An Efficient Chemoenzymatic Approach towards the Synthesis of Rugulactone


Department of Chemistry, University of Crete, Vasilika Vouton, 71003 Heraklion, Crete, Greece
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(3), 640; https://doi.org/10.3390/molecules23030640
Submission received: 13 February 2018
/
Revised: 5 March 2018
/
Accepted: 7 March 2018
/
Published: 12 March 2018
(This article belongs to the Section Bioorganic Chemistry)
Abstract
:Rugulactone is a natural product isolated from the plant Cryptocarya rugulosa. It has shown very important biological activity as an inhibitor of the nuclear factor κB (NF-κB) activation pathway. A new chemoenzymatic approach towards the synthesis of rugulactone is presented here. The chirality, induced to the key intermediate by a stereoselective enzymatic reduction utilizing NADPH-dependent ketoreductase, is described in detail.
1. Introduction
Rugulactone 1 is a secondary metabolite, isolated in 2009 from organic extracts of the plant Cryptocarya rugulosa [1]. It has shown very important biological activity and has been reported to exhibit up to five-fold induction of inhibitor of κB (IκB) proteins of nuclear factor κB (NF-κB) at 25 μg/mL [1]. The transcription factor NF-κB was discovered in 1986 as a nuclear factor regulating the formation of the κ variety of the light chain (one of two subunits from which immunoglobin is built) [2]. Soon afterwards, it became clear that these proteins, which harbor this specific DNA binding activity, are expressed in nearly all cell types. It is well known that NF-κB affects most aspects of cellular physiology—from immunity and inflammation to apoptosis, cell survival, growth, and proliferation [3,4,5,6,7]. Consistent with this role, incorrect regulation of NF-κB has been linked to cancer [8,9] and inflammatory [10,11] and autoimmune [12] diseases. Thus, there is an intensive necessity for the exploration of new biomimetic compounds that can inhibit and control NF-κB activity effectively.
Rugulactone is a rather new dihydro α-pyrone that inhibits the NF–κB activation pathway. This natural product shows an interesting structural pattern with two electrophilic groups of potential Michael acceptors (highlighted in red, Figure 1), which covalently bind to target protein active sites, an α,β-unsaturated γ-lactone together with an α,β-unsaturated ketone. Due to the remarkable inhibitory properties of rugulactone, it has been a challenging subject of research for synthetic organic chemists. Recent studies have verified that many analogues [13,14] and derivatives [8] of rugulactone demonstrate similar or better antibacterial and antifungal activity as compared to the natural product, even with the opposite configuration of the natural product. Several synthetic schemes have been reported where chirality has been induced using various methods [13,14,15,16,17,18,19,20,21,22], such as a chiral pool [20], proline-catalyzed α-aminooxylation of aldehydes [13], Jacobsen’s hydrolytic kinetic resolution of epoxides [16], Keck’s asymmetric allylation [17], allylation of carbonyl compounds with chiral boronic esters [19], or asymmetric catalytic Overman esterification [14]. Also, a chemoenzymatic synthesis was reported by Fadnavis et al. [18], involving an enzymatic kinetic resolution of racemic homoallylic alcohols employing Candida rugosa lipase. However, in this case, the maximum conversion to the desired enantiomer could not exceed 50%. The majority of all of these synthetic routes include several synthetic steps and utilize expensive starting materials and peculiar reagents.
Biocatalysis has long been known as an alternative technology, capable of delivering highly stereo-, chemo-, and regioselective transformations [23,24]. Recent contributions demonstrate the broad diversity of impressive opportunities for chemoenzymatic processes, which underline their potential as valuable solutions for current synthetic challenges in the synthesis of valuable chiral building blocks towards the production of pharmaceuticals and biologically active natural products [23,24,25,26].
As a part of our interest in applications of bioreductions in the synthesis of biologically interesting compounds, we report here a new chemoenzymatic approach towards the synthesis of rugulactone 1.
2. Results and Discussion
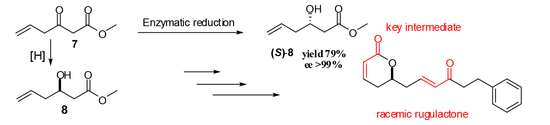
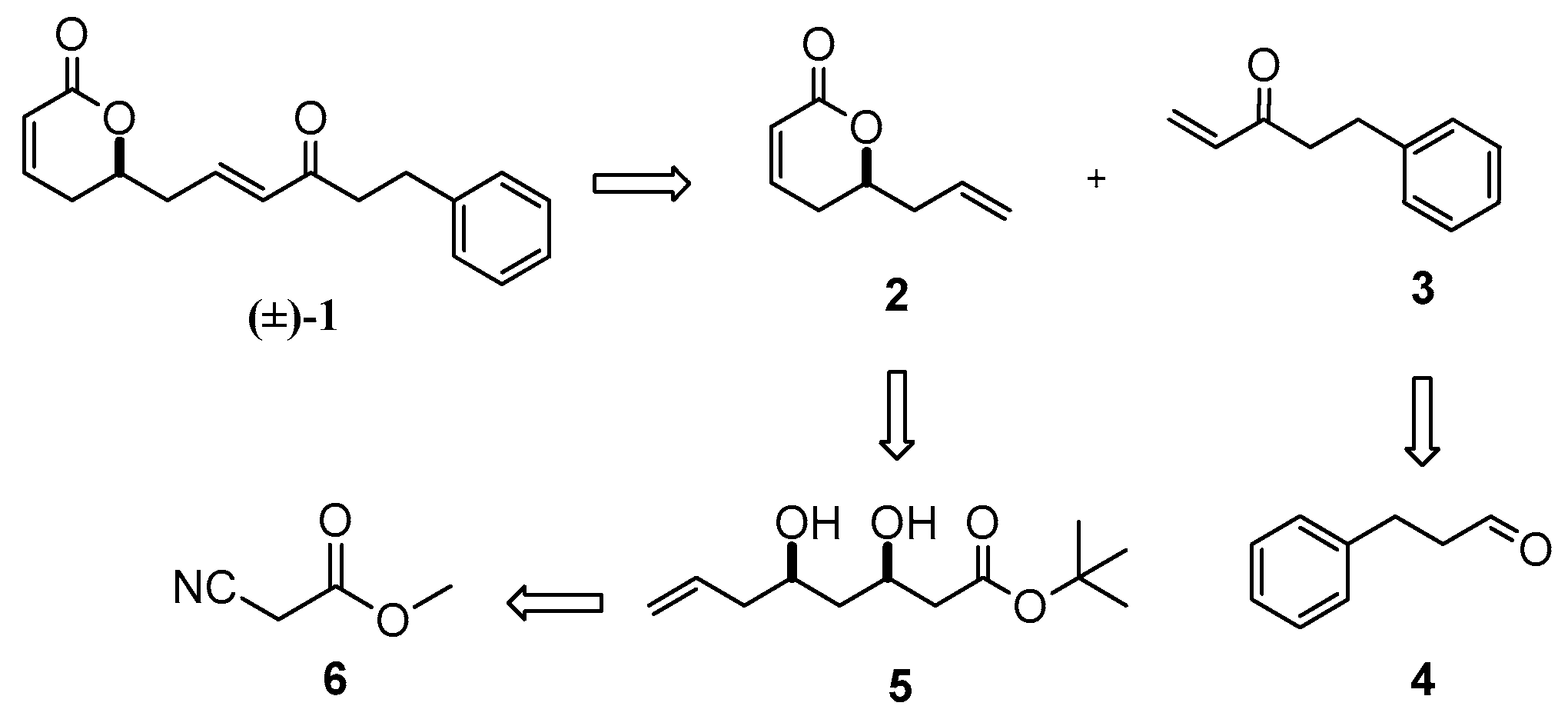
Our synthetic approach consisted of eight simple synthetic steps utilizing inexpensive starting materials. The chirality of the natural product was achieved by a stereoselective enzymatic reduction utilizing NADPH-dependent ketoreductase. Initially, the racemic rugulactone was synthesized according to the retrosynthetic approach shown in Scheme 1.
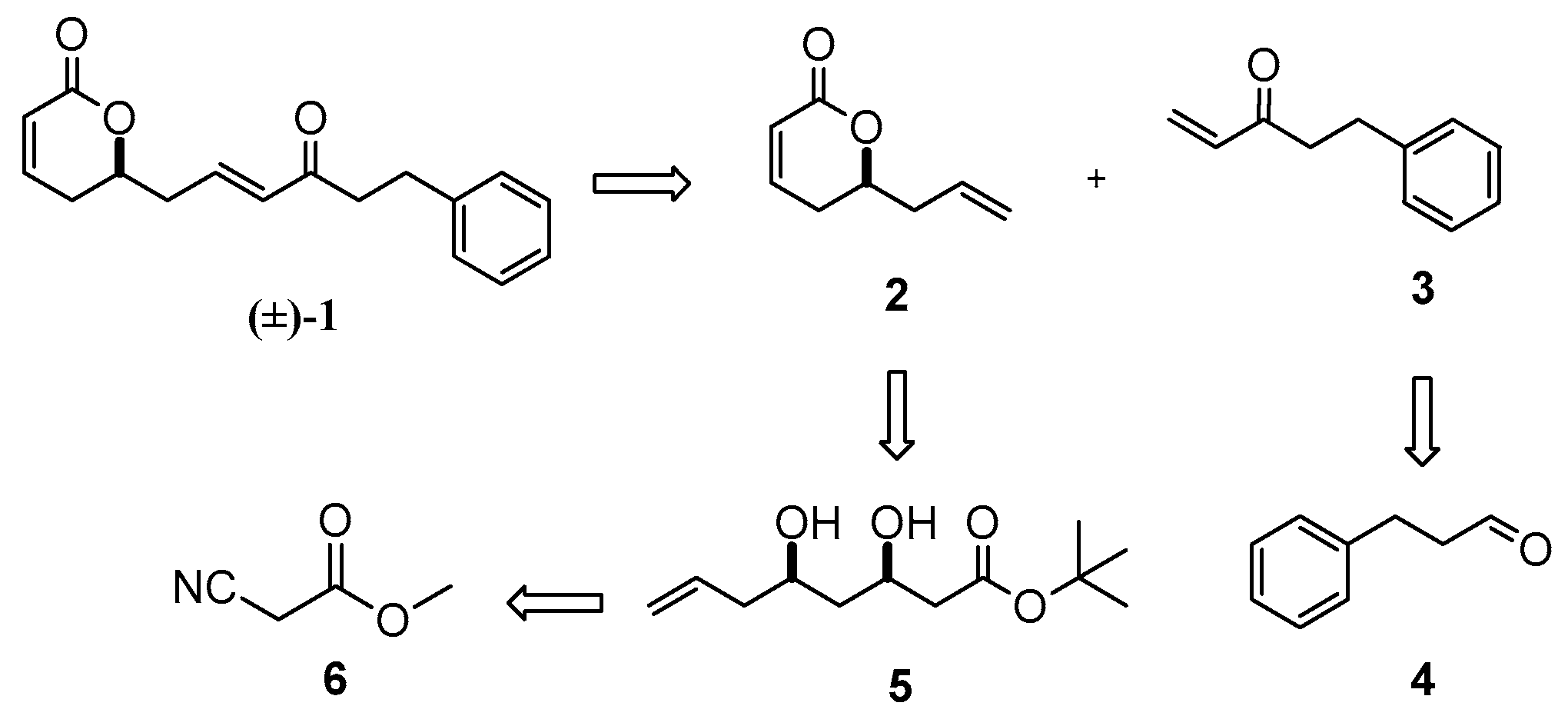
Rugulactone (±)-1 can be derived from a Grubbs’ cross metathesis between the two terminal alkenes 2 and 3. Lactone 2 can be synthesized from the dihydroxy ester 5, which can be derived from the commercially available methyl-2-cyanoacetate 6. On the other hand, enone 3 can be obtained easily from the commercially available aldehyde 4.
Initially, the synthesis of intermediate 5 was achieved through keto ester 7 (Scheme 2). The synthesis of compound 7 was reported many years ago [27,28]. Unfortunately, in our case, the classic C–C coupling involving Claisen or Grignard, as well as Reformatsky and aldol, reactions led to unexpected byproducts. Therefore, these synthetic methods were not useful. Next, we chose an alternative and simpler approach for the synthesis of 7, utilizing an organometallic Barbier reaction [29]. The carbonyl moiety was then reduced by NaBH4 in MeOH to furnish the corresponding hydroxy ester 8. In addition, compound 5 was obtained from a Claisen reaction between the hydroxy ester 8 and commercially available tert-butyl acetate, followed by a carbonyl reduction. Furthermore, a single-step cyclization of dihydroxy compound 5 with simultaneous dehydration, catalyzed by p-toluenesulfonic acid, led to γ-lactone 2 in good yield (Scheme 2).
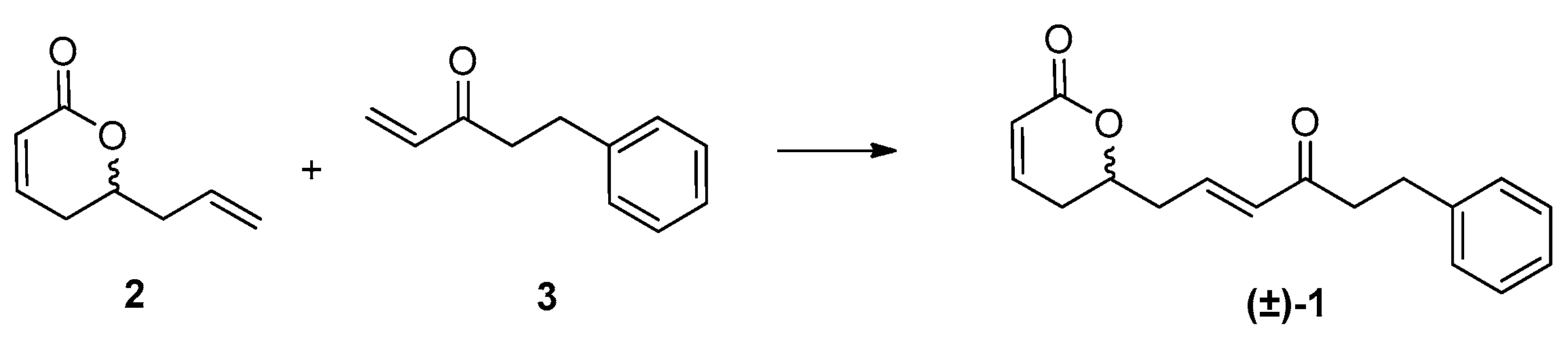
Furthermore, enone 3 was synthesized from commercially available 3-phenylpropanal 4 in two steps involving alkylation of the aldehyde through Grignard vinylation followed by 2-iodoxybenzoic acid (IBX) oxidation of the resulting allylic alcohol 9 (Scheme 2). Finally, the cross-metathesis of the two terminal alkenes—the lactone 2 and the enone 3—catalyzed by Grubbs (II) catalyst [30], led successfully to the formation of (±)-rugulactone 1 in a good reaction yield of 75%, whereas the overall yield of the synthesis was found to be 21% (Scheme 3). The spectroscopic data are identical to those reported in the literature [1].
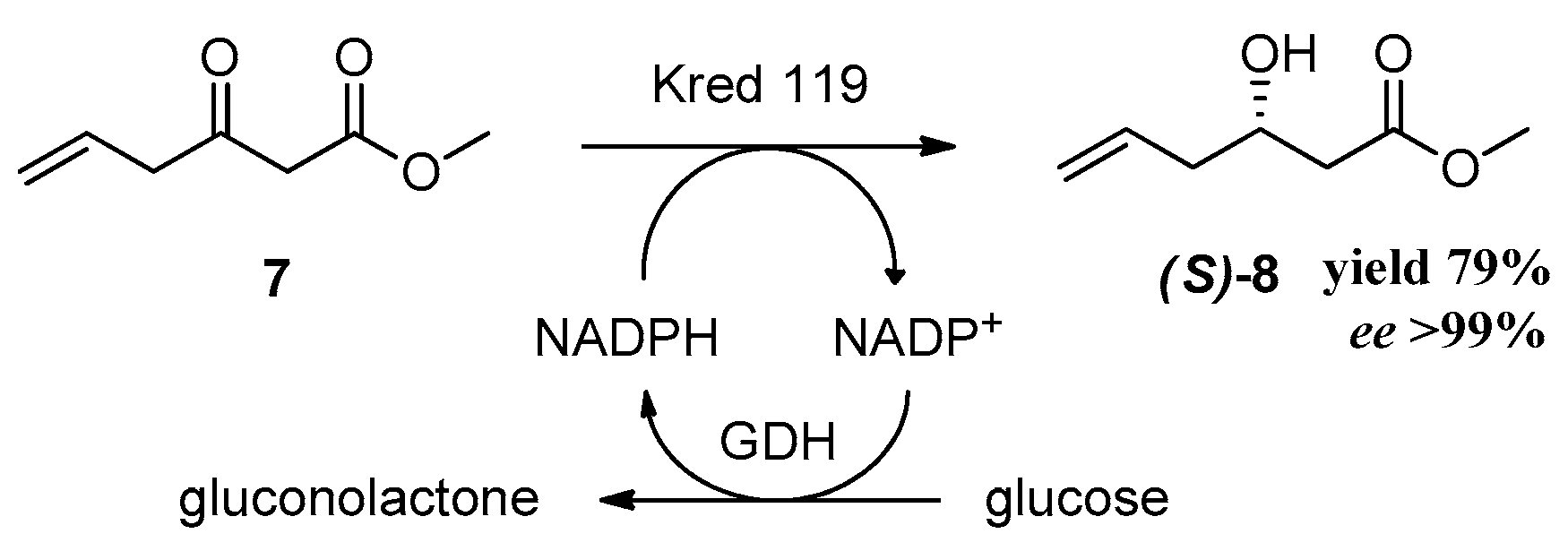
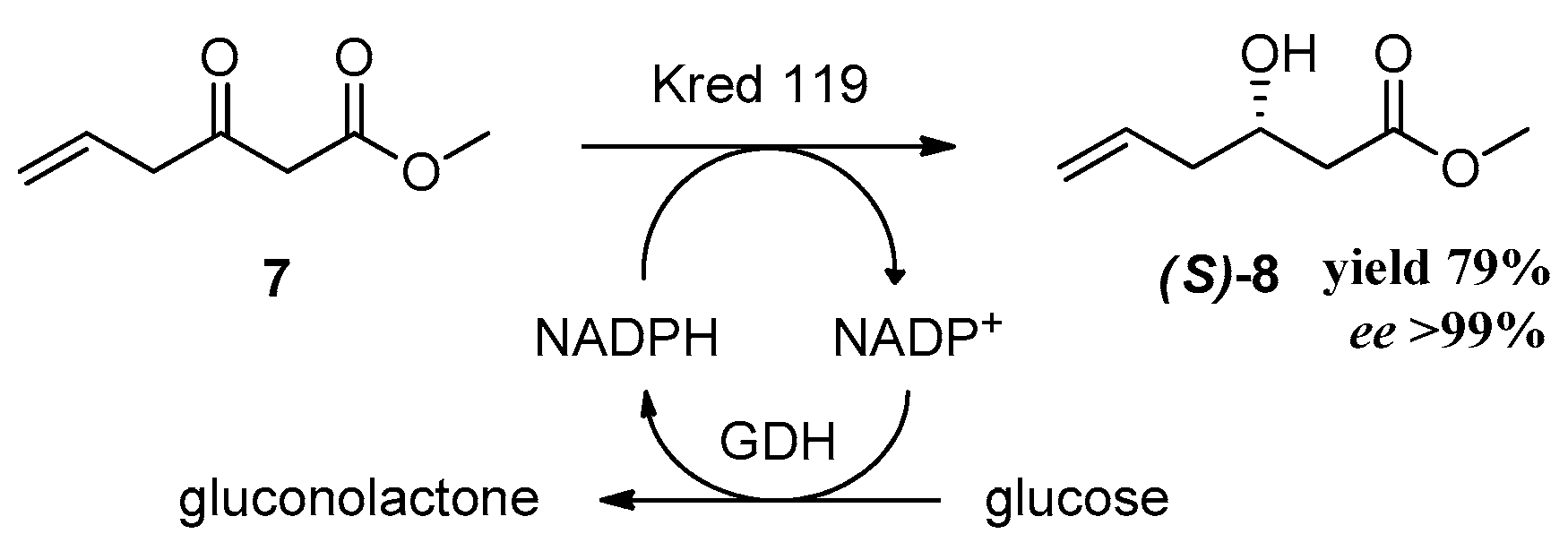
After the successful completion of this attractive synthetic procedure of racemic rugulactone, we applied this methodology for the synthesis of the optically active rugulactone 1 by the intervention of a key enzymatic step. Our research group has lengthy experience in biocatalytic reductions of various carbonyl compounds and their applications for the chemoenzymatic synthesis of natural products with biological activity [31,32]. Moreover, we and other research groups have shown that reductive enzymes, like ketoreductases, are valuable and powerful catalysts in the synthesis of optically active intermediates and precursors for many pharmaceuticals [33,34,35,36,37,38,39,40]. In the present work, the asymmetric reduction of the keto ester 7 using NADPH-dependent ketoreductases was the key step introducing the chirality of the natural product 1. Before conducting the screening for active enzymes, we tested the stability of 7 under the usual enzymatic reaction conditions (phosphate buffer 200 mM, pH 6.9, and 37 °C) [33,34]. Unfortunately, under those conditions, substrate 7 was decomposed, as it was observed by the 1H-NMR spectroscopy. After the clarification of the optimum conditions (phosphate buffer 200 mM, pH 6.9, and temperature 5–8 °C), the stereoselective reduction of 7 was carried out with several ketoreductases to screen and identify the most suitable biocatalyst for the desirable transformation. The enzymes chosen for the screening have displayed high activity in structurally similar carbonyl substrates [32,36,37]. The well-established system for the NADPH recycling was applied in all enzymatic reactions (Scheme 4) [33]. Many enzymes demonstrated activity towards the reduction of 7, and the best results are shown in Table 1.
Ketoreductases (Kreds) A1C, A1D, B1F, 101, and 119 showed excellent activity (>99% conv.) towards the reduction of keto ester 7 (conv > 99%). Kreds A1C, A1D, and 119 displayed excellent enantioselectivity (ee > 99%) (Scheme 4), whereas Kreds B1F and 101 showed reasonable enantioselectivity. All positive enzymes catalyzed efficiently the reduction to form the optically active hydroxy ester 8. The absolute configuration of the hydroxy ester was determined by assigning the stereochemistry of the hydroxy group by the use of methoxy phenyl acetic esters (MPA esters) [41] and was found to be the (S)-8 enantiomer. Compound 8 is the key chiral synthon for the chemoenzymatic synthesis of the (S)-epimer of natural product 1 according to our proposed synthesis (Scheme 2). The synthesis of this epimer is very useful since it has demonstrated high biological activity like many analogues [15] and derivatives [8].
It is worth noting that under the enzymatic reaction conditions, substrate 7 showed a double bond isomerization to form a small amount (6%) of compound 7a, as it was observed by 1H-NMR spectroscopy (Scheme 5).
For this reason, the final product of the enzymatic reduction in every case was a mixture of the desired optically active hydroxy ester 8 and a side product derived from the double bond reduction as well as the carbonyl moiety reduction of the isomer 7a. This observation was in accordance with our previous unpublished results with several ketoreductases, which can also reduce α,β-unsaturated keto moieties together with the double bond reduction. This can most likely be attributed to contaminations of these commercially available enzymes.
3. Materials and Methods
3.1. General
Unless otherwise noted, all solvents and reagents were purchased from Sigma-Aldrich (Munich, Germany) in the highest purity and were used without any further purification. Dry tetrahydrofuran (THF) was used after distillation into a Soxhlet in the presence of metallic Na and benzophenone. Kreds, glucose dehydrogenase, and NADPH were commercially available (Codexis, Redwood City, CA, USA). NMR spectra were generally recorded at room temperature on Bruker Avance series 500 and 300 spectrometers (Billerica, MA, USA). Chemical shifts (δ) are reported in ppm relative to the residual solvent peak (CDCl3, δ: 7.26, 13CDCl3, δ: 77.0), and the multiplicity of each signal is designated by the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. Coupling constants (J) are quoted in Hz. 1H- and 13C-NMR spectra of compounds 1–3, 5, and 7–9, and 1H-NMR of (R)-MPA and (S)-MPA esters of (S)-methyl 3-hydroxyhex-5-enoate can be found in Supplementary Materials. Thermo LTQ-Orbitap XL with an electron transfer dissociation (ETD) ion trap mass spectrometer (Waltham, MA, USA) was used for the high resolution mass spectra (HRMS). Column chromatographic separations were carried out of a flash chromatography system using silica gel and hexane/ethyl acetate or petroleum ether/ethyl acetate solvent mixtures. For thin layer chromatography (TLC), Merck silica gel (grade 60 F245, Merck & Co., Kenilworth, NJ, USA) was used. The progress of the enzymatic reactions and the selectivities were determined by gas chromatography using SHIMADZU GC-2014 gas chromatograph (Shimadzu, Japan) equipped with an flame ionization detector (FID) detector (HP GC column: 30 m × 0.25 mm × 0.25 μm chiral capillary column, 20% permethylated, Cyclodextrin-B, Part No. 19091G-B233).
3.2. Synthetic Procedures
3.2.1. Methyl 3-Oxohex-5-enoate (7)
Aluminum trichloride (160 mg, 1.2 mmol) was added all at once to a solution of zinc powder (780 mg, 12 mmol), methyl 2-cyanoacetate (267 μL, 3 mmol), and crotyl bromide (390 μL, 4.5 mmol) in anhydrous THF (12 mL) at 0 °C (ice-water bath). The reaction mixture was warmed to room temperature and then stirred at room temperature. After the reaction was completed (monitored by TLC), aqueous HCl (2 M, 5 mL) was added to the reaction mixture and was stirred at room temperature for 5 min. The reaction mixture was passed through a short silica gel column and the organic solvent was removed directly under reduced pressure. Further purification was achieved by flash column chromatography (hexane/EtOAc, v/v, 30/1) to give the corresponding ester 7 with 52% isolated yield (222 mg). 1H-NMR (500 MHz; CDCl3; Me4Si): δ 5.87–5.95 (m, 1H), 5.22–5.25 (m,1H), 5.16–5.20 (m, 1H), 3.74 (s, 3H), 3.50 (s, 2H), 3.305 (d, J = 7.0 Hz, 2H). 13C-NMR (500 MHz, CDCl3): δ 200.5, 167.45, 129.5, 119.8, 52.4, 48.3, 47.7.
3.2.2. Methyl 3-Hydroxyhex-5-enoate (8)
Sodium borohydride (0.26 mmol, 10 mg) was added in dry ethanol (10 mL) under nitrogen, and the mixture was cooled to 0 °C. After stirring for 5 min a solution of dry methanol (5 mL) containing methyl 3-oxohex-5-enoate 7 (111 mg, 0.78 mmol) was added dropwise. After stirring for 4 h at 0 °C, the reaction was quenched with saturated ammonium chloride, and methanol was concentrated by rotor evaporator. Then, water (20 mL) was added and extracted twice with EtOAc (2 × 20 mL). The organic layer was dried over MgSO4 and the solvents were evaporated to dryness. The residue was purified by flash column chromatography (hexane/EtOAc, v/v, 20/1) to afford the methyl 3-hydroxyhex-5-enoate 8 with 95% yield (107 mg). 1H-NMR (500 MHz; CDCl3; Me4Si; δ ppm): δ 5.78–5.86 (m, 1H), 5.13–5.16 (m, 1H), 5.11–5.12 (m, 1H), 4.06–4.12 (m, 1H), 3.71 (s, 3H), 2.53 (dd, J1 = 16.6 Hz, J2 = 3.5 Hz, 1H), 2.44 (dd, J1 = 16.6 Hz, J2 = 9 Hz, 1H), 2.23–2.33 (m, 2H). 13C-NMR (500 MHz, CDCl3): δ 173.2, 133.9, 118.2, 67.3, 51.8, 40.9, 40.4.
3.2.3. tert-Butyl 3,5-dihydroxyoct-7-enoate (5)
Under nitrogen atmosphere, dry diisopropyl amine (621 μL, 4.72 mmol) was dissolved in dry THF (5 mL). The mixture was cooled to 0 °C and BuLi 1.6 M in hexane (3.0 mL, 4.4 mmol) was added dropwise. After stirring for 20 min at 0 °C the mixture was cooled to −78 °C and a solution of tert-butyl acetate (439 mg, 2 mmol) in dry THF (2 mL) was added and the mixture was stirred at −78 °C for 20 min. Then, the mixture was cooled to −78 °C and a solution of methyl 3-hydroxyhex-5-enoate 8 (340 mg, 2.36 mmol) in dry THF (3 mL) was added and the reaction mixture was stirred until the completion of the reaction, which was observed by TLC analysis of reaction aliquots. The reaction mixture was quenched with saturated NH4Cl (20 mL) and was extracted with EtOAc (2 × 15 mL). The combined organic layers were dried over MgSO4 and evaporated to dryness. Then, the crude product was dissolved in dry methanol (5 mL) and the methanol mixture was added dropwise to a stirring solution of sodium borohydride (45 mg, 0.87 mmol) in dry methanol (8 mL) under nitrogen at 0 °C. The reaction mixture was stirred until the completion of the reaction monitored by TLC. The reaction was quenched with saturated ammonium chloride, and the methanol was concentrated by rotor evaporator. Then, water (20 mL) was added and the aqueous layer was extracted twice with EtOAc (2 × 20 mL). The organic layer was dried over MgSO4 and the solvents were evaporated to dryness. The residue was purified using silica gel chromatography (hexane/EtOAc, v/v, 15/1) to afford the tert-butyl 3,5-dihydroxyoct-7-enoate 5 with 79% overall yield (363 mg). 1H-NMR (500 MHz; CDCl3; Me4Si): δ 5.78–5.86 (m, 1H), 5.07–5.14 (m, 2H), 4.20–4.42 (m, 1H), 3.90–4.15 (m, 1H), 2.36–2.63 (m, 2H), 2.20–2.36 (m, 2H), 1.52–1.70 (m, 2H), 1.46 (s, 9H). 13C-NMR (500 MHz, CDCl3): δ 172.4, 172.08, 134.6, 134.5, 118, 117.8, 81.5, 71.1, 69.0, 67.5, 65.7, 43.2, 42.5, 42.1, 42.0, 41.5, 41.4, 28.1. HRMS (TOF-ESI) m/z: [M + H]+ Calcd. for C12H22O4+H, 231.1552; Found 231.1592.
3.2.4. Allyl-5,6-dihydro-2H-pyran-2-one (2)
Under nitrogen atmosphere, tert-butyl 3,5-dihydroxyoct-7-enoate 5 (100 mg, 0.46 mmol) was dissolved in dry toluene (8 mL) and p-toluenesulfonic acid monohydrate (45 mg, 0.26 mmol) was added. After stirring the solution at room temperature for 5 min, the reaction mixture was refluxed for 5 h. After completion of the reaction, the reaction mixture was washed twice with sodium carbonate (2 × 8 mL). The organic layer was dried over MgSO4, and the solvent was evaporated to dryness. Pure 6-allyl-5,6-dihydro-2H-pyran-2-one 2 was obtained after silica gel chromatography (hexane/EtOAc v/v, 10/1) with 97% yield (62 mg). 1H-NMR (500 MHz; CDCl3; Me4Si; δ ppm): δ 6.86–6.90 (m, 1H), 6.02 (d, J = 9.7 Hz, 1H), 5.79–5.88 (m, 1H), 5.15–5.19 (m, 2H), 4.46–4.51 (m, 1H), 2.53–2.59 (m, 1H). 2.44–2.50 (m, 1H), 2.33–2.36 (m, 2H) 13C-NMR (500 MHz, CDCl3): δ 164.3, 145.0, 132.2, 121.4, 118.9, 77.0, 39.0, 28.6.
3.2.5. 5-Phenylpent-1-en-3-ol (9)
To an ice-cold solution of 3-phenylpropanal 8 (700 μL, 5.14 mmol) and dry THF (8 mL), vinyl magnesium chloride 1 M (1.5 mL, 1.4 mmol) was added dropwise and the reaction was stirred at room temperature for 3 h under nitrogen dry atmosphere. When the reaction was complete (TLC), it was quenched with saturated aqueous NH4Cl, and the mixture was extracted with Et2O (3 × 50 mL). The combined organic layers were dried (MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography (hexane/EtOAc v/v, 20/1) to produce the corresponding homoallylic alcohol as a clear liquid with 86% isolated yield (195 mg). 1H-NMR (500 MHz; CDCl3; Me4Si): δ 7.27–7.30 (m, 2H), 7.17–7.216 (m, 3H), 5.9 (ddd, J1 = 17.2 Hz, J2 = 10.4 Hz, J3 = 6.2 Hz, 1H), 5.23–5.27 (m, 1H), 5.12–5.15 (m, 1H), 4.11–4.14 (m, 1H), 2.66–2.78 (m, 2H), 1.80–1.91 (m, 2H). 13C-NMR (300 MHz, CDCl3): δ 141.8, 141.0, 128.5, 128.40, 125.9, 115.0, 72.5, 38.5, 31.6.
3.2.6. 5-Phenylpent-1-en-3-one (3)
The homoallylic alcohol 9 obtained above (0.5 mL, 4.92 mmol) was added to a stirred solution of IBX (1.4 g, 5.14 mmol) in dry DMSO (10 mL) at room temperature, and the mixture was stirred for 6 h. When the reaction was complete, the mixture was filtered, diluted with H2O (10 mL), and extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were washed with brine (20 mL), dried (MgSO4), and concentrated in vacuo. Purification by column chromatography (petroleum ether/EtOAc v/v, 25/1) furnished compound 3 with 88% isolated yield (693 mg). 1H-NMR (500 MHz; CDCl3; Me4Si): δ 7.28–7.30 (m, 2H), 7.19–7.21 (m, 3H), 6.36 (dd, J1 = 17.8 Hz, J2 = 10.6 Hz, 1H)), 6.22 (d, J = 17.8 Hz, 1H), 5.83 (d, J = 10.6 Hz, 1H), 2.90–2.98 (m, 4H). 13C-NMR (500MHz, CDCl3): δ 199.8, 141.0, 136.5, 128.5, 128.33, 128.27, 126.1, 41.2, 29.8.
3.2.7. (E)-6-(4-Oxo-6-phenylhex-2-enyl)-5,6-dihydro-2H-pyran-2-one (1)
Under nitrogen atmosphere, Grubbs’ (II) catalyst (15 mg, 5 mol %) was added to a stirred solution of 6-allyl-5,6-dihydro-2H-pyran-2-one 2 (50 mg, 0.36 mmol) and 5-phenylpent-1-en-3-one 3 (174 mg, 1.1 mmol) in dry CH2Cl2 (10 mL), and the reaction mixture was stirred at 45 °C for 12 h. After completion of the reaction, the solvent was evaporated to dryness and the residue was purified using silica gel chromatography (hexane/EtOAc, v/v, 4/1) to afford 1 with 75% isolated yield (73 mg). 1H-NMR (500 MHz; CDCl3; Me4Si): δ 7.26–7.29 (m, 2H), 7.17–7.20 (m, 3H), 6.86–6.90 (m, 1H), 6.77–6.83 (m, 1H), 6.19 (d, J = 15.9 Hz), 6.04 (dt, J1 = 9.8 Hz, J2 = 1.8 Hz, 1H), 4.52–4.57 (m, 1H),2.88–2.96 (m, 4H), 2.58–2.70 (m, 2H), 2.32–2.34 (m, 2H). 13C-NMR (500 MHz, CDCl3): δ 199.0, 163.7, 144.6, 141.0, 140.0, 133.5, 128.5, 128.4, 126.1, 121.5, 76.0, 41.7, 37.5, 29.9, 28.9.
3.3. General Procedure for Enzymatic Reductions
The reductions were performed as follows: In a phosphate buffered solution (1 mL, 200 mM, pH 6.9), the substrate (5 mg, 0.035 mmol), the corresponding Kred (2 mg), glucose (21 mg), glucose dehydrogenase (2 mg), and NADPH (2 mg, 2.5 mM) were added. The reactions were incubated at 3–8 °C. After completion of the reactions, the products were isolated by extracting the crude reaction mixture with EtOAc (3 × 1.5 mL). The combined organic layers were dried over MgSO4 and evaporated to dryness.
(S)-Methyl 3-hydroxyhex-5-enoate, (S)-8
The enzymatic reduction of methyl 3-oxohex-5-enoate 7 catalyzed by Kred-119 was completed after 3 h and the optically active alcohol 8 was produced with 79% yield and > 99% ee. The spectroscopic data are identical to those of compound 8. 1H-NMR (500 MHz; CDCl3; Me4Si): δ 5.78–5.86 (m, 1H), 5.13–5.16 (m, 1H), 5.11–5.12 (m, 1H), 4.06–4.12 (m, 1H), 3.71 (s, 3H), 2.53 (dd, J1 = 16.6 Hz, J2 = 3.5 Hz, 1H), 2.44 (dd, J1 = 16.6 Hz, J2 = 9 Hz, 1H), 2.23–2.33 (m, 2H). 13C-NMR (500 MHz, CDCl3): δ 173.2, 133.9, 118.2, 67.3, 51.8, 40.9, 40.4. GC data: (column: 30 m × 0.25 mm × 0.25 μm chiral capillary column, Cyclodextrin-B 150 °C for 10 min, rate: 0 °C/min; carrier gas: He, press 90 kPa). tR = 2.61 min.
3.4. Preparation of MPA-Esters
3.4.1. General Method for the Synthesis of MPA Esters of Secondary Alcohols
To a solution of the corresponding secondary alcohol (0.1 mmol) in dry CH2Cl2 were added 1.1 equivalent of N,N’-dicyclohexylcarbodiimide (DCC) (0.11 mmol, 23 mg) and 1.1 equivalent of the corresponding (R) or (S) MPA (0.11 mmol, 18 mg) and the reaction mixture was stirred at 0 °C for 4–6 h. After completion of the reaction, the produced urea was filtered and the filtrate was evaporated and then purified by column chromatography with 5/1 Hex/EtOAc. The produced corresponding MPA-ester was isolated with 90% isolated yield.
(R)-MPA Ester of (S)-Methyl 3-hydroxyhex-5-enoate
1H NMR (500 MHz; CDCl3; Me4Si): δ 7.40–7.42 (m, 2H), 7.317–7.36 (m, 3H), 5.41–5.49 (m, 1H), 5.30–5.35 (m, 1H), 4.84–4.90 (m, 2H), 4.72 (s, 1H), 3.60 (s, 3H), 3.41 (s, 3H), 2.52–2.62 (m, 2H), 2.218–2.223 (m, 2H).
(S)-MPA Ester of (S)-Methyl 3-hydroxyhex-5-enoate
1H NMR (500 MHz; CDCl3; Me4Si): δ 7.40–7.42 (m, 2H), 7.31–7.36 (m, 3H), 5.65–5.74 (m, 1H), 5.26–5.35 (m, 1H), 5.05–5.08 (m, 2H), 4.71 (s, 1H), 3.39 (s, 3H), 3.37 (s, 3H), 2.44–2.50 (m, 2H), 2.37–2.41 (m, 2H).
4. Conclusions
In summary, a total synthesis of (±)-rugulactone has been achieved in a highly efficient and concise way—in eight steps with an overall yield of 21%. This flexible synthetic pathway can be applied to the synthesis of a variety of α,β–unsaturated α-pyrones, which bear two potential Michael acceptors. Concerning the chemoenzymatic synthetic approach of the optically active (S) epimer of the natural product 1, we identified through a screening procedure three suitable highly stereoselective ketoreductases, which catalyzed efficiently the formation of the chiral key intermediate 8 as the (S) enantiomer in high yield and excellent ee, using ketoreductases for the first time. Further studies focused on screening other anti-Prelog reductive enzymes for the synthesis of the (R)-8 enantiomer are currently underway.
Supplementary Materials
Supplementary materials are available online at https://www.mdpi.com/1420-3049/23/3/640/s1. 1H- and 13C-NMR of compound 7: S2, 1H- and 13C-NMR compound 8: S3, 1H- and 13C-NMR compound 5: S4, 1H- and 13C-NMR compound 2: S5, 1H- and 13C-NMR compound 9: S6, 1H- and 13C-NMR compound 3: S7, 1H- and 13C-NMR compound 1: S8, 1H-NMR of (R)-MPA and (S)-MPA esters of (S)-methyl 3-hydroxyhex-5-enoate: S9.
Acknowledgments
This work was supported by the Special Account for Research Funds of University of Crete (KA 4453 and KA 4065, SARF UoC). We thank C. Markou for some preliminary experiments. We are also thankful to the PROFI (Proteomics Facility at IMBB-FORTH) for performing the HRMS analysis.
Author Contributions
I.S., T.T.-E. and V.G. conceived and designed the experiments; T.T.-E. and V.G. performed the experiments and analyzed the data; I.S. contributed reagents/materials/analysis tools; I.S., T.T.-E. and V.G. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Meragelman, T.L.; Scudiero, D.A.; Davis, R.E.; Staudt, L.M.; McCloud, T.G.; Cardellina, J.H., II; Shoemaker, R.H. Inhibitors of the NF-κB activation pathway from Cryptocarya rugulosa. J. Nat. Prod. 2009, 72, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Baltimore, D. Pillars article: Multiple nuclear factors interact with the immunoglobulin enhancer sequencies. J. Immunol. 1986, 177, 7485–7496. [Google Scholar]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-κB and rel. proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Dev, A.; Iyer, S.; Razani, B.; Cheng, G. NF-κB and Innate Immunity. In NF-κB in Health and Disease; Current Topics in Microbiology and Immunology; Karin, M., Ed.; Springer-Verlag: Berlin/Heidelberg, Germany, 2011; Volume 349, pp. 115–143. ISBN 978-3-642-16016-5. [Google Scholar]
- Pahl, H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. NF-κB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Karin, M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, K.D.; Reddy, D.S.; Ramaiah, M.J.; Ghosh, S.; Pothula, V.; Lunavath, S.; Thomas, S.; Valli, S.N.C.V.L.P.; Pal Bahdra, M.; Yadav, S.J. Rugulactone derivatives act as inhibitors of NF-κB activation and modulates the transcription of NF-κB dependent genes in MDA-MB-231cells. Bioorg. Med. Chem. Lett. 2014, 24, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Karin, M. The IKK/NF-κB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004, 206, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Baltimore, D. Circuitry of nuclear factor κB signaling. Immunol. Rev. 2006, 210, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Leung, T.H.; Baltimore, D. Genetic analysis of NF-κB/Rel transcription factors defines functional specificities. EMBO J. 2003, 22, 5530–5539. [Google Scholar] [CrossRef] [PubMed]
- Bacher, S.; Schmitz, M.L. The NF-κB pathway as a potential target for autoimmune disease therapy. Curr. Pharm. Des. 2004, 10, 2827–2837. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.K.; Shekhar, V.; Prabhakar, P.; Chinna Babu, B.; Siddhardha, B.; Murthy, U.S.N.; Venkateswarlu, Y. Stereoselective synthesis and biological evaluation of (R)-rugulactone, (6R)-((4R)-hydroxy-6-phenyl-hex-2-enyl)-5,6-dihydro-pyran-2-one and its 4S epimer. Eur. J. Med. Chem. 2010, 45, 4657–4663. [Google Scholar] [CrossRef] [PubMed]
- Nodwell, M.B.; Menz, H.; Kirsh, S.F.; Sieber, S.A. Rugulactone and its analogues exert antibacterial effects through multiple mechanisms including inhibition of thiamine biosynthesis. ChemBioChem 2012, 13, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Cross, F.; Pelotier, B.; Piva, O. Regioselective tandem ring closing/cross metathesis of 1,5-hexadien-3-ol derivatives: Application to the total synthesis of rugulactone. Eur. J. Org. Chem. 2010, 2010, 5063–5070. [Google Scholar] [CrossRef]
- Mohapatra, D.K.; Pragna, P.D.; Reddy, D.S.; Yadav, S.J. First total syntheses and absolute configuration of rugulactone and 6(R)-(40-oxopent-20-enyl)-5,6-dihydro-2H-pyran-2-one. Tetrahedron Lett. 2009, 50, 5941–5944. [Google Scholar] [CrossRef]
- Reddy, D.K.; Shekhar, V.; Reddy, T.S.; Reddy, S.P.; Venkateswarlu, Y. Stereoselective first total synthesis of (R)-rugulactone. Tetrahedron Asymmetry 2009, 20, 2315–2319. [Google Scholar] [CrossRef]
- Reddipalli, G.; Venkataiah, M.; Fadnavis, W.N. Chemo-enzymatic synthesis of both enantiomers of rugulactone. Tetrahedron Asymmetry 2010, 21, 320–324. [Google Scholar] [CrossRef]
- Boese, D.; Fernadez, E.; Pietruszka, J. Stereoselective synthesis of both enantiomers of rugulactone. J. Org. Chem. 2011, 76, 3463–3469. [Google Scholar] [CrossRef] [PubMed]
- Allais, F.; Aouhansou, M.; Majira, A.; Ducro, P.H. Asymmetric total synthesis of rugulactone, an a-pyrone from Cryptocarya rugulosa. Synthesis 2010, 16, 2787–2793. [Google Scholar] [CrossRef]
- Reddy, B.N.; Singh, R.P. A facile stereoselective total synthesis of (R)-rugulactone. ISRN Org. Chem. 2014, 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Goswami, A.; Saikia, P.P.; Saikia, B.; Chaturvedi, D.; Barua, N.C. An improved stereoselective total synthesis of (R)-rugulactone. Tetrahedron Lett. 2011, 52, 5133–5135. [Google Scholar] [CrossRef]
- Albarranan-Velo, J.; Gonzalez-Martınez, D.; Gotor-Fernandez, V. Stereoselective biocatalysis: A mature technology for the asymmetric synthesis of pharmaceutical building blocks. Biocatal. Biotransform. 2017, 36, 102–130. [Google Scholar] [CrossRef]
- Rudroff, F.; Mihovilovic, M.D.; Gröger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and challenges for combining chemo- and biocatalysis. Nat. Catal. 2018, 1, 12–22. [Google Scholar] [CrossRef]
- Gröger, H. Enzyme-catalyzed asymmetric synthesis. In Catalytic Asymmetric Synthesis, 3rd ed.; Ojima, I., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 269–342. ISBN 978-0-470-17577-4. [Google Scholar]
- Gröger, H. Cluster issue on chemoenzymatic synthesis. ChemCatChem 2011, 3, 239–240. [Google Scholar] [CrossRef]
- Anderson, G.W.; Halverstadt, I.F.; Miller, W.H.; Roblin, R.O., Jr. Studies in chemotherapy. X. Antithyroid compounds. synthesis of 5- and 6-substituted 2-thiouracils from oxoesters and thiourea. J. Am. Chem. Soc. 1945, 67, 2197–2200. [Google Scholar] [CrossRef] [PubMed]
- Hamana, H.; Sugasawa, T. Electrophilic reaction of allyltrimethylsilane with nitriles in the presence of boron trichloride. Chem. Lett. 1985, 14, 921–924. [Google Scholar] [CrossRef]
- Lee, A.S.-Y.; Lin, L.S. Synthesis of allyl ketone via Lewis acid promoted Barbier-type reaction. Tetrahedron Lett. 2000, 41, 8803–8806. [Google Scholar] [CrossRef]
- Fu, G.C.; Grubbs, R.H. Synthesis of cycloalkenes via alkylidene-mediated olefin metathesis and carbonyl olefination. J. Am. Chem. Soc. 1993, 115, 3800–3801. [Google Scholar] [CrossRef]
- Kallergi, M.; Kalaitzakis, D.; Smonou, I. Enzymatic total synthesis of banana volatile (S)-2-pentyl (R)-3-hydroxyhexanoate. Eur. J. Org. Chem. 2011, 2011, 3946–3950. [Google Scholar] [CrossRef]
- Kalaitzakis, D.; Smonou, I. Chemoenzymatic synthesis of stegobinone and stegobiol, components of the natural sex pheromone of the drugstore beetle (Stegobium paniceum L.). Eur. J. Org. Chem. 2012, 2012, 43–46. [Google Scholar] [CrossRef]
- Kalaitzakis, D.; Rozzell, J.D.; Kambourakis, S.; Smonou, I. Highly stereoselective reductions of α-alkyl-1,3-diketones and α-alkyl-β-keto esters catalyzed by isolated NADPH-dependent ketoreductases. Org. Lett. 2005, 7, 4799–4801. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzakis, D.; Rozzell, J.D.; Kambourakis, S.; Smonou, I. Synthesis of valuable chiral intermediates by isolated ketoreductases: application in the synthesis of α-alkyl-β-hydroxy ketones and 1,3-diols. Adv. Synth. Catal. 2006, 348, 1958–1969. [Google Scholar] [CrossRef]
- Kalaitzakis, D.; Smonou, I. Highly diastereoselective synthesis of 2-substituted-1,3-diols catalyzed by ketoreductases. Tetrahedron 2010, 66, 9431–9439. [Google Scholar] [CrossRef]
- Kalaitzakis, D.; Smonou, I. A Two-step, one-pot enzymatic synthesis of 2-substituted 1,3-diols. J. Org. Chem. 2010, 75, 8658–8661. [Google Scholar] [CrossRef] [PubMed]
- Bariotaki, A.; Kalaitzakis, D.; Smonou, I. Enzymatic reductions for the regio- and stereoselective synthesis of hydroxy-keto esters and dihydroxy esters. Org. Lett. 2012, 14, 1792–1795. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzakis, D.; Rozzell, D.J.; Kambourakis, S.; Smonou, I. A Two-Step Chemoenzymatic Synthesis of the Natural Pheromone (+)-Sitophilure Utilizing isolated, NADPH-dependent ketoreductases. Eur. J. Org. Chem. 2006, 2309–2313. [Google Scholar] [CrossRef]
- Kalaitzakis, D.; Kambourakis, S.; Rozzell, D.J.; Smonou, I. Stereoselective chemoenzymatic synthesis of sitophilate: A natural pheromone. Tetrahedron Asymmetry 2007, 18, 2418–2426. [Google Scholar] [CrossRef]
- Fryszkowska, A.; Peterson, J.; Davies, N.L.; Dewar, C.; Evans, G.; Bycroft, M.; Triggs, N.; Fleming, T.; Gorantla, S.S.C.; Hoge, G.; et al. Development of a chemoenzymatic process for dehydroepiandrosterone acetate synthesis. Org. Proce. Res. Dev. 2016, 20, 1520–1528. [Google Scholar] [CrossRef]
- Seco, J.M.; Quinoa, E.; Riguera, R. The assignment of absolute configuration by NMR. Chem. Rev. 2004, 104, 17–117. [Google Scholar] [CrossRef]
Sample Availability: Not available. |
Figure 1.
Rugulactone.

Scheme 1.
Retrosynthetic analysis of rugulactone.
Scheme 2.
(A) Reagents and conditions: (a) allyl bromide, Zn powder, tetrahydrofuran (THF), 0 °C–r.t., 52%; (b) NaBH4, MeOH, 0 °C, 95%; (c) (i) lithium diisopropylamide (LDA), tert-butyl acetate, THF, −78 °C (ii) NaBH4, MeOH, 0 °C, 79% over two steps; (d) p-toluenesulfonic acid (PTSA), toluene, reflux, 97%; (B) (e) vinyl magnesium chloride, THF, 0 °C–r.t., 86%; (f) 2-iodoxybenzoic acid (IBX), dimethyl sulfoxide (DMSO), r.t., 88%.
Scheme 2.
(A) Reagents and conditions: (a) allyl bromide, Zn powder, tetrahydrofuran (THF), 0 °C–r.t., 52%; (b) NaBH4, MeOH, 0 °C, 95%; (c) (i) lithium diisopropylamide (LDA), tert-butyl acetate, THF, −78 °C (ii) NaBH4, MeOH, 0 °C, 79% over two steps; (d) p-toluenesulfonic acid (PTSA), toluene, reflux, 97%; (B) (e) vinyl magnesium chloride, THF, 0 °C–r.t., 86%; (f) 2-iodoxybenzoic acid (IBX), dimethyl sulfoxide (DMSO), r.t., 88%.

Scheme 3.
Reagents and conditions: Grubbs(II) catalyst (5 mol %), dry CH2Cl2, 45 °C, 75%.
Scheme 4.
Asymmetric enzymatic reduction of 7 using Kred 119.
Scheme 5.
Isomerization of keto ester 7.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Enzymatic reduction of methyl 3-oxohex-5-enoate 7.
| Substrate | Kred a | Conv. b (time) | Yield c | ee d | Product | |
|---|---|---|---|---|---|---|
| 7 | A1C | >99% | (12 h) | 62% | >99% | (S)-8 |
| A1D | >99% | (12 h) | 64% | 98% | (S)-8 | |
| 119 | >99% | (12 h) | 79% | >99% | (S)-8 | |
| B1F | >99% | (12 h) | 62% | 58.6% | (S)-8 | |
| 101 | >99% | (12 h) | 83% | 72% | (S)-8 | |
a Ketoreductase; b Conversions were derived from 1H-NMR spectra; c Isolated; d Enantiomeric excess was derived from chiral column GC analysis.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tyrikos-Ergas, T.; Giannopoulos, V.; Smonou, I. An Efficient Chemoenzymatic Approach towards the Synthesis of Rugulactone. Molecules 2018, 23, 640. https://doi.org/10.3390/molecules23030640
AMA Style
Tyrikos-Ergas T, Giannopoulos V, Smonou I. An Efficient Chemoenzymatic Approach towards the Synthesis of Rugulactone. Molecules. 2018; 23(3):640. https://doi.org/10.3390/molecules23030640
Chicago/Turabian StyleTyrikos-Ergas, Theodore, Vasileios Giannopoulos, and Ioulia Smonou. 2018. "An Efficient Chemoenzymatic Approach towards the Synthesis of Rugulactone" Molecules 23, no. 3: 640. https://doi.org/10.3390/molecules23030640