Probing the Inhibitor versus Chaperone Properties of sp2-Iminosugars towards Human β-Glucocerebrosidase: A Picomolar Chaperone for Gaucher Disease

 , and
, and
Abstract
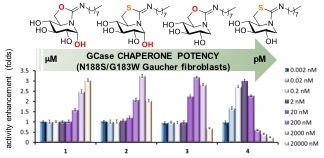
:
1. Introduction
2. Results
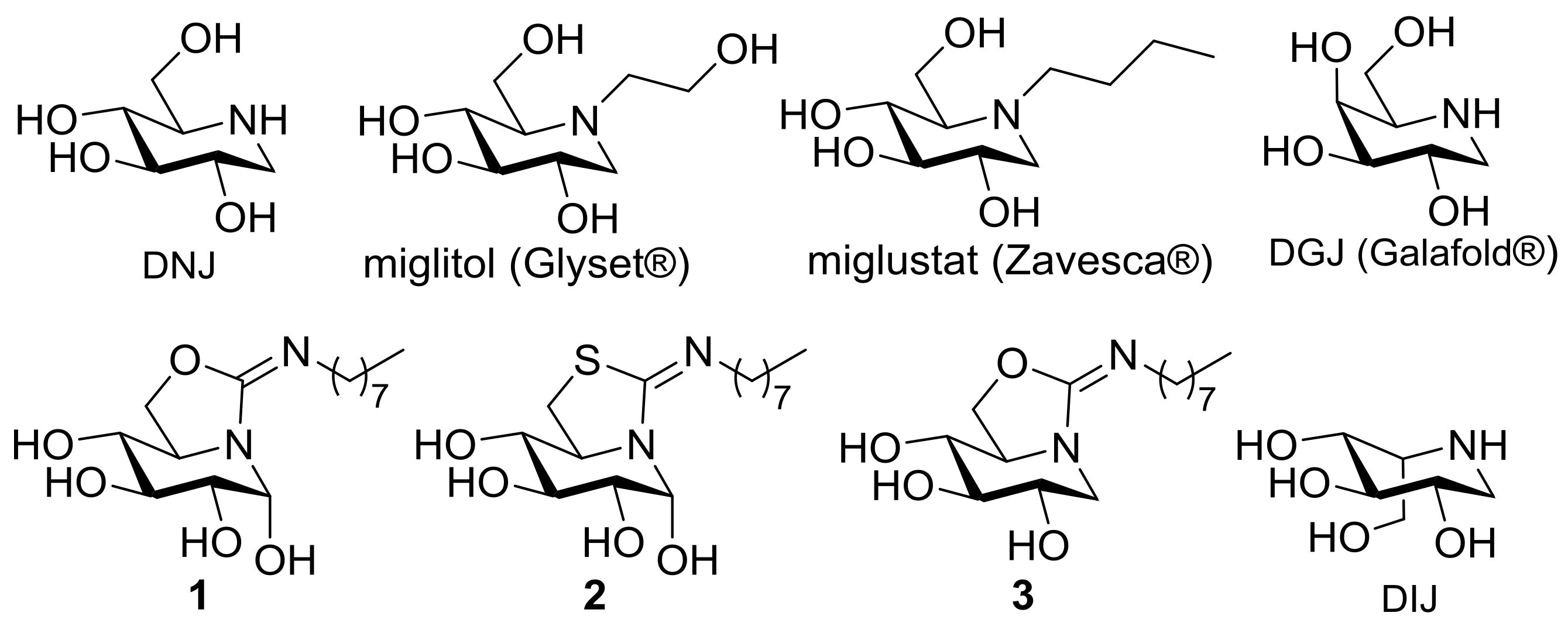
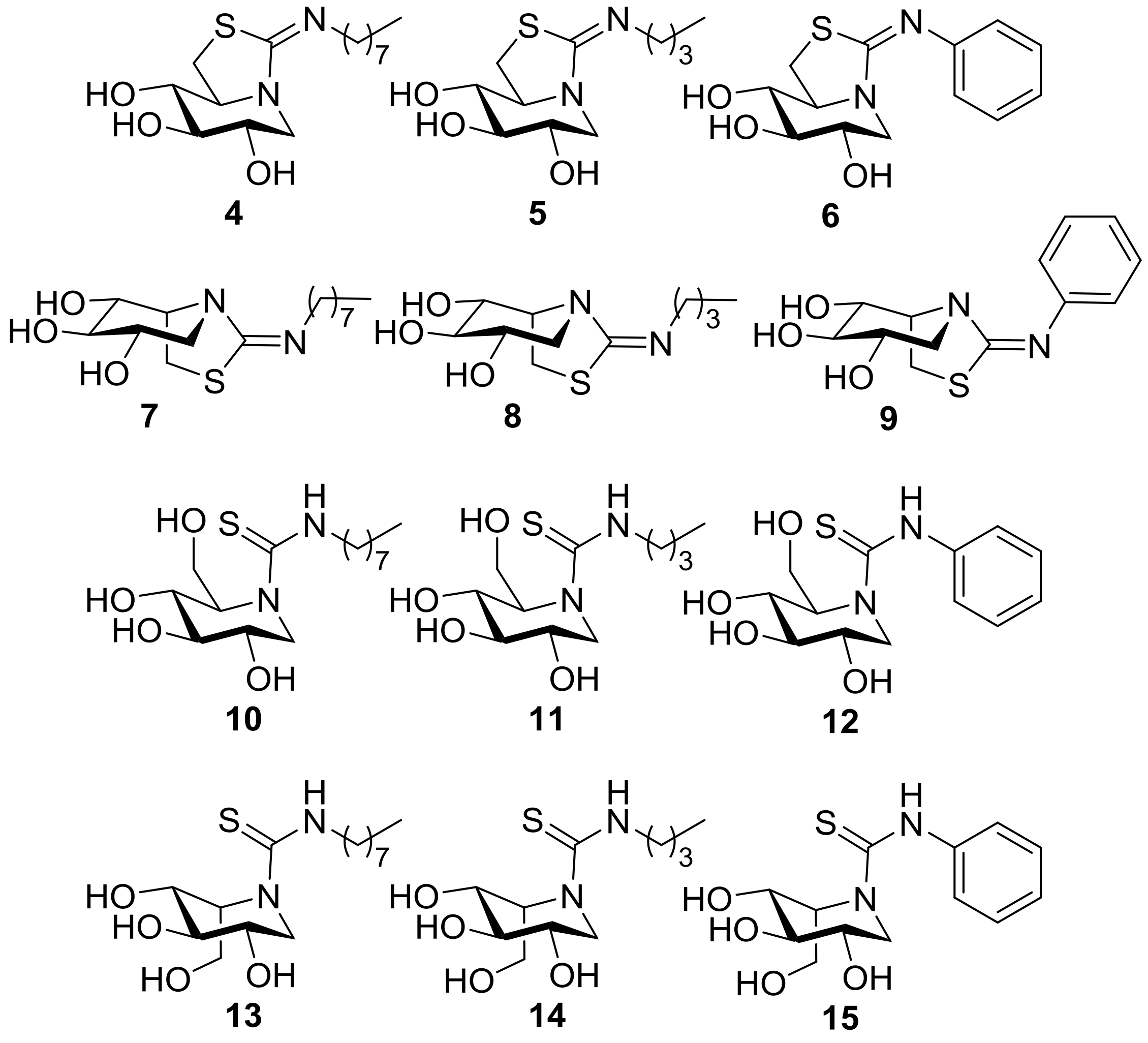
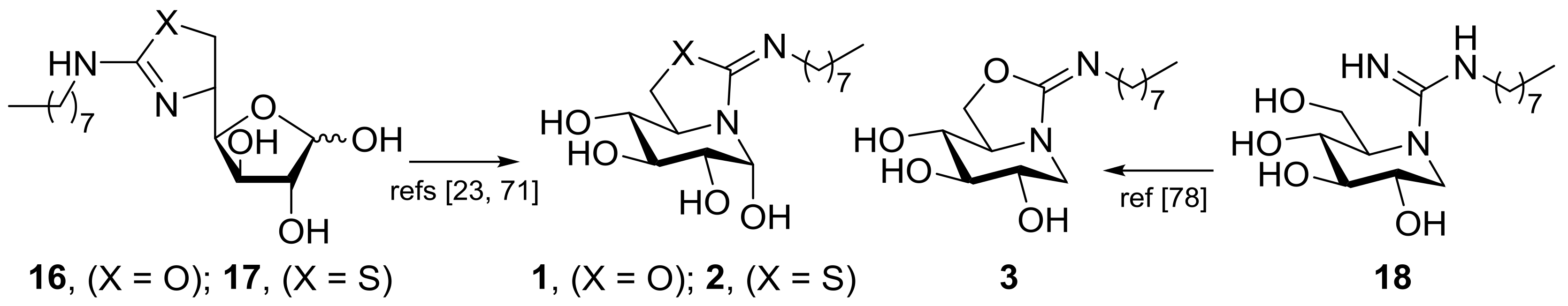
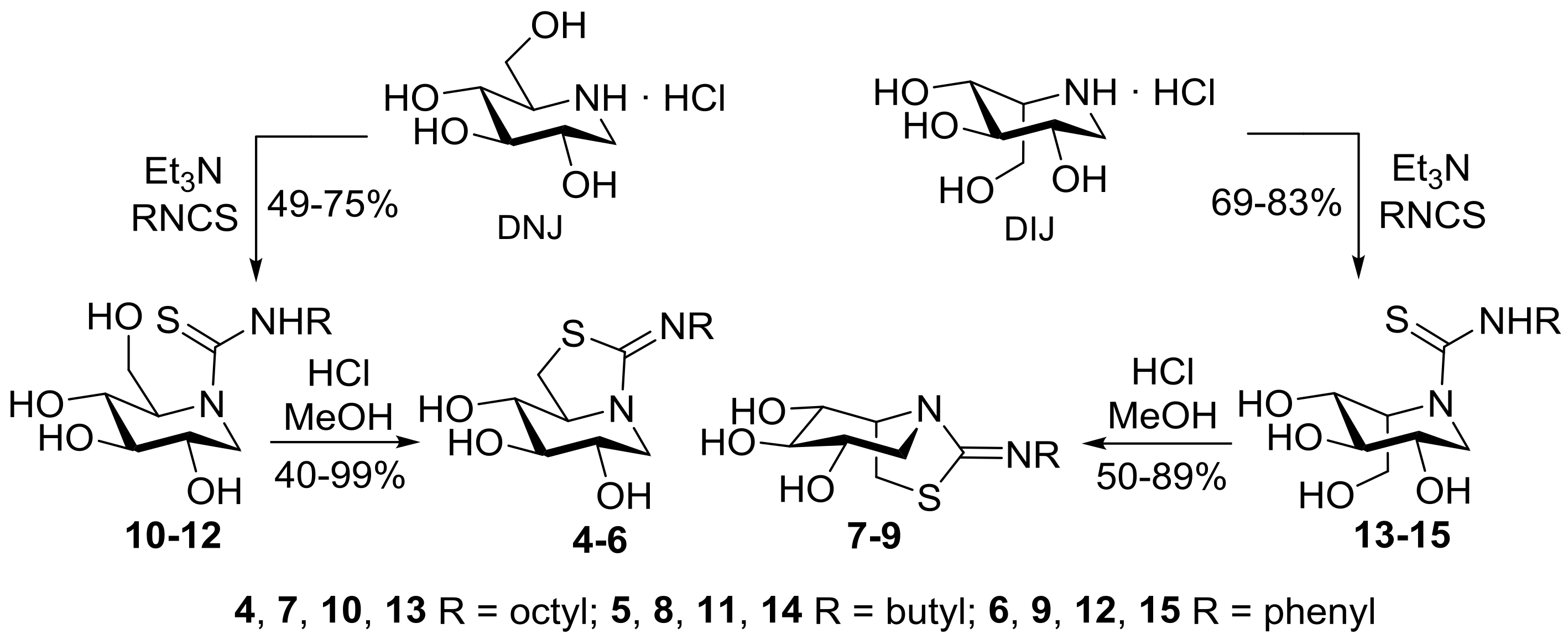
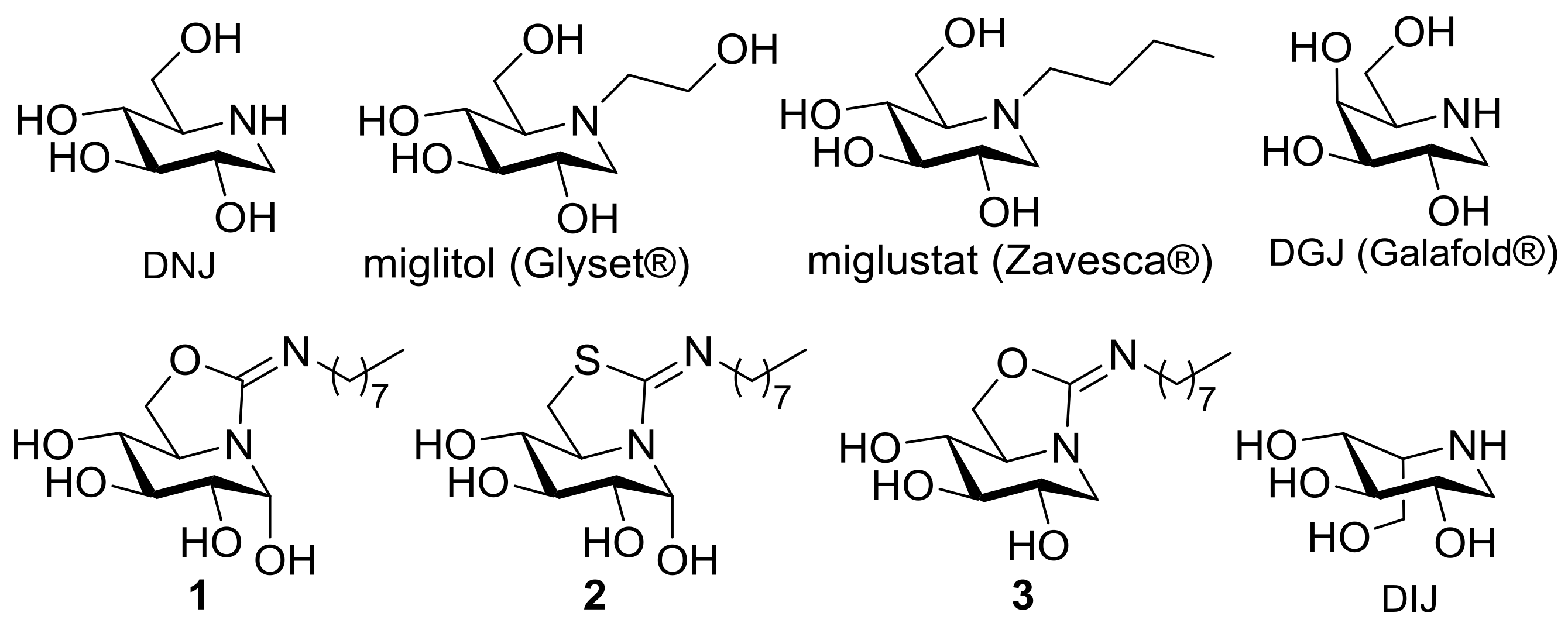
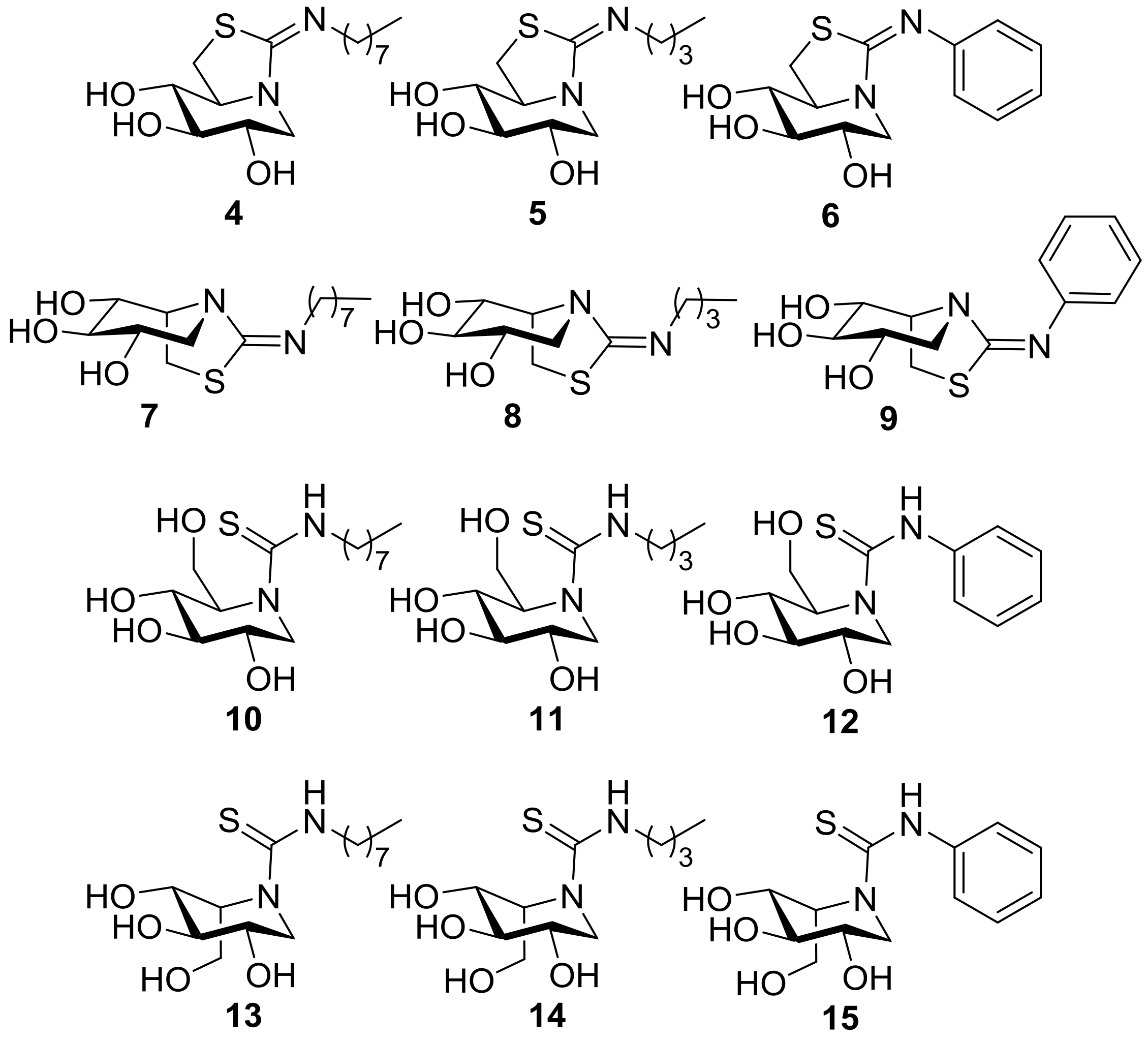
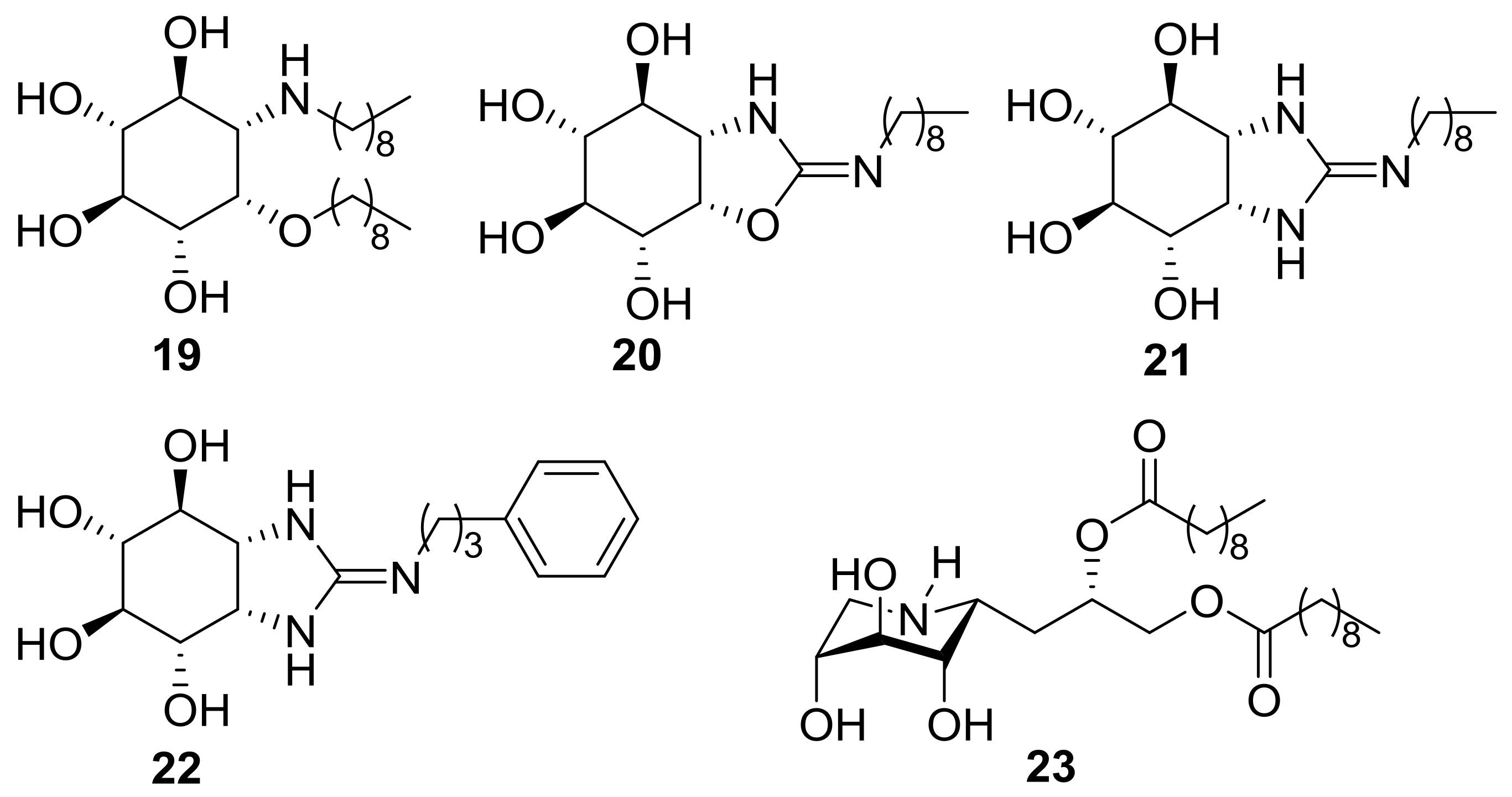
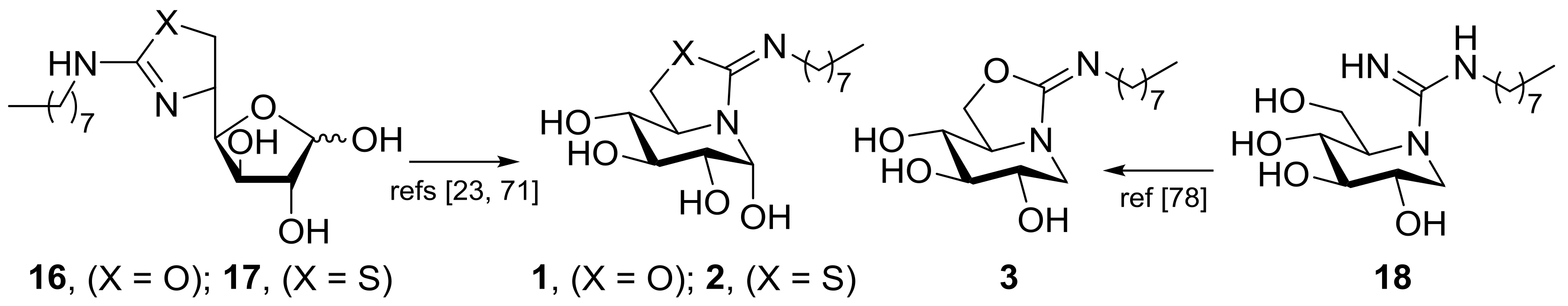
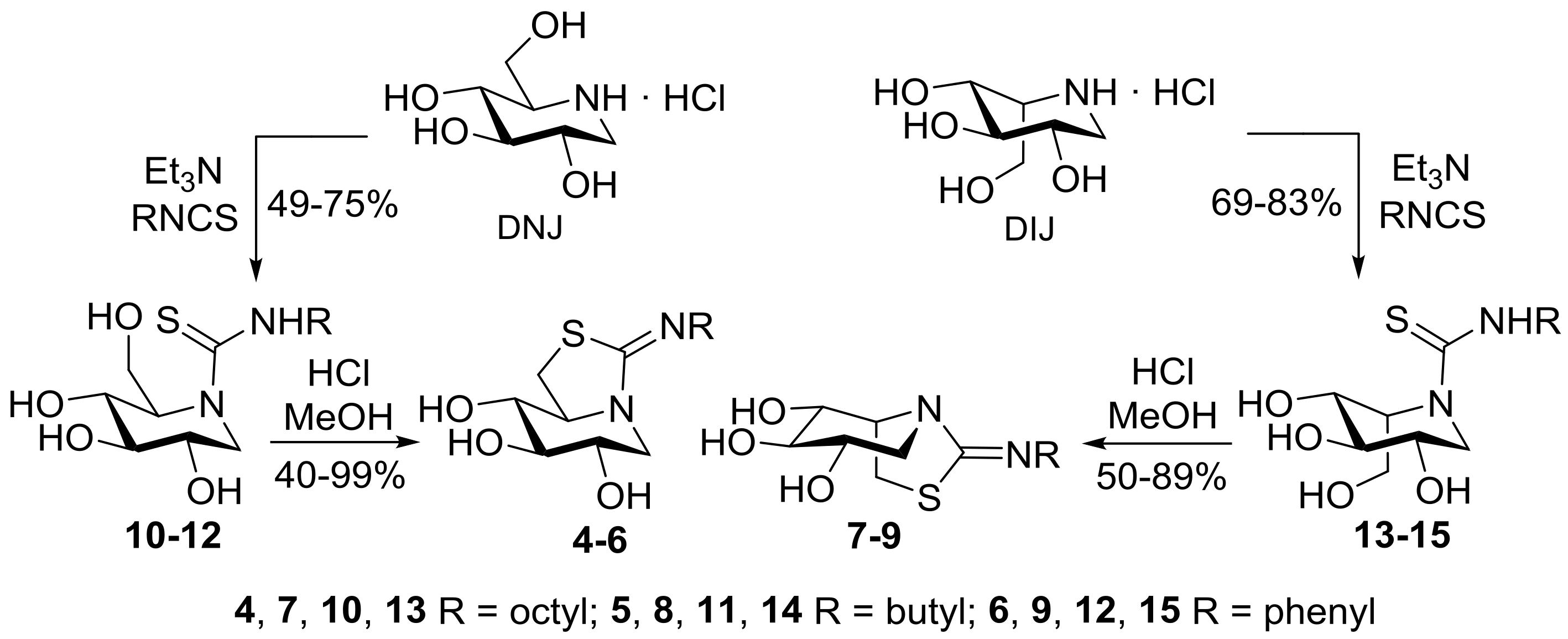
2.1. Synthesis
2.2. Inhibitory Properties against Commercial Enzymes
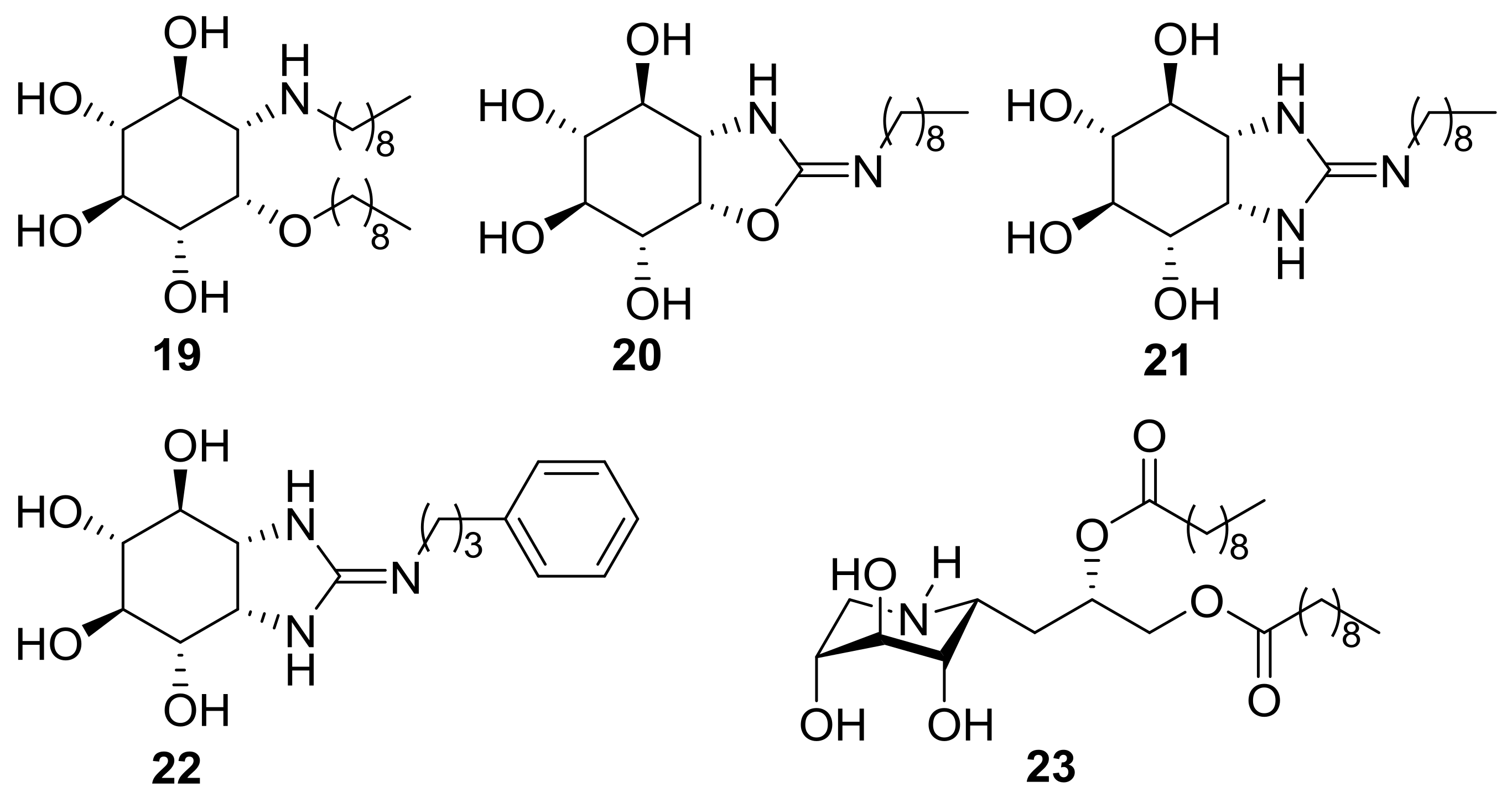
2.3. Inhibition Properties against Human GCase
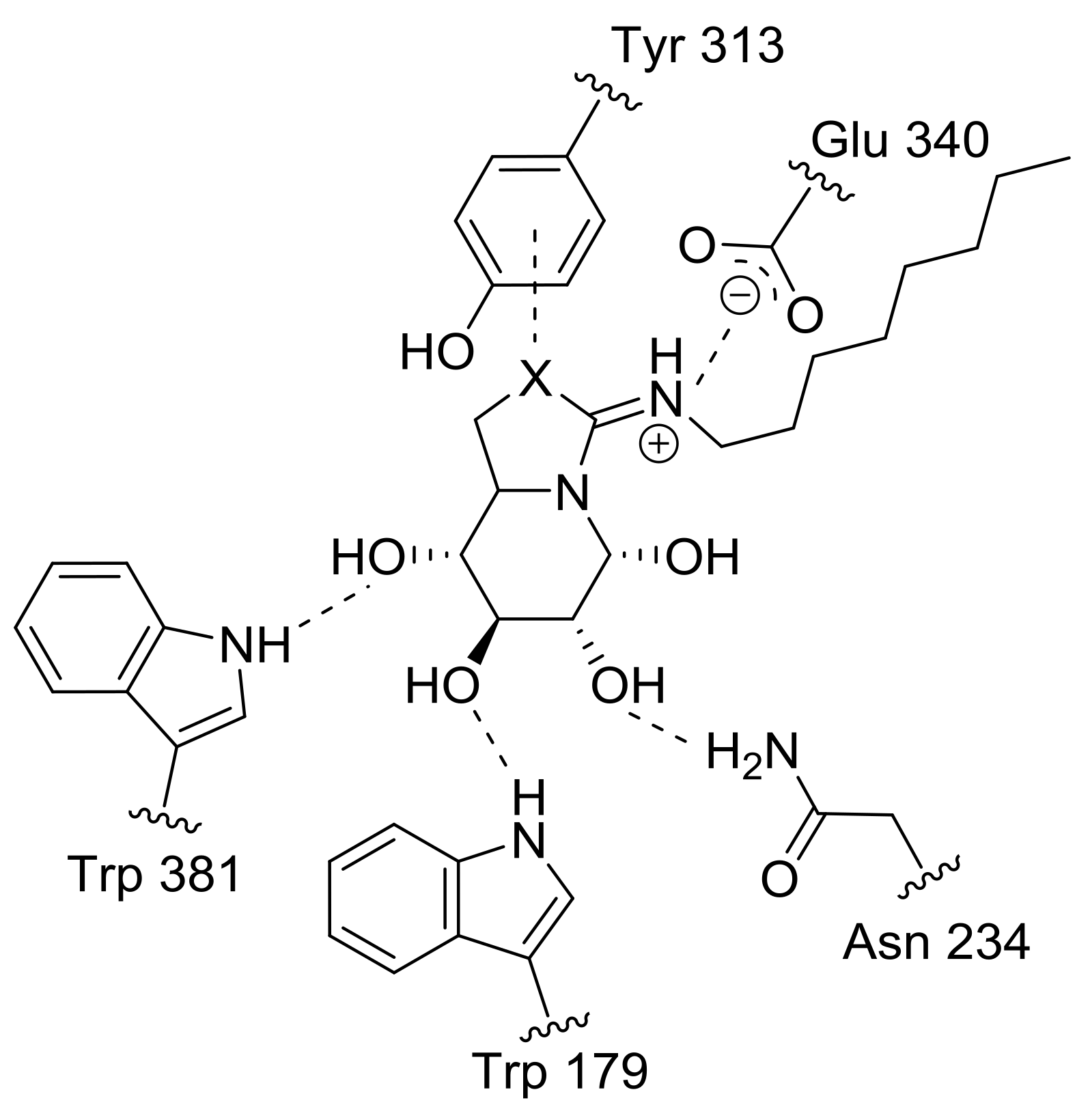
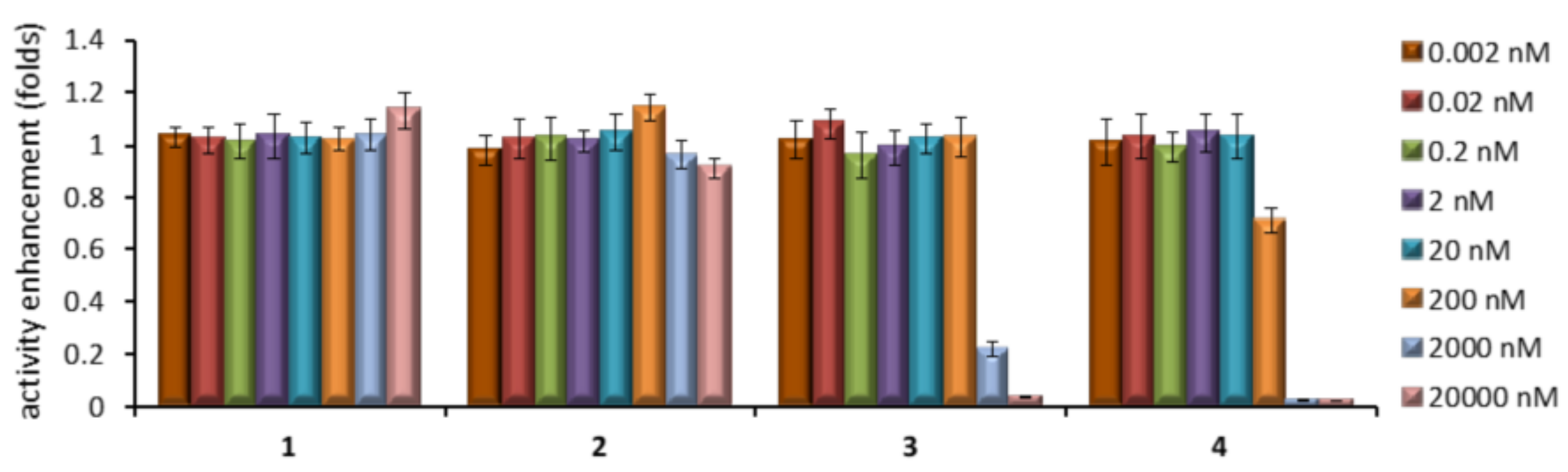
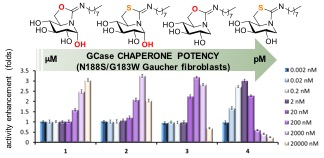
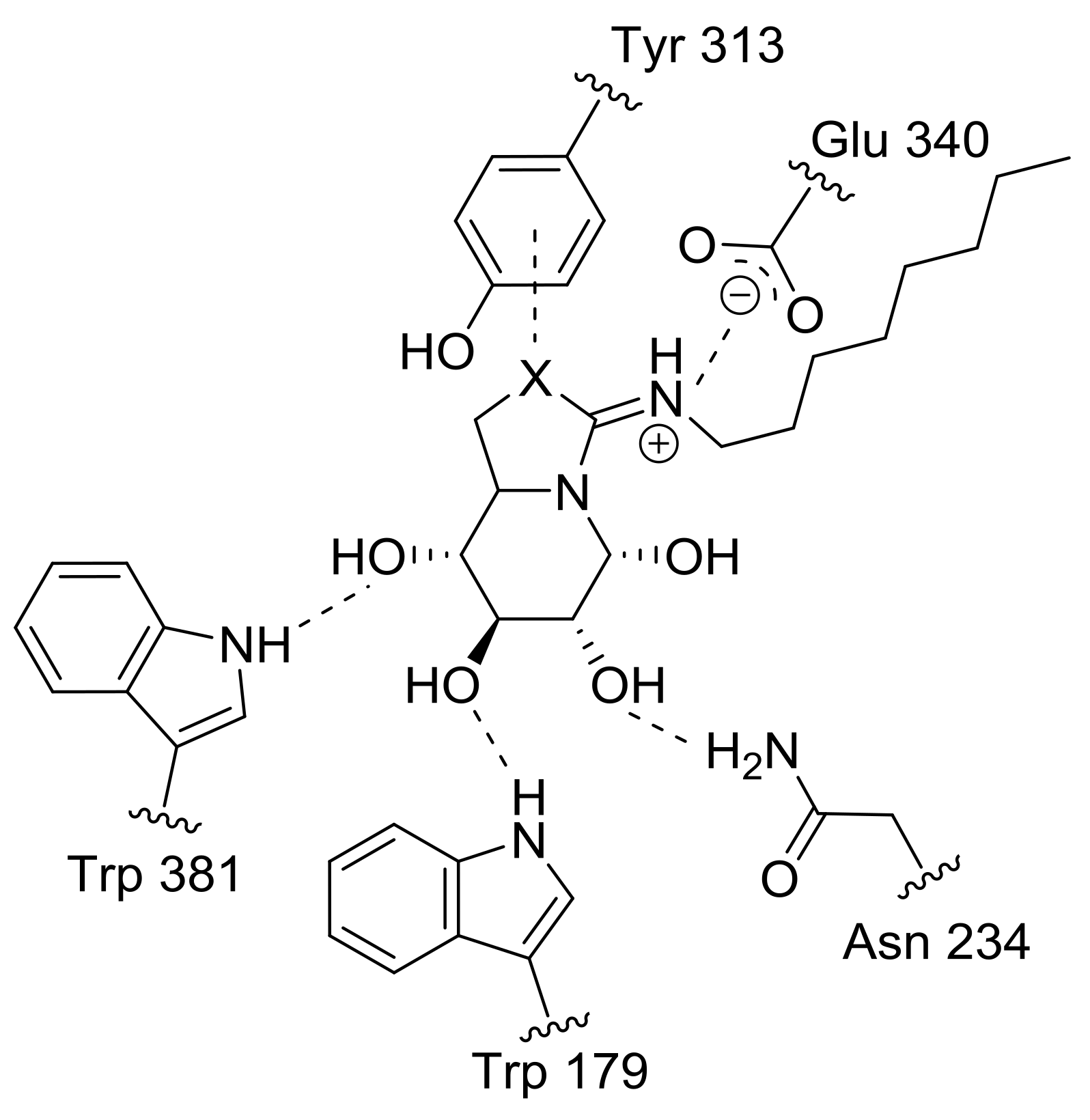
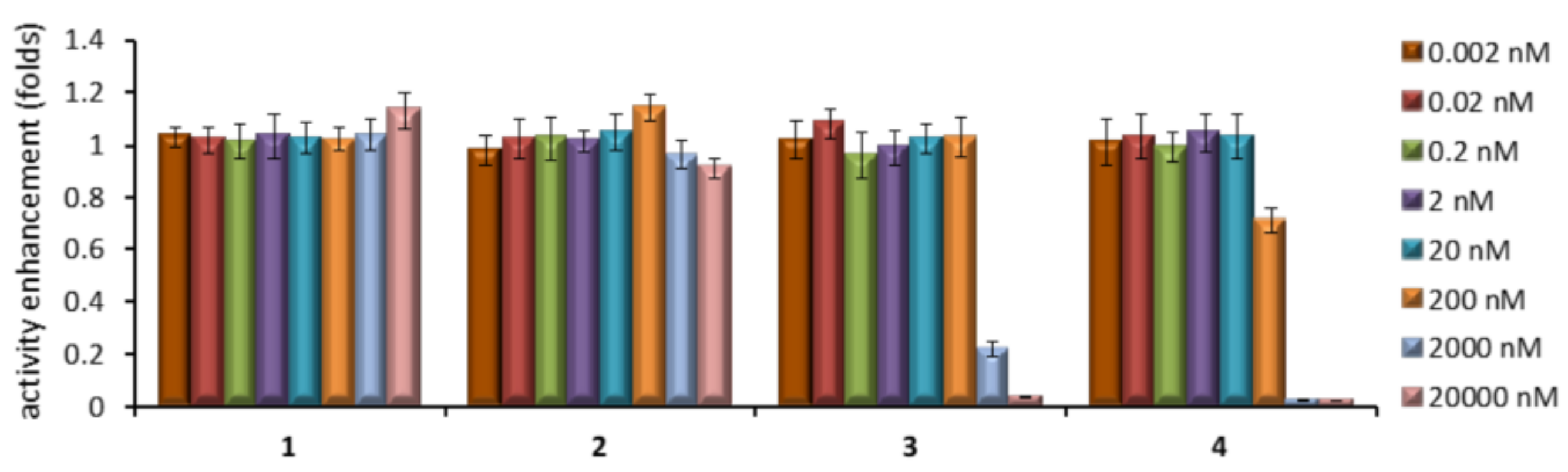
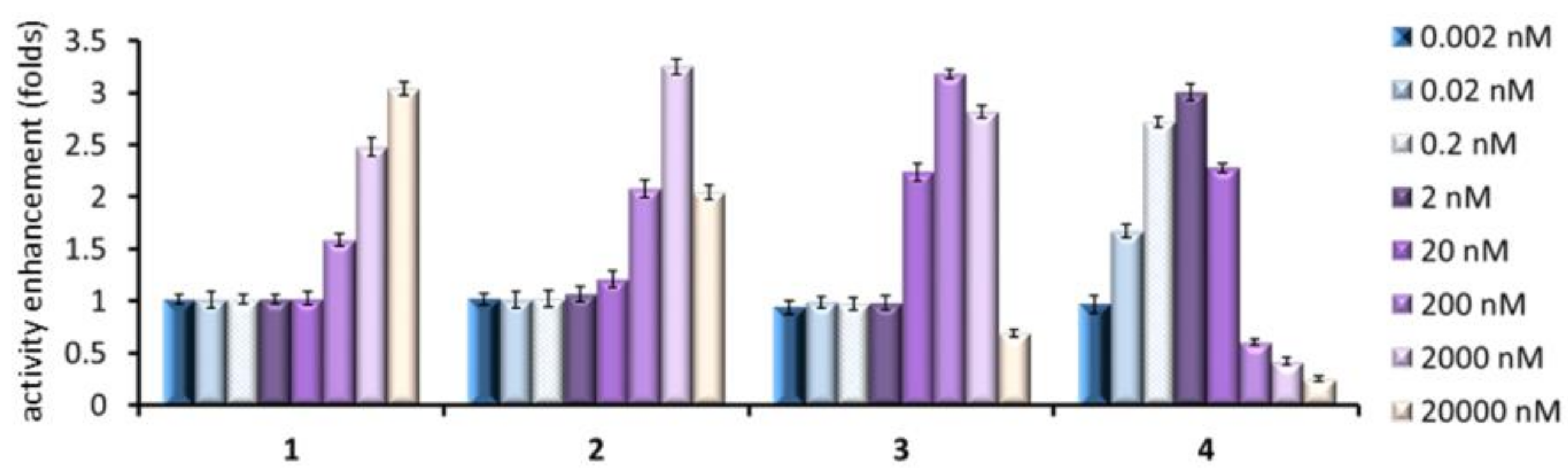
2.4. GCase Chaperoning Capabilities
3. Materials and Methods
3.1. General Methods
3.2. Commercial Enzyme Inhibition Assays
3.3. Measurement of Purified Human GCase Inhibition Activities In Vitro
3.4. Cell Cultures, Chaperone Tests, and Toxicity Assays
3.5. Statistical Analysis
3.6. General Procedure for the Synthesis of the DNJ-Related Thioureas 10–12
3.7. General Procedure for the Synthesis of the DNJ-Related Bicyclic Isothioureas 4–6
3.8. General Procedure for the Synthesis of the DIJ-Related Thioureas 13–15
3.9. General Procedure for the Synthesis of the DIJ-Related Bicyclic Isothioureas 7–9
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ernst, B.; Magnani, J.L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov. 2009, 8, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Horne, G.; Wilson, F.X.; Tinsley, J.; Williams, D.H.; Storer, R. Iminosugars past, present and future: Medicines for tomorrow. Drug Discov. Today 2011, 16, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Nash, R.J.; Kato, A.; Yu, C.-Y.; Fleet, G.W.J. Iminosugars as therapeutic agents: Recent advances and promising trends. Future Med. Chem. 2011, 3, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Horne, G. Iminosugars: Therapeutic Applications and Synthetic Considerations. In Carbohydrate as Drugs, 1st ed.; Seeberger, P.H., Rademacher, C., Eds.; Topics in Medicinal Chemistry; Springer International Publishing: Basel, Switzerland, 2014; Volume 12, pp. 23–52. ISBN 978-3-319-08674-3. [Google Scholar]
- Campbell, L.K.; Baker, D.E.; Campbell, R.K. Miglitol: Assessment of its Role in the Treatment of Patients with Diabetes Mellitus. Ann. Pharmacother. 2000, 34, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Migalastat: First Global Approval. Drugs 2016, 76, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassman, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-Month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.R.; Della Valle, M.C.; Wu, X.; Katz, E.; Pruthi, F.; Bond, S.; Bronfin, B.; Williams, H.; Yu, J.; Bichet, D.G.; et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet. Med. 2017, 19, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Valentao, P.; Andrade, P.B. Tuning protein folding in lysosomal storage diseases: The chemistry behind pharmacological chaperones. Chem. Sci. 2018, 9, 1740–1752. [Google Scholar] [CrossRef]
- Sánchez Fernández, E.M.; García Fernández, J.M.; Ortiz Mellet, C. Glycomimetic-based pharmacological chaperones for lysosomal storage disorders: Lessons from Gaucher, GM1-gangliosidosis and Fabry diseases. Chem. Commun. 2016, 52, 5497–5515. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Benito, J.M.; García Fernández, J.M.; Ortiz Mellet, C. Pharmacological chaperone therapy for Gaucher disease: A patent review. Expert Opin. Ther. Pat. 2011, 26, 885–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Fu, H. Protein Misfolding Diseases. Ann. Rev. Biochem. 2017, 86, 21–26. [Google Scholar] [CrossRef]
- Convertino, M.; Das, J.; Dokholyan, N.V. Pharmacological Chaperones: Design and Development of New Therapeutic Strategies for the Treatment of Conformational Diseases. ACS Chem. Biol. 2016, 11, 1471–1489. [Google Scholar] [CrossRef] [PubMed]
- Mena-Barragán, T.; Narita, A.; Matias, D.; Tiscornia, G.; Nanba, E.K.; Ohno, K.; Suzuki, Y.; Higaki, K.; García Fernández, J.M.; Ortiz Mellet, C. pH-Responsive Pharmacological Chaperones for Rescuing Mutant Glycosidase. Angew. Chem. Int. Ed. 2015, 54, 11696–11700. [Google Scholar] [CrossRef] [PubMed]
- Sevšek, A.; Sastre Toraño, J.; van Ufford, L.Q.; Moret, E.E.; Pieters, R.J.; Martin, N.I. Orthoester functionalized N-guanidino derivatives of 1,5-dideoxy-1,5-imino-d-xylitol as pH-responsive inhibitors of β-glucocerebrosidase. Med. Chem. Commun. 2017, 8, 2050–2054. [Google Scholar] [CrossRef]
- García Fernández, J.M.; Jiménez-Blanco, J.L.; Ortiz Mellet, C.; Fuentes, J.; Díaz-Perez, V.M. N-Thiocarbonyl azasugars: A new family of carbohydrate mimics with controlled anomeric configuration. Chem. Commun. 1997, 1969–1970. [Google Scholar] [CrossRef]
- García-Moreno, M.I.; Díaz-Pérez, P.; Ortiz Mellet, C.; García Fernández, J.M. Castanospermine-trehazoline hybrids: A new family of glycomimetics with tuneable glycosidase inhibitory properties. Chem. Commun. 2002, 848–849. [Google Scholar] [CrossRef]
- Luan, Z.; Higaki, K.; Aguilar-Moncayo, M.; Ninomiya, H.; Ohno, K.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; Suzuki, Y. Chaperone Activity of Bicyclic Nojirimycin Analogues for Gaucher Mutations in Comparison with N-(n-nonyl)-Deoxynojirimycin. ChemBioChem 2009, 10, 2780–2792. [Google Scholar] [CrossRef] [PubMed]
- Luan, Z.; Higaki, K.; Aguilar-Moncayo, M.; Li, L.; Ninomiya, H.; Nanba, E.; Ohno, K.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; et al. A Fluorescent sp2-Iminosugar with Pharmacological Chaperone Activity for Gaucher Disease: Synthesis and Intracellular Distribution Studies. ChemBioChem 2010, 11, 2453–2464. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, P.; Andreu, V.; Pino-Ángeles, A.; Moya-García, A.A.; García-Moreno, M.I.; Rodríguez-Rey, J.C.; Sánchez-Jiménez, F.; Pocoví, M.; Ortiz Mellet, C.; García Fernández, J.M.; et al. Bicyclic derivatives of l-idonojirimycin as pharmacological chaperones for neuronopathic forms of Gaucher disease. ChemBioChem 2013, 14, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Tiscornia, G.; Lorenzo Vivas, E.; Matalonga, L.; Berniakovich, I.; Barragán Monasterio, M.; Eguizábal Argaiz, C.; Gort, L.; González, F.; Ortiz Mellet, C.; García Fernández, J.M.; et al. Neuronopathic Gaucher’s disease: Induced pluripotent stem cells for disease modelling and testing chaperone activity of small compounds. Hum. Mol. Genet. 2013, 22, 633–645. [Google Scholar] [CrossRef] [PubMed]
- De la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; Garrido-Maraver, J.; Cordero, M.D.; Villanueva Paz, M.; Delgado Pavón, A.; Alcocer-Gómez, E.; de Lavera, I.; Ybot-González, P.; et al. Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant β-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Mena-Barragán, T.; Higaki, K.; Johnson, J.; Drury, J.; Lieberman, R.; Nakasone, N.; Ninomiya, H.; Tsukimura, T.; Sakuraba, H.; et al. Molecular basis of 1-deoxygalactonojirimycin arylthiourea binding to human α-galactosidase: Pharmacological chaperoning efficacy on Fabry disease mutants. ACS Chem. Biol. 2014, 9, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Takai, T.; Higaki, K.; Aguilar-Moncayo, M.; Mena-Barragán, T.; Hirano, Y.; Yura, K.; Yu, L.; Ninomiya, H.; García-Moreno, M.I.; Sakakibara, Y.; et al. A Bicyclic 1-Deoxygalactonojirimycin Derivative as Novel Pharmacological Chaperone for GM1 Gangliosidosis. Mol. Ther. 2013, 21, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Ohto, U.; Higaki, K.; Mena-Barragan, T.; Aguilar-Moncayo, M.; Ortiz Mellet, C.; Nanba, E.; García Fernández, J.M.; Suzuki, Y.; Shimizu, T. Structural basis of pharmacological chaperoning for human β-galactosidase. J. Biol. Chem. 2014, 289, 14560–14568. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, A.; Rísquez Cuadro, R.; Verdaguer, X.; García Fernández, J.M.; Nanba, K.; Higaki, K.; Ortiz Mellet, C.; Riera, A. Efficient Stereoselective Synthesis of 2-Acetamido-1,2-dideoxyallonojirimycin (DAJNAc) and sp2-Iminosugar Conjugates: Novel Hexosaminidase Inhibitors with Discrimination Capabilities between the Mature and Precursor Forms of the Enzyme. Eur. J. Med. Chem. 2016, 121, 926–938. [Google Scholar] [CrossRef] [PubMed]
- García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Polyhydroxylated N-(thio)carbamoyl piperidines: Nojirimycin-type glycomimetics with controlled anomeric configuration. Tetrahedron Asymmetry 1999, 10, 4271–4275. [Google Scholar] [CrossRef]
- García-Moreno, M.I.; Benito-Hernández, J.M.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis and evaluation of calystegine B2 analogues as glycosidase inhibitors. J. Org. Chem. 2001, 66, 7604–7614. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Moncayo, M.; Díaz-Pérez, P.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis and biological evaluation of guanidine-type iminosugars. J. Org. Chem. 2008, 73, 1995–1998. [Google Scholar] [CrossRef] [PubMed]
- Mena Barragán, T.; García Moreno, M.I.; Nanba, E.; Higaki, K.; Lisa Concia, A.; Clapés, P.; García Fernández, J.M.; Ortiz Mellet, C. Inhibitor versus chaperone behaviour of fagomine, DAB and LAB sp2-iminosugar conjugates against glycosidases: A structure-activity relationship study in Gaucher fibroblasts. Eur. J. Med. Chem. 2016, 121, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Sevšek, A.; Šrot, L.; Rihter, J.; Čelan, M.; van Ufford, L.Q.; Moret, E.E.; Martin, N.I.; Pieters, R.J. N-Guanidino Derivatives of 1,5-Dideoxy-1,5-imino-d-xylitol are Potent, Selective, and Stable Inhibitors of β-Glucocerebrosidase. ChemMedChem 2017, 12, 483–486. [Google Scholar] [CrossRef] [PubMed]
- García Fernández, J.M.; Ortiz Mellet, C.; Benito-Hernández, J.M.; Fuentes-Mota, J. Synthesis of calystegine B2 analogs by tandem tautomerization-intramolecular glycosylation of thioureidosugars. Synlett 1998, 3, 316–318. [Google Scholar] [CrossRef]
- Díaz-Pérez, P.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis of (1S,2S,3R,8S,8aR)-1,2,3,8-tetrahydroxy-6-oxa-5-thioindolizidine: A stable reducing swainsonine analog with controlled anomeric configuration. Synlett 2003, 3, 341–344. [Google Scholar]
- García-Moreno, M.I.; Rodríguez-Lucena, D.; Ortiz Mellet, C.; García Fernández, J.M. Pseudoamide-type pyrrolidine and pyrrolizidine glycomimetics and their inhibitory activities against glycosidases. J. Org. Chem. 2004, 69, 3578–3581. [Google Scholar] [CrossRef] [PubMed]
- Benltifa, M.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; Wadouachi, A. Synthesis and evaluation of sulfamide-type indolizidines as glycosidase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2805–2808. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Moncayo, M.; Gloster, T.M.; García-Moreno, M.I.; Ortiz Mellet, C.; Davies, G.J.; Llebaria-Soldevilla, A.; Casas-Brugulat, J.; Egido-Gabás, M.; García Fernández, J.M. Molecular basis for β-glucosidase inhibition by ring-modified calystegine analogues. ChemBioChem 2008, 9, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Moncayo, M.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis of thiohydantoine-castanospermine glycomimetics as glycosidase inhibitors. J. Org. Chem. 2009, 74, 3595–3598. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Sánchez-Fernández, E.M.; Ortiz Mellet, C.; Tatibouët, A.; Rauter, A.P.; Rollin, P. N-thiocarbonyl iminosugars: Synthesis and evaluation of castanospermine analogues bearing oxazole-2(3H)-thione moieties. Eur. J. Org. Chem. 2013, 2013, 7941–7951. [Google Scholar] [CrossRef]
- Sánchez-Fernández, E.M.; Álvarez, E.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis of Multibranched Australine Derivatives from Reducing Castanospermine Analogues through the Amadori Rearrangement of gem-Diamine Intermediates: Selective Inhibitors of β-Glucosidase. J. Org. Chem. 2014, 79, 11722–11728. [Google Scholar] [CrossRef] [PubMed]
- García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis of calystegine B-2, B-3, and B-4 analogues: Mapping the structure-glycosidase inhibitory activity relationships in the 1-deoxy-6-oxacalystegine series. Eur. J. Org. Chem. 2004, 8, 1803–1819. [Google Scholar] [CrossRef]
- García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis and biological evaluation of 6-oxa-nor-tropane glycomimetics as glycoside inhibitors. Tetrahedron 2007, 63, 7879–7884. [Google Scholar] [CrossRef]
- Aguilar-Moncayo, M.; García-Moreno, M.I.; Trapero, A.; Egido-Gabás, M.; Llebaria, A.; García Fernández, J.M.; Ortiz Mellet, C. Bicyclic (galacto)nojirimycin analogues as glycosidase inhibitors: Effect of structural modifications in their pharmacological chaperone potential towards β-glucocerebrosidase. Org. Biomol. Chem. 2011, 9, 3698–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Moncayo, M.; Díaz-Pérez, P.; García Fernández, J.M.; Ortiz Mellet, C.; García-Moreno, M.I. Synthesis and glycosidase inhibitory activity of isourea-type bicyclic sp2-iminosugars related to galactonojirimycin and allonojirimycin. Tetrahedron 2012, 68, 681–689. [Google Scholar] [CrossRef]
- Aguilar-Moncayo, M.; García-Moreno, M.I.; Stütz, A.E.; García Fernández, J.M.; Wrodnigg, T.M.; Ortiz Mellet, C. Fluorescent-tagged sp2-iminosugars with potent β-glucosidase inhibitory activity. Bioorg. Med. Chem. 2010, 18, 7439–7445. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Moncayo, M.; Takai, T.; Higaki, K.; Mena-Barragán, T.; Hirano, Y.; Yura, K.; Li, L.; Yu, Y.; Ninomiya, H.; García-Moreno, M.I.; et al. Tuning glycosidase inhibition through aglycone interactions: Pharmacological chaperones for Fabry disease and GM1 gangliosidosis. Chem. Commun. 2012, 48, 6514–6516. [Google Scholar] [CrossRef] [PubMed]
- García-Moreno, M.I.; de la Mata, M.; Sánchez-Fernández, E.M.; Benito, J.M.; Díaz-Quintana, A.; Fustero, S.; Nanba, E.; Higaki, K.; Sánchez-Alcázar, J.A.; García Fernández, J.M.; et al. Fluorinated Chaperone—β-Cyclodextrin Formulations for β-Glucocerebrosidase Activity Enhancement in Neuronopathic Gaucher Disease. J. Med. Chem. 2017, 60, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, J.; Lutz, M.; Zuilhof, H.; Wennekes, T. Exploring the Chemistry of Bicyclic Isoxazolidines for the Multicomponent Synthesis of Glycomimetic Building Blocks. J. Org. Chem. 2016, 81, 8826–8836. [Google Scholar] [CrossRef] [PubMed]
- Zoidl, M.; Gonzalez Santana, A.; Torvisco, A.; Tysoe, C.; Siriwardena, A.; Withers, S.G.; Wrodnigg, T.M. The Staudinger/aza-Wittig/Grignard reaction as key step for the concise synthesis of 1-C-Alkyl-iminoalditol glycomimetics. Carbohydr. Res. 2016, 429, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Bergeron-Brlek, M.; Meanwell, M.; Britton, R. Direct synthesis of imino-C-nucleoside analogues and other biologically active iminosugars. Nat. Commun. 2015, 6, 6903. [Google Scholar] [CrossRef] [PubMed]
- Biela-Banas, A.; Gallienne, E.; Front, S.; Martin, O.R. Stereoselective synthesis of 1-C-alkyl iminogalactitol derivatives, potential chaperones for galactosidase-linked LSDs: A real challenge. Tetrahedron Lett. 2014, 55, 838–841. [Google Scholar] [CrossRef]
- Sánchez-Fernández, E.M.; Rísquez-Cuadro, R.; Aguilar-Moncayo, M.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Generalized anomeric effect in gem-diamines: Stereoselective synthesis of α-N-linked disaccharide mimics. Org. Lett. 2009, 11, 3306–3309. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, E.M.; Rísquez-Cuadro, R.; Chasseraud, M.; Ahidouch, A.; Ortiz Mellet, C.; Ouadid-Ahidouch, H.; García Fernández, J.M. Synthesis of N-, S-, and C-Glycoside castanospermine analogues with selective neutral α-glucosidase inhibitory activity as antitumor agents. Chem. Commun. 2010, 46, 5328–5330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez Fernández, E.M.; Rísquez-Cuadro, R.; Ortiz Mellet, C.; García Fernández, J.M.; Nieto, P.M.; Angulo, J. sp2-Iminosugar O-, S- and N-glycosides as conformational mimics of α-linked disaccharides: Implications for glycosidase inhibition. Chem. Eur. J. 2012, 18, 8527–8539. [Google Scholar] [CrossRef] [PubMed]
- Allan, G.; Ouadid-Ahidouch, H.; Sánchez Fernández, E.M.; Rísquez Cuadro, R.; García Fernández, J.M.; Ortiz Mellet, C.; Ahidouch, A. New Castanospermine glycoside analogues inhibit breast cancer cell proliferation and induce apoptosis without affecting normal cells. PLoS ONE 2013, 8, e76411. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Alcalde-Estevez, E.; García-Ramírez, M.; Cazzoni, D.; de la Villa, P.; Sánchez Fernández, E.M.; Ortiz Mellet, C.; García Fernández, J.M.; Hernández, C.; Simo, R.; et al. Modulation of microglia polarization dynamics during diabetic retinopathy in db/db mice. BBA Mol. Basis Dis. 2016, 1862, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Fernández, E.M.; Navo, C.; Martínez-Saez, N.; Gonçalves-Pereira, R.; Somovilla, V.J.; Avenoza, A.; Busto, J.H.; Bernardes, G.J.L.; Jiménez-Osés, G.; Corzana, F.; et al. Tn Antigen Mimics based on sp2-Iminosugars with Affinity for an anti-MUC1 Antibody. Org. Lett. 2016, 18, 3890–3893. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, E.M.; Gonçalves-Pereira, R.; Rísquez-Cuadro, R.; Plata, G.B.; Padrón, J.M.; García Fernández, J.M.; Ortiz Mellet, C. Influence of the configurational pattern of sp2-iminosugar pseudo N-, S-, O- and C-glycosides on their glycoside inhibitory and antitumor properties. Carbohydr. Res. 2016, 429, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Allan, G.; Gueder, N.; Telliez, M.-S.; Hague, F.; Sánchez Fernández, E.M.; García Fernández, J.M.; Ortiz Mellet, C.; Ahidouch, A.; Ouadid-Ahidouch, H. sp2-Iminosugar α-glucosidase inhibitor 1-C-octyl-2-oxa-3-oxocastanospermine specifically affected breast cancer cell migration through Stim1, β1-integrin, and FAK signaling pathways. J. Cell. Physiol. 2017, 232, 3631–3640. [Google Scholar] [CrossRef]
- Alcalde-Estevez, E.; Arroba, A.I.; Sánchez Fernández, E.M.; Ortiz Mellet, C.; García Fernández, J.M.; Masgrau, L.; Valverde, A.M. The sp2-iminosugar glycolipid 1-dodecylsulfonyl-5N,6O-oxomethylidenenojirimycin (DSO2-ONJ) as selective anti-inflammatory agent by modulation of hemeoxygenase-1 in Bv.2 microglial cells and retinal explants. Food Chem. Toxicol. 2018, 111, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, M. Amphiphilic glycoconjugates as potential anti-cancer chemotherapeutics. Eur. J. Med. Chem. 2018, 143, 1208–1253. [Google Scholar] [CrossRef] [PubMed]
- Rísquez-Cuadro, R.; García Fernández, J.M.; Nierengarten, J.-F.; Ortiz Mellet, C. Fullerene-sp2-iminosugar balls as multimodal ligands for lectins and glycosidases: A mechanistic hypothesis for the inhibitory multivalent. Chem. Eur. J. 2013, 19, 16791–16803. [Google Scholar] [CrossRef] [PubMed]
- Abellán Flos, M.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; Nierengarten, J.-F.; Vincent, S.P. Potent glycosidase inhibition with heterovalent fullerenes: Unveiling the binding modes triggering multivalent inhibition. Chem. Eur. J. 2016, 22, 11450–11460. [Google Scholar] [CrossRef] [PubMed]
- García-Moreno, M.I.; Ortega-Caballero, F.; Rísquez-Cuadro, R.; Ortiz Mellet, C.; García Fernández, J.M. The Impact of Heteromultivalency in Lectin Recognition and Glycosidase Inhibition: An Integrated Mechanistic Study. Chem. Eur. J. 2017, 23, 6295–6304. [Google Scholar] [CrossRef] [PubMed]
- García Fernández, J.M.; Nierengarten, J.-F.; Ortiz Mellet, C. Multivalency as an action principle in multimodal lectin recognition and glycosidase inhibition: A paradigm shift driven by carbon-based glyconanomaterials. J. Mater. Chem. B 2017, 5, 6428–6436. [Google Scholar] [CrossRef]
- Díaz-Pérez, V.; García-Moreno, M.I.; Ortiz Mellet, C.; Fuentes-Mota, J.; Díaz-Arribas, J.C.; Cañada, J.; García Fernández, J.M. Generalized anomeric effect in action: Synthesis and evaluation of stable reducing indolizidine glycomimetics as glycosidase inhibitors. J. Org. Chem. 2000, 65, 136–143. [Google Scholar] [CrossRef] [PubMed]
- García-Moreno, M.I.; Díaz-Pérez, P.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis and evaluation of isourea-type glycomimetics related to the indolizidines and trehazolin glycosidase inhibitor families. J. Org. Chem. 2003, 68, 8890–8901. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Pérez, P.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis and comparative glycosidase inhibitory properties of reducing castanospermine analogues. Eur. J. Org. Chem. 2005, 2903–2913. [Google Scholar] [CrossRef]
- Aguilar-Moncayo, M.; Gloster, T.M.; Turkenburg, J.P.; García-Moreno, M.I.; Ortiz Mellet, C.; Davies, G.J.; García Fernández, J.M. Glycosidase inhibition by ring-modified castanospermine analogues: Tackling enzyme selectivity by inhibitor tailoring. Org. Biomol. Chem. 2009, 7, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lavado, J.; de la Mata Fernández, M.; Jiménez Blanco, J.L.; García-Moreno, M.I.; Benito, J.M.; Díaz-Quintana, A.; Sánchez Alcázar, J.A.; Higaki, K.; Nanba, E.; Ohno, K.; et al. Targeted Delivery of Pharmacological Chaperones for Gaucher Disease to Macrophages by a Mannosylated Cyclodextrin Carrier. Org. Biomol. Chem. 2014, 12, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Brumshtein, B.; Aguilar-Moncayo, M.; Benito, J.M.; García Fernandez, J.M.; Silman, I.; Shaaltiel, Y.; Aviezer, D.; Sussman, J.L.; Futerman, A.H.; Ortiz Mellet, C. Cyclodextrin-mediated crystallization of acid β-glucosidase in complex with amphiphilic bicyclic nojirimycin analogues. Org. Biomol. Chem. 2011, 9, 4160–4167. [Google Scholar] [CrossRef] [PubMed]
- Brumshtein, B.; Aguilar-Moncayo, M.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; Silman, I.; Shaaltiel, J.; Aviezer, D.; Sussman, J.L.; Futerman, A.H. 6-Amino-6-deoxy-5,6-di-N-(N′-octyliminomethylidene)nojirimycin: Synthesis, biological evaluation, and crystal structure in complex with acid beta-glucosidase. ChemBioChem 2009, 10, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Sevšek, A.; Čelan, M.; Erjavec, B.; van Ufford, L.Q.; Sastre Toraño, J.; Moret, E.E.; Pieters, R.J.; Martin, N.I. Bicyclic isoureas derived from 1-deoxynojirimycinare potent inhibitors of β-glucocerebrosidase. Org. Biomol. Chem. 2016, 14, 8670–8673. [Google Scholar] [CrossRef] [PubMed]
- Wennekes, T.; van den Berg, R.J.B.H.N.; Boltje, T.J.; Wilma, E.; Donker-Koopman, W.E.; Kuijper, B.; van der Marel, G.A.; Strijland, A.; Verhagen, C.P.; Aerts, J.M.F.G.; et al. Synthesis and Evaluation of Lipophilic Aza-C-glycosides as Inhibitors of Glucosylceramide Metabolism. Eur. J. Org. Chem. 2010, 1258–1283. [Google Scholar] [CrossRef]
- Compain, P. Searching for Glycomimetics That Target Protein Misfolding in Rare Diseases: Successes, Failures, and Unexpected Progress Made in Organic Synthesis. Synlett 2014, 25, 1215–1240. [Google Scholar] [CrossRef]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; PenningtonL, D.; Meanwell, N.A. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.O. Discovering Protein-Ligand Chalcogen Bonding in the Protein Data Bank Using Endocyclic Sulfur-Containing Heterocycles as Ligand Search Subsets. J. Mol. Model. 2017, 23, 287. [Google Scholar] [CrossRef] [PubMed]
- Motherwell, W.B.; Moreno, R.B.; Pavlakos, I.; Arendorf, J.R.T.; Arif, T.; Tizzard, G.J.; Coles, S.J.; Aliev, A.E. Noncovalent Interactions of π Systems with Sulfur: The Atomic Chameleon of Molecular Recognition. Angew. Chem. Int. Ed. Engl. 2018, 26, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Daeffler, K.N.-M.; Lester, H.A.; Dougherty, D.A. Functionally Important Aromatic–Aromatic and Sulfur−π Interactions in the D2 Dopamine Receptor. J. Am. Chem. Soc. 2012, 134, 14890–14896. [Google Scholar] [CrossRef] [PubMed]
- Sawkar, A.R.; Cheng, W.-C.; Beutler, E.; Wong, C.-H.; Balch, W.E.; Kelly, J.W. Chemical chaperones increase the cellular activity of N370S β-glucosidase: A therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci. USA 2002, 99, 15428–15433. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.K.; Shen, D.L.; Shan, X.; He, X.; Kermode, A.R.; Vocadlo, D.J. Fluorescence-quenched substrates for live cell imaging of human glucocerebrosidase activity. J. Am. Chem. Soc. 2015, 137, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Trapero, A.; González-Bulnes, P.; Butters, T.D.; Llebaria, A. Potent Aminocyclitol Glucocerebrosidase Inhibitors are Subnanomolar Pharmacological Chaperones for Treating GaucherDisease. J. Med. Chem. 2012, 55, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Trapero, A.; Alfonso, I.; Butters, T.D.; Llebaria, A. Polyhydroxylated Bicyclic Isoureas and Guanidines Are Potent Glucocerebrosidase Inhibitors and Nanomolar Enzyme Activity Enhancers in Gaucher Cells. J. Am. Chem. Soc. 2011, 133, 5474–5484. [Google Scholar] [CrossRef] [PubMed]
- Schönemann, W.; Gallienne, E.; Ikeda-Obatake, K.; Asano, N.; Nakagawa, S.; Kato, A.; Adachi, I.; Górecki, M.; Frelek, J.; Martin, O.R. Glucosylceramide Mimics: Highly Potent GCase Inhibitors and Selective Pharmacological Chaperones for Mutations Associated with Types 1 and 2 Gaucher Disease. ChemMedChem 2013, 8, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R.; Soska, R.; Lun, Y.; Feng, J.; Frascella, M.; Young, B.; Brignol, N.; Pellegrino, L.; Sitaraman, S.A.; Desnick, R.J.; et al. The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. Mol. Ther. 2010, 18, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Mauer, M.; Sokolovskiy, A.; Barth, J.A.; Castelli, J.P.; Williams, H.N.; Benjamin, E.R.; Najafian, B. Reduction of podocyte globotriaosylceramide content in adult male patients with Fabry disease with amenable GLA mutations following 6 months of migalastat treatment. J. Med. Genet. 2017, 54, 781–786. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 4–15 are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | α-Glcase1 | α-Glcase2 | α-Glcase3 | β-Glcase1 | β-Glcase2 | α-Galase | β-Galase | α-Manase |
|---|---|---|---|---|---|---|---|---|
| 4 | n.i. 1 | n.i. | n.i. | 0.045 2 | 0.1 ± 0.02 | n.i. | n.i. | n.i. |
| 5 | n.i. | n.i. | n.i. | 1.1 ± 0.1 | 5.8 ± 0.5 | n.i. | n.i. | n.i. |
| 6 | 406 ± 20 | n.i. | 44 ± 3 | 48 ± 4 | 15 ± 1 | n.i. | n.i. | n.i. |
| 7 | n.i. | n.i. | n.i. | 3.9 ± 0.3 | 15 ± 2 | 504 ± 32 | n.i. | n.i. |
| 8 | n.i. | n.i. | n.i. | 19 ± 9 | 185 ± 14 | n.i. | n.i. | n.i. |
| 9 | 262 ± 15 | n.i. | n.i. | 309 ± 19 | 255 ± 20 | 772 ± 35 | n.i. | n.i. |
| 10 | 481 ± 27 | 116 ± 8 | 129 ± 10 | 1.3 ± 0.1 | 1.3 ± 0.1 | n.i. | n.i. | n.i. |
| 11 | 568 ± 30 | 121 ± 9 | 213 ± 18 | 20 ± 2 | 12.7 ± 1 | 847 ± 42 | n.i. | n.i. |
| 12 | 60 ± 4 | 10 ± 1 | 18 ± 2 | 23 ± 3 | 71 ± 8 | 122 ± 9 | n.i. | 294 ± 18 |
| 13 | n.i. | n.i. | n.i. | 227 ± 13 | 2.3 ± 0.2 | 60 ± 15 | n.i. | n.i. |
| 14 | n.i. | n.i. | n.i. | 51 ± 13 | 11 ± 1 | n.i. | n.i. | n.i. |
| 15 | 293 ± 18 | 443 ± 22 | 271 ± 17 | 294 ± 18 | 66 ± 15 | 77 ± 16 | n.i. | n.i. |
| pH | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 7 | 15.1 ± 0.97 | 0.26 ± 0.013 | 1.7 ± 0.066 | 0.013 ± 0.0011 |
| 5 | 54.4 ± 0.38 | 1.05 ± 0.068 | 6.3 ± 0.16 | 0.059 ±0.0035 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mena-Barragán, T.; García-Moreno, M.I.; Sevšek, A.; Okazaki, T.; Nanba, E.; Higaki, K.; Martin, N.I.; Pieters, R.J.; Fernández, J.M.G.; Mellet, C.O. Probing the Inhibitor versus Chaperone Properties of sp2-Iminosugars towards Human β-Glucocerebrosidase: A Picomolar Chaperone for Gaucher Disease. Molecules 2018, 23, 927. https://doi.org/10.3390/molecules23040927
Mena-Barragán T, García-Moreno MI, Sevšek A, Okazaki T, Nanba E, Higaki K, Martin NI, Pieters RJ, Fernández JMG, Mellet CO. Probing the Inhibitor versus Chaperone Properties of sp2-Iminosugars towards Human β-Glucocerebrosidase: A Picomolar Chaperone for Gaucher Disease. Molecules. 2018; 23(4):927. https://doi.org/10.3390/molecules23040927
Chicago/Turabian StyleMena-Barragán, Teresa, M. Isabel García-Moreno, Alen Sevšek, Tetsuya Okazaki, Eiji Nanba, Katsumi Higaki, Nathaniel I. Martin, Roland J. Pieters, José M. García Fernández, and Carmen Ortiz Mellet. 2018. "Probing the Inhibitor versus Chaperone Properties of sp2-Iminosugars towards Human β-Glucocerebrosidase: A Picomolar Chaperone for Gaucher Disease" Molecules 23, no. 4: 927. https://doi.org/10.3390/molecules23040927