | 0.254 0.246 | 1.253 1.521 | P C O | 0.736 −0.318 −0.909 |
| 3aap | P–C P–O | 1.664 1.644 | 2.238 0.819 | 2.101 1.030 | 0.204 0.165 | 0.251 0.251 | 1.235 1.523 | P C O | 0.726 −0.311 −0.912 |
| 4a | P–C P–O C–O | 1.672 1.773 1.873 | 2.005 0.642 0.520 | 1.917 0.794 0.472 | 0.198 0.126 1 | 0.251 0.164 1 | 1.265 1.306 1 | P C O | 0.589 −0.302 −0.775 |
| 5a | P–C P–O | 1.562 1.475 | 2.421 1.401 | 2.341 1.903 | 0.206 0.233 | 0.322 0.518 | 1.561 2.225 | P C O | 1.540 −0.867 −0.917 |
| 6a | P–C C–O | 1.683 1.152 | 1.674 2.022 | 1.719 2.212 | 0.163 0.477 | 0.248 1.014 | 1.522 2.128 | P C O | 0.049 0.346 −0.424 |
| 7a | P–C C–O | 1.547 1.299 | 2.696 1.115 | 2.833 1.184 | 0.202 0.333 | 0.380 0.450 | 1.878 1.350 | P C O | 0.327 −0.184 −0.641 |
| 8a | P–O C–O | 1.591 1.249 | 0.928 1.309 | 1.197 1.359 | 0.152 0.328 | 0.323 0.737 | 2.125 2.249 | P C O | 0.421 0.084 −0.639 |
| 9a | P–O C–O | 1.919 1.145 | 0.410 1.908 | 0.550 2.079 | 0.074 0.463 | 0.077 1.059 | 1.039 2.289 | P C O | 0.081 0.538 −0.558 |
| syn-6at | P–C C–O | 1.941 1.169 | 0.851 1.978 | 0.965 2.202 | 0.130 1.113 | 0.043 0.506 | 0.332 0.454 | P C O | 0.298 0.223 −0.454 |
| 6at | P–C C–O | 1.963 1.167 | 0.815 1.991 | 0.950 2.199 | 0.125 0.458 | 0.038 0.952 | 0.308 2.077 | P C O | 0.297 0.225 −0.460 |
| 12at | P–C C–O | 1.846 1.212 | 1.016 1.821 | 1.142 1.994 | 0.161 0.418 | 0.103 0.736 | 0.640 1.759 | P C O | 0.339 0.014 −0.473 |
| 14 | P–C P–O C–O | 1.820 1.967 1.278 | 1.471 0.728 1.370 | 1.572 0.735 1.270 | 0.146 1 0.351 | 0.086 1 0.537 | 0.589 1 1.530 | P C O | −0.249 −0.123 −0.628 |
| 15 | P–C C–O | 1.618 1.199 | 2.241 1.638 | 2.581 1.876 | 0.178 0.426 | 0.307 0.768 | 1.718 1.802 | P C O | −0.436 0.088 −0.652 |
| 16 | P–C P–O | 1.597 1.519 | 2.847 1.156 | 2.727 1.571 | 0.185 0.211 | 0.261 0.430 | 1.414 2.036 | P C O | 1.146 −1.078 −1.068 |
| 17 | P–O C–O | 1.756 1.179 | 0.647 1.662 | 0.782 1.784 | 0.100 0.408 | 0.168 0.955 | 1.685 2.338 | P C O | −0.528 0.110 −0.583 |
| 15t | P–C C–O | 1.792 1.216 | 1.377 1.639 | 1.544 1.859 | 0.152 0.405 | 0.142 0.735 | 0.933 1.815 | P C O | −0.350 −0.037 −0.613 |
| RSERC4 | RSERC5 | |
|---|---|---|
| 1a | 49.90 | 49.08 |
| 2a | 7.53 | 4.59 |
| 14 | 36.83 | 41.99 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rey, A.; Espinosa Ferao, A.; Streubel, R. Quantum Chemical Calculations on CHOP Derivatives—Spanning the Chemical Space of Phosphinidenes, Phosphaketenes, Oxaphosphirenes, and COP− Isomers. Molecules 2018, 23, 3341. https://doi.org/10.3390/molecules23123341
Rey A, Espinosa Ferao A, Streubel R. Quantum Chemical Calculations on CHOP Derivatives—Spanning the Chemical Space of Phosphinidenes, Phosphaketenes, Oxaphosphirenes, and COP− Isomers. Molecules. 2018; 23(12):3341. https://doi.org/10.3390/molecules23123341
Chicago/Turabian StyleRey, Alicia, Arturo Espinosa Ferao, and Rainer Streubel. 2018. "Quantum Chemical Calculations on CHOP Derivatives—Spanning the Chemical Space of Phosphinidenes, Phosphaketenes, Oxaphosphirenes, and COP− Isomers" Molecules 23, no. 12: 3341. https://doi.org/10.3390/molecules23123341