Discovering Structural Motifs in miRNA Precursors from the Viridiplantae Kingdom


Abstract
:1. Introduction
2. Results
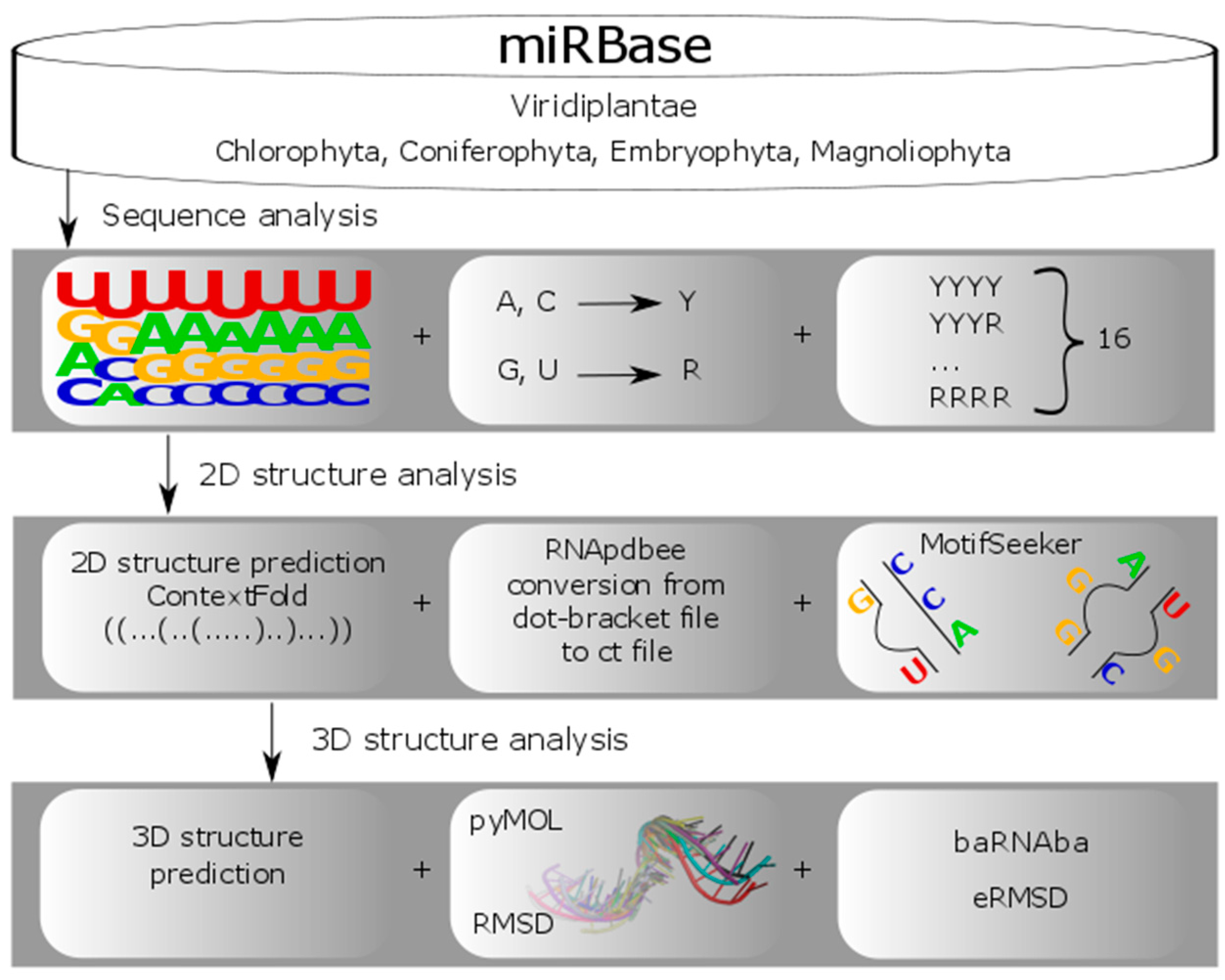
2.1. A Scheme of Data Processing
2.2. Dataset Preparation
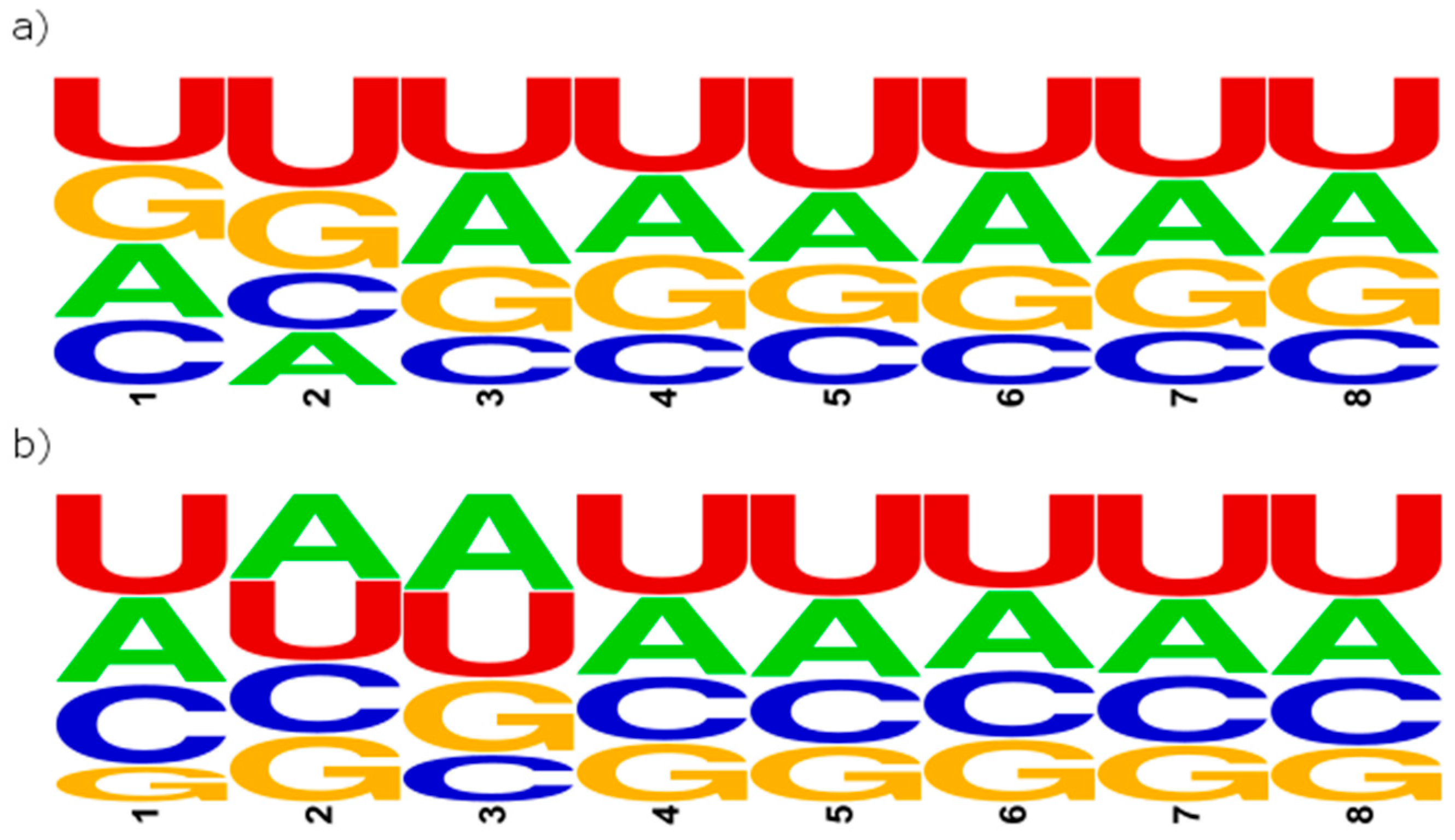
2.3. Primary Structure-Based Analysis
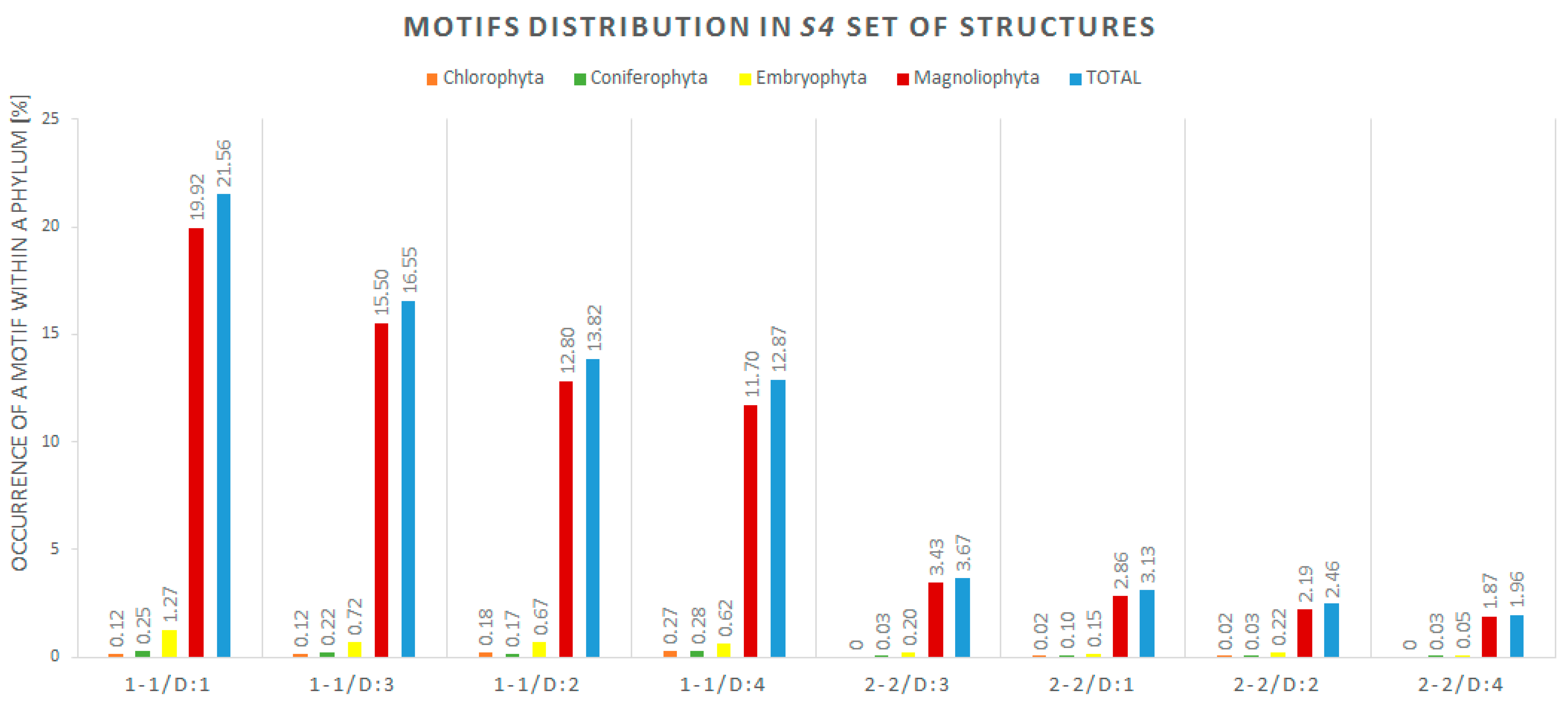
2.4. Secondary Structure-Based Analysis
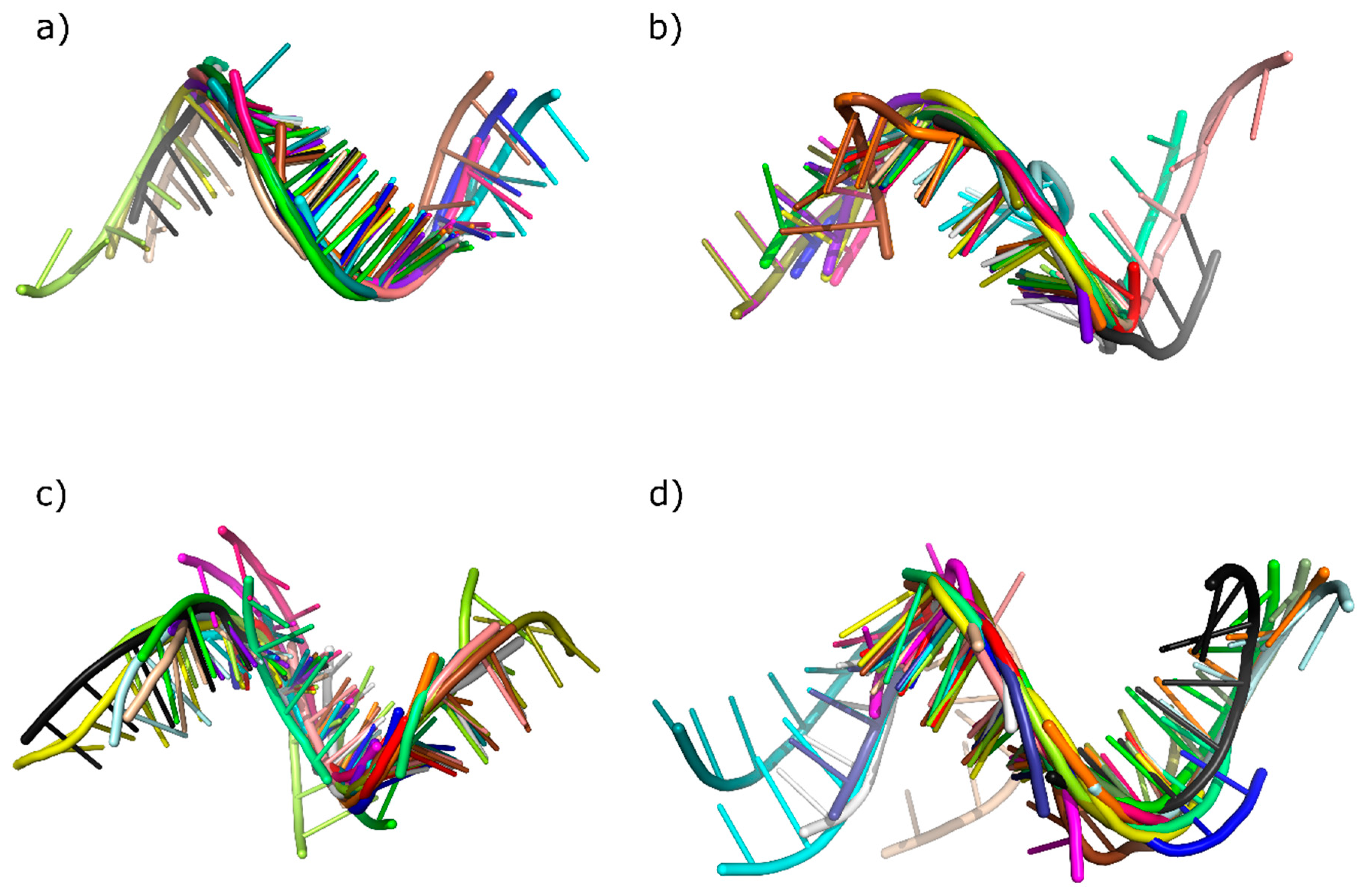
2.5. Tertiary Structure-Based Analysis
3. Discussion
4. Materials and Methods
4.1. WebLogo
4.2. Purine–Pyrimidine Patterns
4.3. ContextFold
4.4. RNApdbee
4.5. MotifSeeker
4.6. RNAComposer
| #zma_MIR168a >example GAAGCCGCGCCGCCUCGGGCUCGCUUGGUGCAGAUCGGGACCCGCCGCCCGGCCGACGGGACGGAUCCCGCCUUGCACCAAGUGAAUCGGAGCCGGCGGAGCGA ContextFold |
4.7. PyMOL
4.8. BaRNAba
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Iorio, M.V.; Croce, C.M. microRNA involvement in human cancer. Carcinogenesis 2012, 33, 1126–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutar, Y. miRNA and cancer; computational and experimental approaches. Curr. Pharm. Biotechnol. 2014, 15, 429. [Google Scholar] [CrossRef] [PubMed]
- Rogers, K.; Chen, X. Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Stepien, A.; Knop, K.; Dolata, J.; Taube, M.; Bajczyk, M.; Barciszewska-Pacak, M.; Pacak, A.; Jarmolowski, A.; Szweykowska-Kulinska, Z. Posttranscriptional coordination of splicing and miRNA biogenesis in plants. Wiley Interdiscip. Rev. RNA 2017, 8, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNA Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J. Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [PubMed]
- Mickiewicz, A.; Rybarczyk, A.; Sarzynska, J.; Figlerowicz, M.; Blazewicz, J. AmiRNA Designer—New method of artificial miRNA design. Acta Biochim. Pol. 2016, 63, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Miskiewicz, J.; Tomczyk, K.; Mickiewicz, A.; Sarzynska, J.; Szachniuk, M. Bioinformatics Study of Structural Patterns in Plant MicroRNA Precursors. BioMed Res. Int. 2017, 2017, 6783010. [Google Scholar] [CrossRef] [PubMed]
- Achkar, N.P.; Cambiagno, D.A.; Manavella, P.A. miRNA Biogenesis: A Dynamic Pathway. Trends Plant Sci. 2016, 21, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.K.; Ryu, M.Y.; Shah, P.; Poulsen, C.P.; Yang, S.W. Post-Translational Regulation of miRNA Pathway Components, AGO1 and HYL1, in Plants. Mol. Cells 2016, 39, 581–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Chávez Montes, R.A.; de Fátima Rosas-Cárdenas, F.; De Paoli, E.; Accerbi, M.; Rymarquis, L.A.; Mahalingam, G.; Marsch-Martínez, N.; Meyers, B.C.; Green, P.J.; de Folter, S. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat. Commun. 2014, 5, 3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarver, J.E.; Donoghue, P.C.; Peterson, K.J. Do miRNAs have a deep evolutionary history? BioEssays 2012, 34, 857–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drusin, S.I.; Suarez, I.P.; Gauto, D.F.; Rasia, R.M.; Moreno, D.M. dsRNA-protein interactions studied by molecular dynamics techniques. Unravelling dsRNA recognition by DCL1. Arch. Biochem. Biophys. 2016, 15, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Dolata, J.; Bajczyk, M.; Bielewicz, D.; Niedojadlo, K.; Niedojadlo, J.; Pietrykowska, H.; Walczak, W.; Szweykowska-Kulinska, Z.; Jarmolowski, A. Salt stress Reveals a New Role for ARGONAUTE1 in miRNA Biogenesis at the Transcriptional and Posttranscriptional Levels. Plant Physiol. 2016, 172, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Conrad, T.; Orom, U.A. Insight into miRNA biogenesis with RNA sequencing. Oncotarget 2015, 6, 26546–26547. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhou, Y.; Castillo-González, C.; Lu, A.; Ge, C.; Zhao, Y.T.; Duan, L.; Li, Z.; Axtell, M.J.; Wang, X.J.; et al. Bidirectional processing of pri-miRNAs with branched terminal loops by Arabidopsis Dicer-like1. Nat. Struct. Mol. Biol. 2013, 20, 1106–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starega-Roslan, J.; Krol, J.; Koscianska, E.; Kozlowski, P.; Szlachcic, W.J.; Sobczak, K.; Krzyzosiak, W.J. Structural basis of microRNA length variety. Nucleic Acids Res. 2011, 39, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Origin, Biogenesis, and Activity of Plant MicroRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mickiewicz, A.; Sarzynska, J.; Milostan, M.; Kurzynska-Kokorniak, A.; Rybarczyk, A.; Lukasiak, P.; Kulinski, T.; Figlerowicz, M.; Blazewicz, J. Modeling of the catalytic core of Arabidopsis thaliana Dicer-like 4 protein and its complex with double-stranded RNA. Comput. Biol. Chem. 2017, 66, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Beezhold, K.J.; Castranova, V.; Chen, F. Microprocessor of microRNAs: Regulation and potential for therapeutic intervention. Mol. Cancer 2010, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Søkilde, R.; Newie, I.; Persson, H.; Borg, Å.; Rovira, C. Passenger strand loading in overexpression experiments using microRNA mimics. RNA Biol. 2015, 12, 787–791. [Google Scholar]
- Zha, X.; Xia, Q.; Yuan, Y.A. Structural insights into small RNA sorting and mRNA target binding by Arabidopsis Argonaute Mid domains. FEBS Lett. 2012, 586, 3200–3207. [Google Scholar] [CrossRef] [PubMed]
- Starega-Roslan, J.; Galka-Marciniak, P.; Krzyzosiak, W.J. Nucleotide sequence of miRNA precursor contributes to cleavage site selection by Dicer. Nucleic Acids Res. 2015, 43, 10939–10951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Jasso, C.F.; Arenas-Huertero, C.; Reyes, J.L.; Contreras-Cubas, C.; Covarrubias, A.; Vaca, L. First step in pre-miRNAs processing by human Dicer. Acta Pharmacol. Sin. 2009, 30, 1177–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Nucleotide Ambiguity Code (IUPAC). Available online: http://www.dnabaser.com/articles/IUPAC%20ambiguity%20codes.html (accessed on 1 February 2017).
- Zakov, S.; Goldberg, Y.; Elhadad, M.; Ziv-Ukelson, M. Rich parameterization improves RNA structure prediction. J. Comput. Biol. 2011, 18, 1525–1542. [Google Scholar] [CrossRef] [PubMed]
- Puton, T.; Kozlowski, L.P.; Rother, K.M.; Bujnicki, J.M. CompaRNA: A server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res. 2013, 41, 4307–4323. [Google Scholar] [CrossRef] [PubMed]
- Antczak, M.; Zok, T.; Popenda, M.; Lukasiak, P.; Adamiak, R.W.; Blazewicz, J.; Szachniuk, M. RNApdbee—A webserver to derive secondary structures from pdb files of knotted and unknotted RNAs. Nucleic Acids Res. 2014, 42, W368–W372. [Google Scholar] [CrossRef] [PubMed]
- Rybarczyk, A.; Szostak, N.; Antczak, M.; Zok, T.; Popenda, M.; Adamiak, R.W.; Blazewicz, J.; Szachniuk, M. New in silico approach to assessing RNA secondary structures with non-canonical base pairs. BMC Bioinform. 2015, 16, 276. [Google Scholar] [CrossRef] [PubMed]
- Zok, T.; Antczak, M.; Zurkowski, M.; Popenda, M.; Blazewicz, J.; Adamiak, R.W.; Szachniuk, M. RNApdbee 2.0: Multifunctional tool for RNA structure annotation. Nucleic Acids Res. 2018, 46. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, J.; Milostan, M. StructAnalyzer—A tool for sequence versus structure similarity analysis. Acta Biochim. Pol. 2016, 63, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, J.; Zok, T.; Milostan, M.; Szachniuk, M. LCS-TA to identify similar fragments in RNA 3D structures. BMC Bioinform. 2017, 18, 456. [Google Scholar] [CrossRef] [PubMed]
- Blazewicz, J.; Szachniuk, M.; Wojtowicz, A. RNA tertiary structure determination: NOE pathways construction by tabu search. Bioinformatics 2005, 21, 2356–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antczak, M.; Popenda, M.; Zok, T.; Sarzynska, J.; Ratajczak, T.; Tomczyk, K.; Adamiak, R.W.; Szachniuk, M. New functionality of RNAComposer: An application to shape the axis of miR160 precursor structure. Acta Biochim. Pol. 2016, 63, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Purzycka, K.J.; Popenda, M.; Szachniuk, M.; Antczak, M.; Lukasiak, P.; Blazewicz, J.; Adamiak, R.W. Automated 3D RNA structure prediction using the RNAComposer method for riboswitches. Methods Enzymol. 2015, 553, 3–34. [Google Scholar] [PubMed]
- The PyMOL Molecular Graphics System, version 1.8; Schrodinger, LLC: New York, NY, USA, 2015.
- Bottaro, S.; Palma, F.D.; Bussi, G. The role of nucleobase interactions in RNA structure and dynamics. Nucleic Acids Res. 2014, 42, 13306–13314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kufareva, I.; Abagya, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [PubMed]
- Almeidaa, M.I.; Reisb, R.M.; Calin, G.A. MicroRNA history: Discovery, recent applications, and next frontiers. Mutat. Res. 2011, 717, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varani, G. Twenty years of RNA: The discovery of microRNAs. RNA 2015, 21, 751–752. [Google Scholar] [CrossRef] [PubMed]
- Context Fold 1.00. Available online: https://www.cs.bgu.ac.il/~negevcb/contextfold/readme.pdf (accessed on 1 February 2017).
- eRMSD. Available online: https://plumed.github.io/doc-master/user-doc/html/_e_r_m_s_d.html (accessed on 1 February 2017).
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | A [%] | C [%] | G [%] | U [%] | R [%] | Y [%] |
|---|---|---|---|---|---|---|
| 1 | 24.48 | 21.87 | 25.62 | 28.03 | 50.10 | 49.90 |
| 2 | 17.78 | 19.28 | 26.56 | 36.38 | 44.34 | 55.66 |
| 3 | 30.32 | 16.67 | 22.37 | 30.64 | 52.69 | 47.31 |
| 4 | 25.68 | 17.31 | 25.46 | 31.54 | 51.15 | 48.85 |
| 5 | 23.18 | 19.56 | 20.26 | 36.99 | 43.45 | 56.55 |
| 6 | 30.21 | 17.23 | 22.11 | 30.45 | 52.32 | 47.68 |
| 7 | 25.17 | 18.12 | 23.54 | 33.17 | 48.71 | 51.29 |
| 8 | 26.71 | 18.75 | 23.76 | 30.78 | 50.48 | 49.52 |
| Position | A [%] | C [%] | G [%] | U [%] | R [%] | Y [%] |
|---|---|---|---|---|---|---|
| 1 | 27.98 | 26.51 | 12.19 | 33.32 | 40.17 | 59.83 |
| 2 | 27.90 | 23.37 | 22.09 | 26.64 | 49.99 | 50.01 |
| 3 | 31.48 | 15.92 | 24.14 | 28.46 | 55.62 | 44.38 |
| 4 | 25.56 | 21.45 | 19.72 | 33.28 | 45.27 | 54.73 |
| 5 | 25.84 | 21.67 | 18.73 | 33.76 | 44.57 | 55.43 |
| 6 | 25.73 | 22.26 | 20.81 | 31.20 | 46.54 | 53.46 |
| 7 | 24.57 | 22.54 | 19.00 | 33.89 | 43.57 | 56.43 |
| 8 | 25.31 | 22.81 | 18.15 | 33.73 | 43.46 | 56.54 |
| Pattern | VS4-5′ [%] | VS4-3′ [%] | Total [%] |
|---|---|---|---|
| RRYR | 4.36 | 3.82 | 4.09 |
| YRYR | 4.41 | 4.57 | 4.49 |
| RYYR | 6.22 | 3.90 | 5.06 |
| RRRY | 5.43 | 5.92 | 5.67 |
| RYRY | 6.08 | 5.33 | 5.71 |
| RRYY | 6.13 | 5.30 | 5.71 |
| YRYY | 4.98 | 6.78 | 5.88 |
| RYYY | 6.90 | 4.98 | 5.94 |
| YYYR | 6.77 | 5.45 | 6.11 |
| RYRR | 7.50 | 5.29 | 6.39 |
| RRRR | 7.43 | 5.64 | 6.53 |
| YYRY | 6.77 | 6.67 | 6.72 |
| YRRR | 6.38 | 7.40 | 6.89 |
| YRRY | 4.83 | 10.10 | 7.46 |
| YYYY | 7.29 | 9.17 | 8.23 |
| YYRR | 8.55 | 9.68 | 9.11 |
| Motif/Distance | Number of Motifs | Number of Structures with at Least One Motif |
|---|---|---|
| 1-1/D:1 | 1397 | 1288 |
| 1-1/D:3 | 1043 | 989 |
| 1-1/D:2 | 861 | 826 |
| 1-1/D:4 | 807 | 769 |
| 2-2/D:3 | 221 | 219 |
| 2-2/D:1 | 190 | 187 |
| 2-2/D:2 | 149 | 147 |
| 2-2/D:4 | 118 | 117 |
| Fragment Id | RMSD [Å] | |||
|---|---|---|---|---|
| Chlorophyta | Coniferophyta | Embryophyta | Magnoliophyta | |
| 1 | 2.112 | 0.463 | 1.882 | 2.245 |
| 2 | 0.278 | 0.430 | 0.290 | 2.270 |
| 3 | 0.256 | 1.194 | 2.058 | 1.135 |
| 4 | 0.117 | 0.381 | 1.626 | 0.679 |
| 5 | 0.467 | 0.258 | 2.351 | 0.352 |
| 6 | 2.209 | 1.228 | 1.810 | 0.567 |
| 7 | 0.257 | 0.469 | 1.966 | 0.123 |
| 8 | 0.560 | 1.226 | 1.587 | 0.449 |
| 9 | 0.142 | 1.018 | 1.773 | 2.171 |
| 10 | 0.864 | 0.412 | 1.247 | 1.672 |
| 11 | 0.502 | 0.461 | 0.910 | 0.845 |
| 12 | 0.547 | 0.444 | 1.573 | 0.607 |
| 13 | 0.034 | 1.377 | 0.974 | 1.171 |
| 14 | 0.389 | 0.846 | 1.546 | 0.963 |
| 15 | 1.155 | 1.036 | 0.944 | 0.836 |
| 16 | 0.139 | 0.481 | 0.837 | 1.094 |
| 17 | 0.686 | 1.210 | 1.839 | 0.597 |
| 18 | 0.637 | 0.390 | 1.730 | 1.344 |
| 19 | 2.159 | 0.266 | 0.330 | 2.304 |
| Average | 0.711 | 0.715 | 1.435 | 1.128 |
| Fragment Id | eRMSD [Å] | |||
|---|---|---|---|---|
| Chlorophyta | Coniferophyta | Embryophyta | Magnoliophyta | |
| 1 | 0.459 | 0.765 | 0.802 | 0.554 |
| 2 | 0.788 | 0.771 | 0.434 | 0.503 |
| 3 | 0.587 | 0.436 | 0.725 | 0.730 |
| 4 | 0.291 | 1.047 | 0.776 | 1.101 |
| 5 | 0.477 | 1.047 | 0.868 | 0.325 |
| 6 | 0.432 | 0.746 | 0.858 | 0.444 |
| 7 | 0.561 | 1.025 | 0.868 | 0.832 |
| 8 | 0.442 | 0.799 | 0.817 | 0.365 |
| 9 | 0.459 | 0.675 | 0.767 | 0.455 |
| 10 | 0.438 | 0.800 | 0.842 | 0.643 |
| 11 | 0.386 | 0.749 | 1.080 | 0.390 |
| 12 | 0.251 | 0.753 | 0.841 | 0.398 |
| 13 | 0.605 | 0.745 | 0.906 | 0.457 |
| 14 | 0.410 | 0.680 | 0.791 | 0.447 |
| 15 | 0.410 | 0.891 | 0.883 | 0.394 |
| 16 | 0.463 | 0.729 | 0.901 | 0.467 |
| 17 | 0.564 | 1.023 | 0.788 | 0.331 |
| 18 | 0.528 | 1.058 | 0.764 | 0.604 |
| 19 | 0.453 | 0.712 | 0.810 | 0.604 |
| Average | 0.474 | 0.813 | 0.817 | 0.529 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miskiewicz, J.; Szachniuk, M. Discovering Structural Motifs in miRNA Precursors from the Viridiplantae Kingdom. Molecules 2018, 23, 1367. https://doi.org/10.3390/molecules23061367
Miskiewicz J, Szachniuk M. Discovering Structural Motifs in miRNA Precursors from the Viridiplantae Kingdom. Molecules. 2018; 23(6):1367. https://doi.org/10.3390/molecules23061367
Chicago/Turabian StyleMiskiewicz, Joanna, and Marta Szachniuk. 2018. "Discovering Structural Motifs in miRNA Precursors from the Viridiplantae Kingdom" Molecules 23, no. 6: 1367. https://doi.org/10.3390/molecules23061367