Theoretical Investigations on Mechanisms and Pathways of C2H5O2 with BrO Reaction in the Atmosphere


Abstract
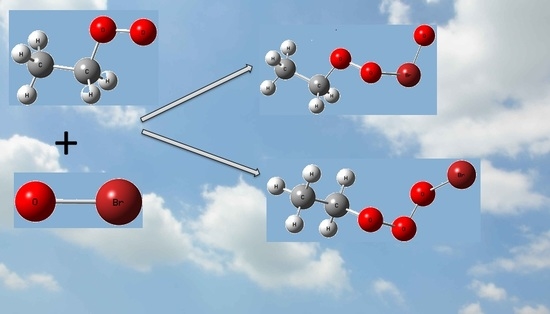
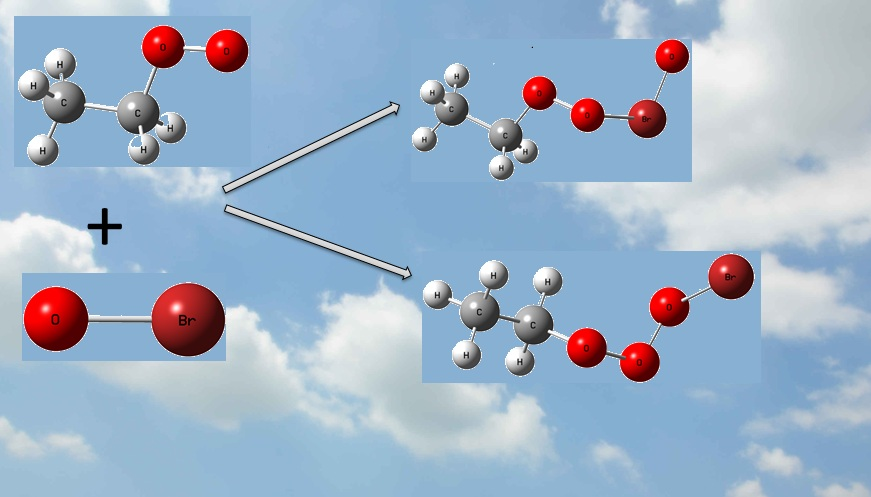
:
1. Introduction
2. Results
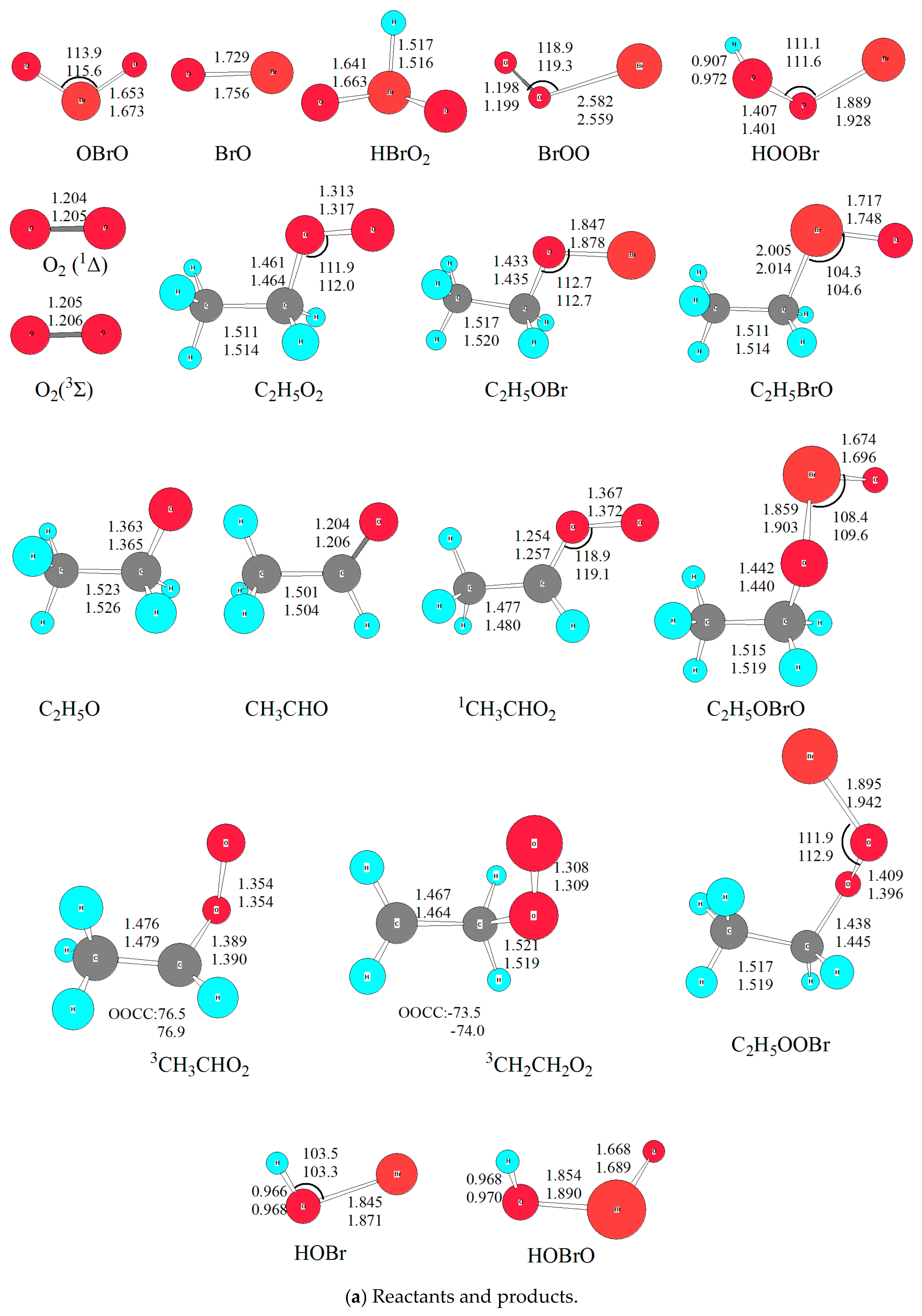
2.1. Reliability of Theoretical Methods
2.2. Reaction Channels of the C2H5O2 + BrO Reaction
2.2.1. The Formation of Intermediates in the C2H5O2 + BrO Reaction
2.2.2. The Reaction Pathways on the Singlet PES
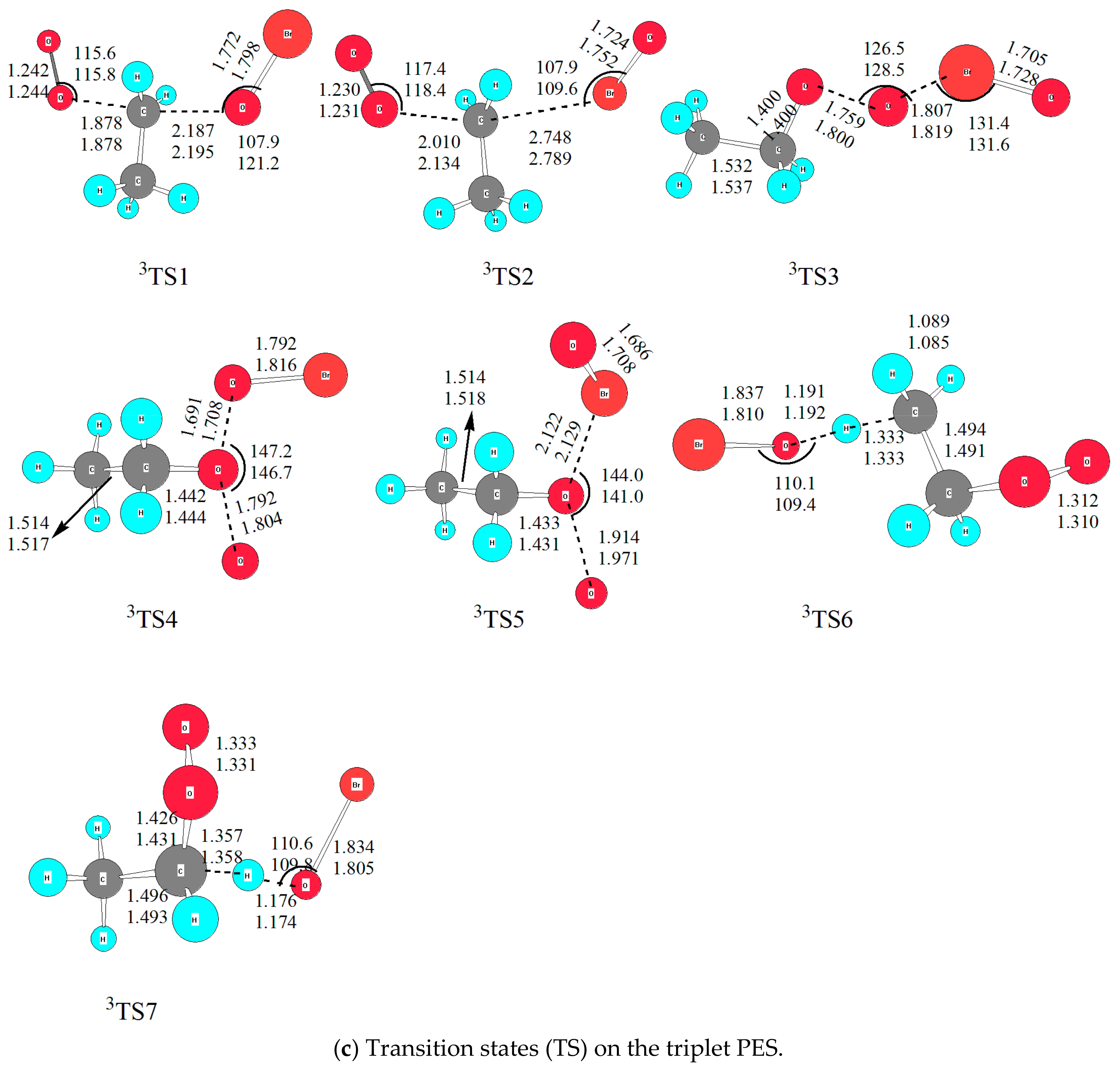
2.2.3. The Substitution and Abstraction Channels on the Triplet PES
2.2.4. Vertical Excitation Energy TV of C2H5O3Br, C2H5O2BrO and C2H5OBrO2
3. Materials and Methods
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Watson, R.T.; Albritton, D.L.; Anderson, S.O.; Lee-Bapty, S. Methyl bromide: Its atmospheric science, technology and economics. In Montreal Protocol Assessment Supplement; United Nations Environmental Programme: Nairobi, Kenya, 1993; p. 243. [Google Scholar]
- Montzka, S.A.; Reimann, S.; Engel, A.; Kruger, K.; O’ Doherty, S.J.; Sturges, W.T. Ozone-depleting substances (ODSs) and related chemicals. In Scientific Assessment of Ozone Depletion: 2010; Proceedings of the Global Ozone Research and Monitoring Project Report No. 52; World Meteorological Organization: Geneva, Switzerland, 2011; pp. 1–112. [Google Scholar]
- Elrod, M.J.; Meads, R.F.; Lipson, J.B.; Seeley, J.V.; Molina, M.J. Temperature dependence of the rate constant for the HO2 + BrO reaction. J. Phys. Chem. 1996, 100, 5808–5812. [Google Scholar] [CrossRef]
- Bedjanian, Y.; Riffault, V.; Poulet, G. Kinetic study of the reactions of BrO radicals with HO2 and DO2. J. Phys. Chem. A 2001, 105, 3167–3175. [Google Scholar] [CrossRef]
- Bridier, I.; Veyret, B.; Lesclaux, R. Flash photolysis kinetic study of reactions of the BrO radical with BrO and HO2. Chem. Phys. Lett. 1993, 201, 563–568. [Google Scholar] [CrossRef]
- Poulet, G.; Pirre, M.; Maguin, F.; Ramaroson, R.; LeBras, G. Role of the BrO + HO2 reaction in the stratospheric chemistry of bromine. Geophys. Res. Lett. 1992, 19, 2305–2308. [Google Scholar] [CrossRef]
- Bloss, W.J.; Rowley, D.M.; Cox, R.A.; Jones, R.L. Rate Coefficient for the BrO + HO2 Reaction at 298 K. Phys. Chem. Chem. Phys. 2002, 4, 3639–3647. [Google Scholar] [CrossRef]
- Salisbury, G.; Monks, P.S.; Bauguitte, S.; Bandy, B.J.; Penkett, S.A. A seasonal comparison of ozone photochemistry in clean and polluted air masses at Mace Head Ireland. J. Atmos. Chem. 2002, 41, 163–187. [Google Scholar] [CrossRef]
- Creasey, D.J.; Halford-Maw, P.A.; Heard, D.E.; Pilling, M.J.; Whitaker, B.J. Implementation and initial deployment of a field instrument for measurement of OH and HO2 in the troposphere by laser-induced fluorescence. J. Chem. Soc. Faraday Trans. 1997, 93, 2907–2913. [Google Scholar] [CrossRef]
- Sommariva, R.; Osthoff, H.D.; Brown, B.B.; Bates, T.S.; Ravishankara, A.R.; Trainer, M. Radicals in the marine boundary layer during NEAQS 2004: A model study of day-time and night-time sources and sinks. Atmos. Chem. Phys. 2009, 9, 3075–3093. [Google Scholar] [CrossRef]
- Aranda, A.; LeBras, G.; LaVerdet, G.L.; Poulet, G. The BrO + CH3O2 reaction: Kinetics and role in the atmospheric ozone budget. Geophys. Res. Lett. 1997, 24, 2745–2748. [Google Scholar] [CrossRef]
- Enami, S.; Yamanaka, T.; Nakayama, T.; Hashimoto, S.; Kawasaki, M.; Shallcross, D.E.; Nakano, Y.; Ishiwata, T. A gas-phase kinetic study of the reaction between bromine monoxide and methylperoxy radicals at atmospheric temperatures. J. Phys. Chem. A 2007, 111, 3342–3348. [Google Scholar] [CrossRef] [PubMed]
- Larichev, M.; Maguin, F.; LeBras, G.; Poulet, G. Kinetics and Mechanism of the BrO + HO2 Reaction. J. Phys. Chem. 1995, 99, 15911–15918. [Google Scholar] [CrossRef]
- Guha, S.; Francisco, J.S. An ab initio study of the pathways for the reaction between CH3O2 and BrO radicals. J. Chem. Phys. 2003, 118, 1779–1793. [Google Scholar] [CrossRef]
- Shallcross, D.E.; Leather, K.E.; Back, A.; Xiao, P.; Lee, E.P.F.; Ng, M.; Mok, D.K.W.; Dyke, J.M.; Hossaini, R.; Chipperfield, M.P.; et al. Reaction between CH3O2 and BrO radicals: A new source of upper troposphere lower stratosphere hydroxyl radicals. J. Chem. Phys. 2015, 119, 4618–4632. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Yamano, D.; Nakayama, T.; Hashimoto, S.; Kawasaki, M.; Wallington, T.J.; Miyano, S.; Tonokura, K.; Takahashi, K. Atmospheric chemistry of BrO radicals: Kinetics of the reaction with C2H5O2 radicals at 233–333 K. J. Phys. Chem. A 2009, 113, 10231–10237. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.X.; Hou, H.; Wang, B.S. Theoretical study of the mechanisms and kinetics of the reactions of hydroperoxy (HO2) radicals with hydroxymethylperoxy (HOCH2O2) and methoxymethylperoxy (CH3OCH2O2) radicals. Phys. Chem. Chem. Phys. 2014, 16, 22805–22814. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.N.; Ou-Yang, S.L.; Li, L. Theoretical study of ClOO + NO reaction: Mechanism and kinetics. Molecules 2017, 22, 2121. [Google Scholar] [CrossRef] [PubMed]
- De Souza, G.L.C.; Brown, A. The ground and excited states of HBrO2 [HOOBr, HOBrO, and HBr(O)O] and HBrO3 (HOOOBr and HOOBrO) isomers. Theor. Chem. Acc. 2016, 135, 178. [Google Scholar] [CrossRef]
- Vereecken, L.; Francisco, J.S. Theoretical studies of atmospheric reaction mechanisms in the troposphere. Chem. Soc. Rev. 2012, 41, 6259–6293. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory using reduced order perturbation theory. J. Chem. Phys. 2007, 127, 124105. [Google Scholar] [CrossRef] [PubMed]
- Pople, J.A.; Head-Gordon, M.; Raghavachari, K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; von Rague Schleyer, R. Efficient diffuse function-augmented basis-sets for anion calculations. Ⅲ. The 3-21+G basis set for 1st-row elements, Li-F. J. Comp. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Binning, R.C., Jr.; Curtiss, L.A. Compact contracted basis-sets for third-row atoms: Ga-Kr. J. Comp. Chem. 1990, 11, 1206–1216. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Head-Gordon, T. Analytic MP2 frequencies without fifth order storage: Theory and application to bifurcated hydrogen bonds in the water hexamer. Chem. Phys. Lett. 1994, 220, 122–128. [Google Scholar] [CrossRef]
- Wong, M.W.; Frisch, M.J.; Wiberg, K.B. Solvent effects 1. The mediation of electrostatic effects by solvents. J. Am. Chem. Soc. 1991, 113, 4776–4782. [Google Scholar] [CrossRef]
- Fang, W.H. A CASSCF study on photodissociation of acrolein in the Gas Phase. J. Am. Chem. Soc. 1999, 121, 8376–8384. [Google Scholar] [CrossRef]
- Fantacci, S.; Migani, A.; Olivucci, M. CASPT2//CASSCF and TDDFT//CASSCF mapping of the excited state isomerization path of a minimal model of the retinal chromophore. J. Phys. Chem. A 2004, 108, 1208–1213. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Comparative DFT study of van der Waals complexes: Rare-gas dimers, alkaline-earth dimers, zinc dimer, and zinc-rare-gas dimers. J. Phys. Chem. 2006, 110, 5121–5129. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N. Understanding the kinetics of spin-forbidden chemical reactions. Phys. Chem. Chem. Phys. 2007, 9, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N. Spin-forbidden reactions: Computational insight into mechanisms and kinetics. Comp. Mol. Sci. 2014, 4, 1–14. [Google Scholar] [CrossRef]
- Schnappinger, T.; Kölle, P.; Marazzi, M.; Monari, A.; González, L.; de Vivie-Riedle, R. Ab initio molecular dynamics of thiophene: The interplay of internal conversion and intersystem crossing. Phys. Chem. Chem. Phys. 2017, 19, 25662–25670. [Google Scholar] [CrossRef] [PubMed]
- Marazzi, M.; Wibowo, M.; Gattuso, H.; Dumont, E.; Roca-Sanjuán, D.; Monari, A. Hydrogen abstraction by photoexcited benzophenone: Consequences for DNA photosensitization. Phys. Chem. Chem. Phys. 2016, 18, 7829–7836. [Google Scholar] [CrossRef] [PubMed]
- Li, H.W.; Tang, Y.Z.; Wang, R.S. Atmospheric chemistry of CF3O2: A theoretical study on mechanisms and pathways of the CF3O2 + IO reaction. Phys. Chem. Chem. Phys. 2013, 15, 5936–5944. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Z.; Sun, H.F.; Sun, J.Y.; Zhang, Y.J.; Wang, R.S. Theoretical study on mechanisms and pathways of the CF3O2 + ClO reaction. Atmos. Environ. 2014, 92, 367–375. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J.; Mennucci, B.; Tomasi, J.; Cammi, R.; Barone, V. Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys. 2006, 124, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Gill, P.W.M.; Johnson, B.G.; Robb, M.A.; Cheeseman, J.R.; Keith, T.A.; Petersson, G.A.; Pople, J.A. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
Sample Availability: not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C2H5O2 + BrO Reaction Channels | G4 | QCISD(T) | ||
|---|---|---|---|---|
| ΔH | ΔH a | ΔH b | ΔH c | |
| C2H5O + BrOO | −6.8 | 17.2 | 18.0 | −19.4 |
| C2H5O + OBrO | 30.0 | 39.3 | 38.8 | 36.0 |
| C2H5OBr + O2(3∑) | −190.9 | −186.3 | −189.9 | −194.1 |
| C2H5OBr + O2(1∆) | −72.8 | −62.1 | −62.8 | −67.9 |
| HOBr +CH3CHO2 | −116.7 | −109.1 | −108.9 | −110.5 |
| HOOBr + CH3CHO | −256.4 | −247.9 | −249.1 | −253.8 |
| HBr + CH3CHO + 3O2 | −310.0 | −313.3 | −312.7 | −317.1 |
| Species | QCISDT | B3LYP | |
|---|---|---|---|
| ΔE | ΔH | ZPE | |
| C2H5O2 + BrO | 0 | 0 | 191.6 |
| C2H5O + BrOO | 14.9 | 18.0 | 179.4 |
| C2H5O + OBrO | 37.5 | 38.8 | 179.9 |
| C2H5Obr + O2(3∑) | −190.5 | −189.9 | 170.6 |
| C2H5Obr + O2(1∆) | −63.3 | −62.8 | 192.5 |
| HOBr + 1CH3CHO2 | −109.5 | −108.9 | 188.6 |
| HOOBr + CH3CHO | −249.8 | −249.1 | 188.9 |
| C2H5BrO + O2(3∑) | −3.3 | −1.4 | 187.3 |
| C2H5OOBr + O(3P) | 119.1 | 119.8 | 191.3 |
| C2H5ObrO + O(3P) | 150.2 | 151.8 | 188.9 |
| CH3CHO + HBrO2 | −14.8 | −15.1 | 179.3 |
| CH3CHO + HOBrO | −228.6 | −227.8 | 186.5 |
| HOBr + 3CH3CHO2 | 15.2 | 17.1 | 181.2 |
| HOBr + 3CH2CH2O2 | 19.3 | 21.3 | 181.9 |
| IM1 | −72.3 | 199.5 | |
| IM2 | −19.1 | 197.4 | |
| IM3 | −83.6 | 199.2 | |
| IM4 | 97.7 | 197.1 | |
| TS1 | 34.4 | 196.7 | |
| TS2 | 67.3 | 189.8 | |
| TS3 | 201.3 | 186.0 | |
| TS4 | 47.7 | 184.1 | |
| TS5 | 50.7 | 183.6 | |
| TS6 | 42.8 | 189.5 | |
| TS7 | −17.6 | 185.7 | |
| TS8 | 237.6 | 189.5 | |
| 3TS1 | 95.7 | 187.5 | |
| 3TS2 | 142.4 | 183.7 | |
| 3TS3 | 151.8 | 188.2 | |
| 3TS4 | 193.2 | 188.8 | |
| 3TS5 | 210.6 | 187.2 | |
| 3TS6 | 55.4 | 176.8 | |
| 3TS7 | 49.6 | 176.7 | |
| Excited States | C2H5O3Br | C2H5O2BrO | C2H5OBrO2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| TV | f | λ | TV | f | λ | TV | f | λ | |
| 1 | 3.02 | 0.0002 | 410.2 | 2.23 | 0.0000 | 555.1 | 4.621 | 0.0019 | 268.3 |
| 2 | 3.77 | 0.0020 | 328.5 | 3.94 | 0.0017 | 314.4 | 4.93 | 0.0240 | 251.5 |
| 3 | 4.32 | 0.0046 | 287.0 | 4.07 | 0.1354 | 304.3 | 5.11 | 0.0001 | 242.8 |
| 4 | 4.57 | 0.1277 | 271.2 | 4.15 | 0.0019 | 299.0 | 5.131 | 0.0023 | 241.6 |
| 5 | 5.32 | 0.0142 | 233.1 | 5.74 | 0.0038 | 216.1 | 5.891 | 0.0712 | 210.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.; Tang, Y.; Zhang, W.; Qu, X.; Fu, Z. Theoretical Investigations on Mechanisms and Pathways of C2H5O2 with BrO Reaction in the Atmosphere. Molecules 2018, 23, 1268. https://doi.org/10.3390/molecules23061268
Lu C, Tang Y, Zhang W, Qu X, Fu Z. Theoretical Investigations on Mechanisms and Pathways of C2H5O2 with BrO Reaction in the Atmosphere. Molecules. 2018; 23(6):1268. https://doi.org/10.3390/molecules23061268
Chicago/Turabian StyleLu, Chenggang, Yizhen Tang, Wei Zhang, Xunshuai Qu, and Zhihao Fu. 2018. "Theoretical Investigations on Mechanisms and Pathways of C2H5O2 with BrO Reaction in the Atmosphere" Molecules 23, no. 6: 1268. https://doi.org/10.3390/molecules23061268
APA StyleLu, C., Tang, Y., Zhang, W., Qu, X., & Fu, Z. (2018). Theoretical Investigations on Mechanisms and Pathways of C2H5O2 with BrO Reaction in the Atmosphere. Molecules, 23(6), 1268. https://doi.org/10.3390/molecules23061268