Ruthenium(η6,η1-arene-CH2-NHC) Catalysts for Direct Arylation of 2-Phenylpyridine with (Hetero)Aryl Chlorides in Water

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
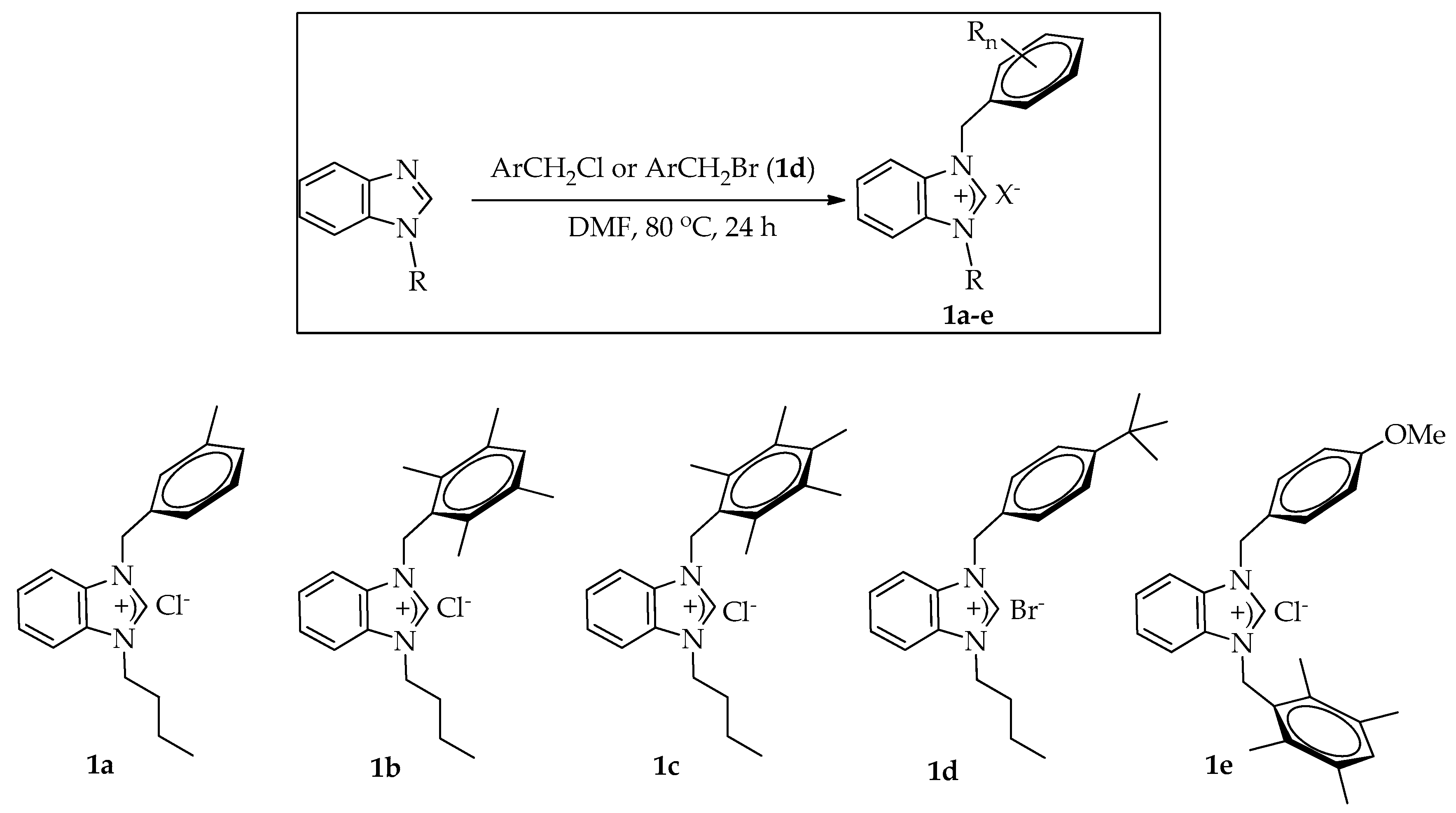
2.1. Preparation of Benzimidazolium Halides
2.2. Preparation of Ruthenium(II)-NHC Complexes Containing the η6,η1–NHC Mixed Chelating Ligand
2.3. Single Crystal X-ray Diffraction and Structure Analysis of Complex 2b
2.4. Optimization Conditions of Direct Arylation of 2-Phenylpyridine with (hetero)Aryl Chlorides with Catalysts 2
2.5. Direct Arylation of 2-Phenylpyridine with (Hetero)Aryl Chlorides
3. Materials and Methods
3.1. General
3.2. General Procedure for the Preparation of Benzimidazolium Halides 1a–e
3.3. General Procedure for the Preparation of Ruthenium(II)NHC Complexes 2a–e
3.4. General Procedure for the Direct Catalytic Arylation of 2-Phenylpyridine with (Hetero)Aryl Chlorides
3.5. Single Crystal X-ray Diffraction and Structure Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl-aryl bond formation one century after the discovery of the Ullmann reaction. Chem. Rev. 2002, 102, 1359–1470. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.W. Comprehensive Organic Synthesis, 1st ed.; Trost, B.M., Fleming, I., Eds.; Pergamon: London, UK, 1991; Volume 3, pp. 481–520. ISBN 0080359299. [Google Scholar]
- Kumada, M. Nickel and palladium complex catalyzed cross-coupling reactions of organometallic reagents with organic halides. Pure Appl. Chem. 1980, 52, 669–679. [Google Scholar] [CrossRef]
- Negishi, E. Palladium- or Nickel-catalyzed cross coupling. A new selective method for carbon-carbon bond formation. Acc. Chem. Res. 1982, 15, 340–348. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Hiyama, T.; Hatanaka, Y. Palladium-catalyzed cross-coupling reaction of organometalloids through activation with fluoride ion. Pure Appl. Chem. 1994, 66, 1471–1478. [Google Scholar] [CrossRef]
- Stille, J.K. The palladium-catalyzed cross-coupling reactions of organotin reagents with organic electrophiles [new synthetic methods (58)]. Angew. Chem. Int. Ed. Engl. 1986, 25, 508–524. [Google Scholar] [CrossRef]
- De Meijere, A.; Diederich, F. (Eds.) Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004; Volume 1, ISBN 978-3-527-30518-6. [Google Scholar]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-catalyzed cross-coupling reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef] [PubMed]
- Shilov, A.E.; Shul’pin, G.B. Activation of C–H bonds by metal complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, F.; Murai, S. Activation of Unreactive Bonds and Organic Synthesis; Murai, S., Ed.; Springer: Berlin, Germany, 1999; pp. 47–79. ISBN 978-3-540-68525-8. [Google Scholar]
- Dyker, G. Transition metal catalyzed coupling reactions under C-H activation. Angew. Chem. Int. Ed. 1999, 38, 1698–1712. [Google Scholar] [CrossRef]
- Robin, B.; Bedford, R.B.; Limmert, M.E. Catalytic intermolecular ortho-arylation of phenols. J. Org. Chem. 2003, 68, 8669–8682. [Google Scholar] [CrossRef]
- Goj, L.A.; Gunnoe, T.B. Developments in catalytic aromatic C–H transformations: Promising tools for organic synthesis. Curr. Org. Chem. 2005, 9, 671–685. [Google Scholar] [CrossRef]
- Campeau, L.-C.; Fagnou, K. Palladium-catalyzed direct arylation of simple arenes in synthesis of biaryl molecules. Chem. Commun. 2006, 12, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Stuart, D.R.; Fagnou, K. The catalytic cross-coupling of unactivated arenes. Science 2007, 316, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, V.S.; Parthasarathy, K.; Cheng, C.H. Synthesis of fluorenones from aromatic aldoxime ethers and aryl halides by palladium-catalyzed dual C–H activation and Heck cyclization. Angew. Chem. Int. Ed. 2008, 47, 9462–9465. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Engle, K.M.; Wang, D.H.; Yu, J.Q. Palladium(II)-catalyzed C–H activation/C–C cross-coupling reactions: Versatility and practicality. Angew. Chem. Int. Ed. 2009, 48, 5094–5115. [Google Scholar] [CrossRef] [PubMed]
- Bellina, F.; Rossi, R. Recent advances in the synthesis of (hetero)aryl-substituted heteroarenes via transition metal-catalysed direct (hetero)arylation of heteroarene C–H bonds with aryl halides or pseudohalides, diaryliodonium salts, and potassium aryltrifluoroborates. Tetrahedron 2009, 65, 10269–10310. [Google Scholar] [CrossRef]
- Ackermann, L.; Vicente, R.; Kapdi, A.R. Transition-metal-catalyzed direct arylation of (hetero)arenes by C-H bond cleavage. Angew. Chem. Int. Ed. 2009, 48, 9792–9826. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, V.S.; Parthasarathy, K.; Cheng, C.H. One-pot synthesis of diarylmethylidenefluorenes and phenanthrenes by palladium-catalyzed multiple C–H bond functionalization. Chem. Eur. J. 2010, 16, 1436–1440. [Google Scholar] [CrossRef] [PubMed]
- Gandeepan, P.; Parthasarathy, K.; Cheng, C.H. Synthesis of phenanthrone derivatives from sec-alkyl aryl ketones and aryl Halides via a palladium-catalyzed dual C–H bond activation and enolate cyclization. J. Am. Chem. Soc. 2010, 132, 8569–8571. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, S.; Davi, M.; Sofack-Kreutzer, J.; Pierre, C.; Kefalidis, C.E.; Clot, E.; Fagnou, K.; Baudoin, O. Intramolecular palladium-catalyzed alkane C–H arylation from aryl chlorides. J. Am. Chem. Soc. 2010, 132, 10706–10716. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xu, Z.P.; Sun, P.P.; Jiang, X.Q.; Fang, M. Synthesis of biphenyl-2-carbonitrile derivatives via a palladium-catalyzed sp2 C–H bond activation using cyano as a directing group. Org. Lett. 2011, 13, 1286–1289. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.N.; Smith, J.F. Catalytic C–C bond formation via ortho-metalated complexes. J. Am. Chem. Soc. 1986, 108, 2728–2735. [Google Scholar] [CrossRef]
- Murai, S.; Kakiuchi, F.; Sekine, S.; Tanaka, Y.; Kamatani, A.; Sonoda, M.; Chatani, N. Efficient catalytic addition of aromatic carbon-hydrogen bonds to olefins. Nature 1993, 366, 529–531. [Google Scholar] [CrossRef]
- Oi, S.; Fukita, S.; Hirata, N.; Watanuki, N.; Miyano, S.; Inoue, Y. Ruthenium complex-catalyzed direct ortho arylation and alkenylation of 2-arylpyridines with organic halides. Org. Lett. 2001, 3, 2579–2581. [Google Scholar] [CrossRef] [PubMed]
- Prades, A.; Poyatos, M.; Peris, E. (η6-Arene)ruthenium(N-heterocyclic carbene) complexes for the chelation-assisted arylation and deuteration of arylpyridines: Catalytic studies and mechanistic insights. Adv. Synth. Catal. 2010, 352, 1155–1162. [Google Scholar] [CrossRef]
- Ackermann, L. Phosphine oxides as preligands in ruthenium-catalyzed arylations via C–H bond functionalization using aryl chlorides. Org. Lett. 2005, 7, 3123–3125. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L. Carboxylate-assisted transition-metal-catalyzed C–H bond functionalizations: Mechanism and scope. Chem. Rev. 2011, 111, 1315–1345. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Vicente, R.; Althammer, A. Assisted ruthenium-catalyzed C–H bond activation: Carboxylic acids as cocatalysts for generally applicable direct arylations in apolar solvents. Org. Lett. 2008, 10, 2299–2302. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Arockiam, P.B.; Fischmeister, C.; Bruneau, C.; Dixneuf, P.H. C–H bond functionalisation with [RuH(codyl)2]BF4 catalyst precursor. Green Chem. 2011, 13, 2315–2319. [Google Scholar] [CrossRef]
- Davies, D.L.; Al-Duaij, O.; Fawcett, J.; Giardiello, M.; Hilton, S.T.; Russell, D.R. Room-temperature cyclometallation of amines, imines and oxazolines with [MCl2Cp*]2 (M = Rh, Ir) and [RuCl2(p-cymene)]2. Dalton Trans. 2003, 21, 4132–4138. [Google Scholar] [CrossRef]
- Stefane, B.; Fabris, J.; Pozgan, F. C–H bond functionalization of arylpyrimidines catalyzed by an in situ generated ruthenium(II) carboxylate system and the construction of tris(heteroaryl)-substituted benzenes. Eur. J. Org. Chem. 2011, 19, 3474–3481. [Google Scholar] [CrossRef]
- Aihara, Y.; Chatani, N. Ruthenium-catalyzed direct arylation of C–H bonds in aromatic amides containing a bidentate directing group: Significant electronic effects on arylation. Chem. Sci. 2013, 4, 664–670. [Google Scholar] [CrossRef]
- Shan, C.; Luo, X.; Qi, X.; Liu, S.; Li, Y.; Lan, Y. Mechanism of ruthenium-catalyzed direct arylation of C–H bonds in aromatic amides: A computational study. Organometallics 2016, 35, 1440–1445. [Google Scholar] [CrossRef]
- Zha, G.F.; Qin, H.L.; Kantchev, E.A.B. Ruthenium-catalyzed direct arylations with aryl chlorides. RSC Adv. 2016, 6, 30875–30885. [Google Scholar] [CrossRef]
- Nareddy, P.; Jordan, F.; Szostak, M. Ruthenium(II)-catalyzed ortho-C–H arylation of diverse N-heterocycles with aryl silanes by exploiting solvent-controlled N-coordination. Org. Biomol. Chem. 2017, 15, 4783–4788. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, I.; Demir, S.; Cetinkaya, B.; Gourlaouen, C.; Maseras, F.; Bruneau, C.; Dixneuf, P.H. Direct arylation of arene C–H bonds by cooperative action of NHCarbene-ruthenium(II) catalyst and carbonate via proton abstraction mechanism. J. Am. Chem. Soc. 2008, 130, 1156–1157. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Özdemir, İ.; Çetinkaya, B. Synthesis and catalytic properties of novel ruthenium N-heterocyclic-carbene complexes. J. Organomet. Chem. 2009, 694, 4025–4031. [Google Scholar] [CrossRef]
- Özdemir, I.; Demir, S.; Gürbüz, N.; Çetinkaya, B.; Toupet, L.; Bruneau, C.; Dixneuf, P.H. Synthesis, characterization and catalytic activity of new N-heterocyclic bis(carbene)ruthenium complexes. Eur. J. Inorg. Chem. 2009, 13, 1942–1949. [Google Scholar] [CrossRef]
- Ackermann, L.; Kapdi, A.R.; Potukuchi, H.K.; Kozhushkov, S.I. Syntheses via C–H Bond Functionalizations. In Handbook of Green Chemistry—Green Processes; Li, C.-J., Ed.; Wiley-VCH: Weinheim, Germany, 2012; pp. 259–305. [Google Scholar]
- Arockiam, P.; Poirier, V.; Fischmeister, C.; Bruneau, C.; Dixneuf, P.H. Diethyl carbonate as a solvent for ruthenium catalysed C–H bond functionalization. Green Chem. 2009, 11, 1871–1875. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Fischmeister, C.; Bruneau, C.; Dixneuf, P.H. C–H bond functionalization in water catalyzed by carboxylato ruthenium(II) systems. Angew. Chem. Int. Ed. 2010, 49, 6629–6632. [Google Scholar] [CrossRef] [PubMed]
- Arockiam, P.B.; Fischmeister, C.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-catalyzed selective monoarylation in water and sequential functionalisations of C–H bonds. Green. Chem. 2013, 15, 67–71. [Google Scholar] [CrossRef]
- Li, B.; Devaraj, K.; Darcel, C.; Dixneuf, P.H. Catalytic C–H bond arylation of aryl imines and oxazolines in water with ruthenium(II)-acetate catalyst. Tetrahedron 2012, 68, 5179–5184. [Google Scholar] [CrossRef]
- Singh, K.S.; Dixneuf, P.H. Direct C-H bond arylation in water promoted by (O,O)- and (O,N)-chelate ruthenium(II) catalysts. ChemCatChem. 2013, 5, 1313–1316. [Google Scholar] [CrossRef]
- Emmanuel, F.F.; Bruneau, B.; Dixneuf, P.H.; Jutand, A. Autocatalysis for C–H bond activation by ruthenium(II) complexes in catalytic arylation of functional arenes. J. Am. Chem. Soc. 2011, 133, 10161–10170. [Google Scholar] [CrossRef]
- Fabre, I.; von Wolff, N.; Le Duc, G.; Flegeau, E.F.; Bruneau, B.; Dixneuf, P.H.; Jutand, A. Autocatalytic intermolecular versus intramolecular deprotonation in C–H bond activation of functional arenes by ruthenium(II) or palladium(II) complexes. Chem. Eur. J. 2013, 19, 7595–7604. [Google Scholar] [CrossRef] [PubMed]
- Günal, S.; Kaloğlu, N.; Özdemir, İ.; Demir, S.; Özdemir, İ. Novel benzimidazolium salts and their silver complexes: Synthesis and antibacterial properties. Inorg. Chem. Commun. 2012, 21, 142–146. [Google Scholar] [CrossRef]
- Demir, S.; Özdemir, İ.; Şahin, O.; Çetinkaya, B.; Büyükgüngör, O. Synthesis and catalytic activity of novel benizimidazlolinylidene-ruthenium(II) complexes. Synlett 2010, 3, 496–500. [Google Scholar] [CrossRef]
- Çetinkaya, B.; Demir, S.; Özdemir, I.; Toupet, L.; Semeril, D.; Bruneau, C.; Dixneuf, H.P. η6-Mesityl,η1-imidazolinylidene–Carbene–Ruthenium(II) complexes: Catalytic activity of their allenylidene derivatives in alkene metathesis and cycloisomerisation reactions. Chem. Eur. J. 2003, 9, 2323. [Google Scholar] [CrossRef] [PubMed]
- Arslan, H.; Vanderveer, D.; Özdemir, I.; Çetinkaya, B.; Demir, S. Crystal structure of [RuCl2[N-(2,4,6-trimethyl-benzyl)N-(n-butyl)]-imidazolidin-2-ylidene] and [RuCl2[N-(2,4,6-trimethyl-benzyl)-N-(2-methoxyethyl)]-imidazolidin-2-ylidene]. J. Chem. Crystallogr. 2005, 35, 491–495. [Google Scholar] [CrossRef]
- Arslan, H.; VanDerveer, D.; Ozdemir, I.; Yasar, S.; Cetinkaya, B. Dichloro[3-(1-naphthylmethyl)-1-(2,4,6-trimethylbenzyl)imidazolidin-2-ylidene]ruthenium. Acta Cryst. E 2005, 61, M1873–M1875. [Google Scholar] [CrossRef]
- Bruker, SMART, version 6.12; Bruker AXS Inc.: Madison, WI, USA, 2012.
- Bruker, SAINT, version 6.12; Bruker AXS Inc.: Madison, WI, USA, 2012.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1a–e and 2a–e are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Formula | Isolated Yield (%) | M.p. (Metlting Point) (°C) | v(CN) (Frequency) (cm−1) | 1H NMR H(2) (ppm) | 13C NMR C(2) (ppm) |
|---|---|---|---|---|---|---|
| 1a | C19H23ClN2 | 78 | 198–199 | 1558 | 11.49 | 143.2 |
| 1b | C22H29ClN2 | 83 | 176–177 | 1553 | 10.86 | 142.9 |
| 1d | C22H29BrN2 | 80 | 184–185 | 1560 | 11.45 | 142.6 |
| 1e | C26H29ClN2O | 87 | 191–192 | 1557 | 11.53 | 143.6 |
| 2a | C19H22Cl2N2Ru | 87 | 231–232 | 1401 | - | 181.3 |
| 2b | C22H28Cl2N2Ru | 80 | 311–312 | 1400 | - | 184.9 |
| 2c | C23H30Cl2N2Ru | 82 | 319–320 | 1406 | - | 186.0 |
| 2d | C22H28Cl2N2Ru | 71 | 298–299 | 1403 | - | 186.1 |
| 2e | C26H28Cl2N2ORu | 76 | 310–311 | 1407 | - | 185.2 |
| Bond Lenght | Bond Angles | Torsion Angle | |||
|---|---|---|---|---|---|
| C(1)–N(2) | 1.360(4) | N(2)–C(1)–N(1) | 105.2(3) | N(2)–C(1)–Ru(1)–Cl(2) | −53.0(3) |
| C(1)–N(1) | 1.361(4) | N(2)–C(1)–Ru(1) | 138.3(2) | N(1)–C(1)–Ru(1)–Cl(2) | 129.7(2) |
| C(1)–Ru(1) | 2.040(3) | N(1)–C(1)–Ru(1) | 116.5(2) | N(2)–C(1)–Ru(1)–Cl(1) | 36.1(3) |
| C(2)–N(1) | 1.386(4) | N(1)–C(12)–C(13) | 106.3(2) | N(1)–C(1)–Ru(1)–Cl(1) | −141.1(2) |
| C(2)–C(7) | 1.394(5) | C(14)–C(13)–Ru(1) | 73.54(17) | N(2)–C(1)–N(1)–C(12) | 176.2(3) |
| C(7)–N(2) | 1.393(4) | C(18)–C(13)–Ru(1) | 73.77(18) | Ru(1)–C(1)–N(1)–C(2) | 177.82(19) |
| C(8)–N(2) | 1.466(4) | C(12)–C(13)–Ru(1) | 116.3(2) | ||
| C(12)–N(1) | 1.466(4) | C(1)–N(1)–C(2) | 111.6(3) | ||
| C(12)–C(13) | 1.517(4) | C(1)–N(1)–C(12) | 121.5(3) | ||
| C(13)–Ru(1) | 2.109(3) | C(2)–N(1)–C(12) | 126.7(3) | ||
| C(14)–Ru(1) | 2.190(3) | C(1)–N(2)–C(7) | 111.1(3) | ||
| C(15)–C(16) | 1.426(5) | C(1)–N(2)–C(8) | 124.5(3) | ||
| C(15)–C(20) | 1.513(4) | C(7)–N(2)–C(8) | 124.3(3) | ||
| C(15)–Ru(1) | 2.241(3) | ––– | |||
| C(16)–Ru(1) | 2.274(3) | ––– | |||
| C(17)–Ru(1) | 2.267(3) | ––– | |||
| C(18)–Ru(1) | 2.196(3) | ––– | |||
| Cl(1)–Ru(1) | 2.4267(9) | ––– | |||
| Cl(2)–Ru(1) | 2.4222(8) | ––– |
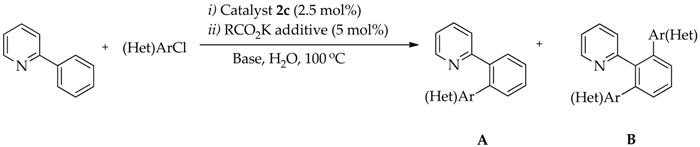
| Entry | (Het)ArCl | Additive | Base | Time (h) | Conversion (%) | Yield (%) | |
|---|---|---|---|---|---|---|---|
| A | B | ||||||
| 1 | 2-Chlorothiophene | KOPiv | Cs2CO3 | 5 | 9 | 100 | - |
| 2 | 2-Chlorothiophene | KOPiv | K2CO3 | 5 | - | - | - |
| 3 | 2-Chlorothiophene | KOPiv | Na2CO3 | 5 | - | - | - |
| 4 | 2-Chlorothiophene | KOAc | Cs2CO3 | 5 | 15 | 100 | - |
| 5 | 2-Chlorothiophene | KOAc | Cs2CO3 | 10 | 47 | 100 | - |
| 6 | 2-Chlorothiophene | KOAc | Cs2CO3 | 20 | 95 | 100 | - |
| 7 | 2-Chlorothiophene | KOAc | Cs2CO3 | 24 | 97 | 100 | - |
| 8 | 4-Chlorotoluene | KOAc | Cs2CO3 | 20 | 100 | 100 | - |
| 9 | 4-Chlorotoluene | KOAc | Cs2CO3 | 10 | 100 | 100 | - |
| 10 | 4-Chlorotoluene | KOAc | Cs2CO3 | 5 | 100 | 100 | - |

| Entry | Ru-NHC | (Het)ArCl | Time (h) | Conversion (%) | Yield (%) | |
|---|---|---|---|---|---|---|
| A | B | |||||
| 1 | 2a | 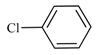 | 1 | 100 | 100 | - |
| 2 | 2b | 1 | 100 | 100 | - | |
| 3 | 2c | 1 | 80 | 80 | 20 | |
| 4 | 2d | 1 | 90 | 90 | 10 | |
| 5 | 2e | 1 | 85 | 87 | 13 | |
| 6 | 2a | 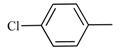 | 5 | 100 | 100 | - |
| 7 | 2b | 5 | 100 | 100 | - | |
| 8 | 2c | 5 | 100 | 100 | - | |
| 9 | 2d | 5 | 100 | 88 | 12 | |
| 10 | 2e | 5 | 90 | 95 | 5 | |
| 11 | 2a | 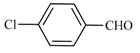 | 5 | 100 | 70 | 30 |
| 12 | 2b | 5 | 100 | 80 | 20 | |
| 13 | 2c | 5 | 100 | 72 | 28 | |
| 14 | 2d | 5 | 100 | 92 | 8 | |
| 15 | 2e | 5 | 100 | 86 | 14 | |
| 16 | 2a |  | 5 | 76 | 76 | 24 |
| 17 | 2b | 5 | 72 | 72 | 28 | |
| 18 | 2c | 5 | 71 | 71 | 29 | |
| 19 | 2d | 5 | 70 | 70 | 30 | |
| 20 | 2e | 5 | 80 | 85 | 15 | |
| 21 | 2a |  | 5 | 72 | 72 | 28 |
| 22 | 2b | 5 | 77 | 77 | 23 | |
| 23 | 2c | 5 | 70 | 70 | 30 | |
| 24 | 2d | 5 | 79 | 79 | 21 | |
| 25 | 2e | 5 | 74 | 75 | 25 | |
| 26 | 2a | 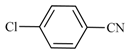 | 5 | 76 | 76 | 24 |
| 27 | 2b | 5 | 71 | 71 | 29 | |
| 28 | 2c | 5 | 88 | 88 | 22 | |
| 29 | 2d | 5 | 85 | 85 | 15 | |
| 30 | 2e | 5 | 89 | 90 | 10 | |
| 31 | 2a |  | 20 | 90 | 90 | 10 |
| 32 | 2b | 20 | 88 | 95 | 5 | |
| 33 | 2c | 20 | 95 | 100 | - | |
| 34 | 2d | 20 | 89 | 97 | 3 | |
| 35 | 2e | 20 | 83 | 85 | 15 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaloğlu, N.; Özdemir, İ.; Gürbüz, N.; Arslan, H.; Dixneuf, P.H. Ruthenium(η6,η1-arene-CH2-NHC) Catalysts for Direct Arylation of 2-Phenylpyridine with (Hetero)Aryl Chlorides in Water. Molecules 2018, 23, 647. https://doi.org/10.3390/molecules23030647
Kaloğlu N, Özdemir İ, Gürbüz N, Arslan H, Dixneuf PH. Ruthenium(η6,η1-arene-CH2-NHC) Catalysts for Direct Arylation of 2-Phenylpyridine with (Hetero)Aryl Chlorides in Water. Molecules. 2018; 23(3):647. https://doi.org/10.3390/molecules23030647
Chicago/Turabian StyleKaloğlu, Nazan, İsmail Özdemir, Nevin Gürbüz, Hakan Arslan, and Pierre H. Dixneuf. 2018. "Ruthenium(η6,η1-arene-CH2-NHC) Catalysts for Direct Arylation of 2-Phenylpyridine with (Hetero)Aryl Chlorides in Water" Molecules 23, no. 3: 647. https://doi.org/10.3390/molecules23030647