Synthesis, Characterization, and In Vitro Cancer Cell Growth Inhibition Evaluation of Novel Phosphatidylcholines with Anisic and Veratric Acids

 ,
, 
Abstract
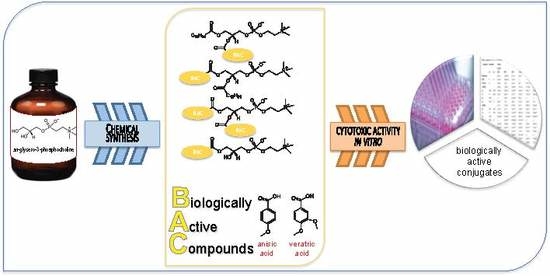
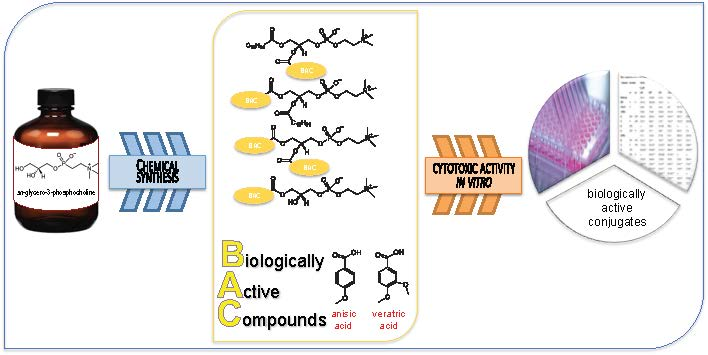
:
1. Introduction
2. Results and Discussion
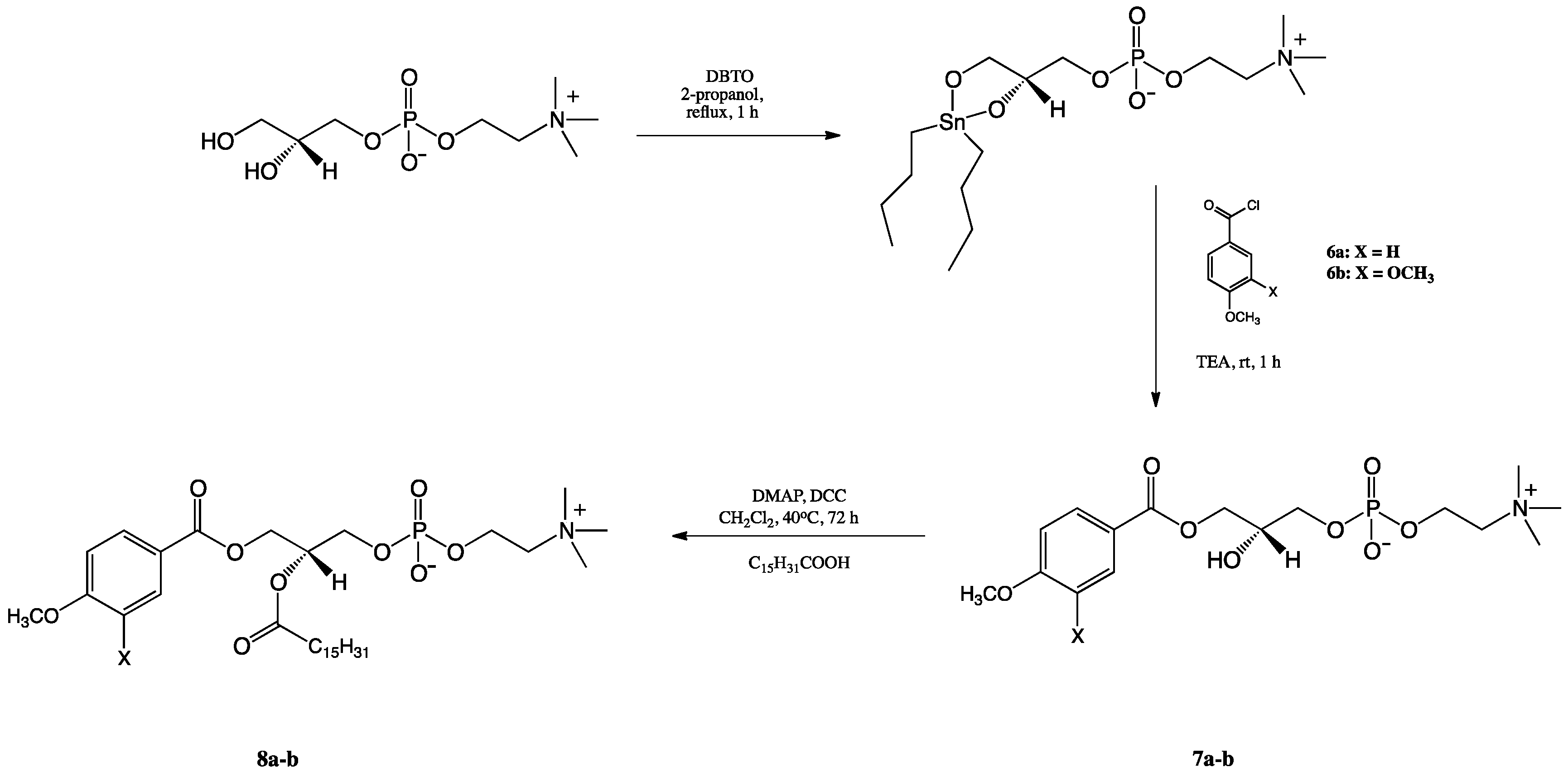
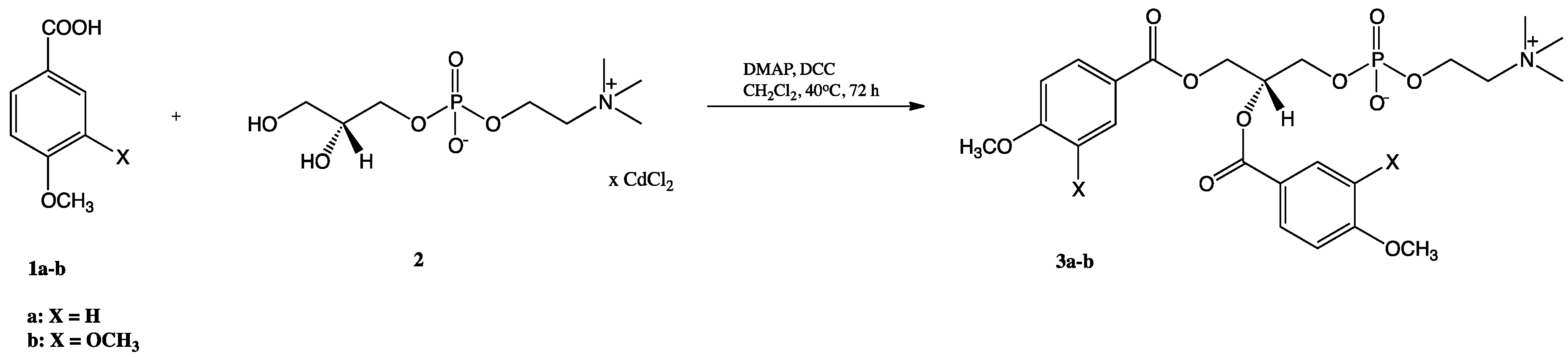
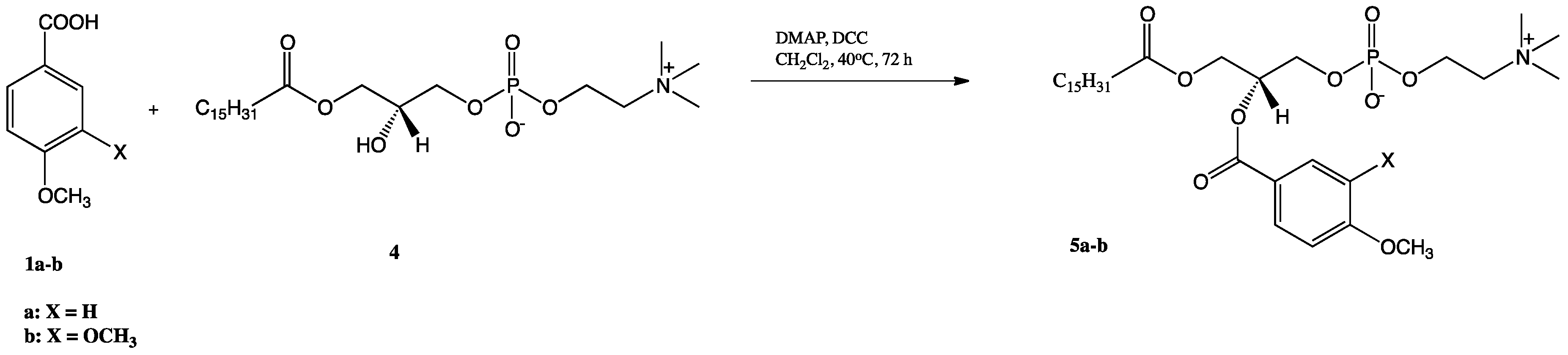
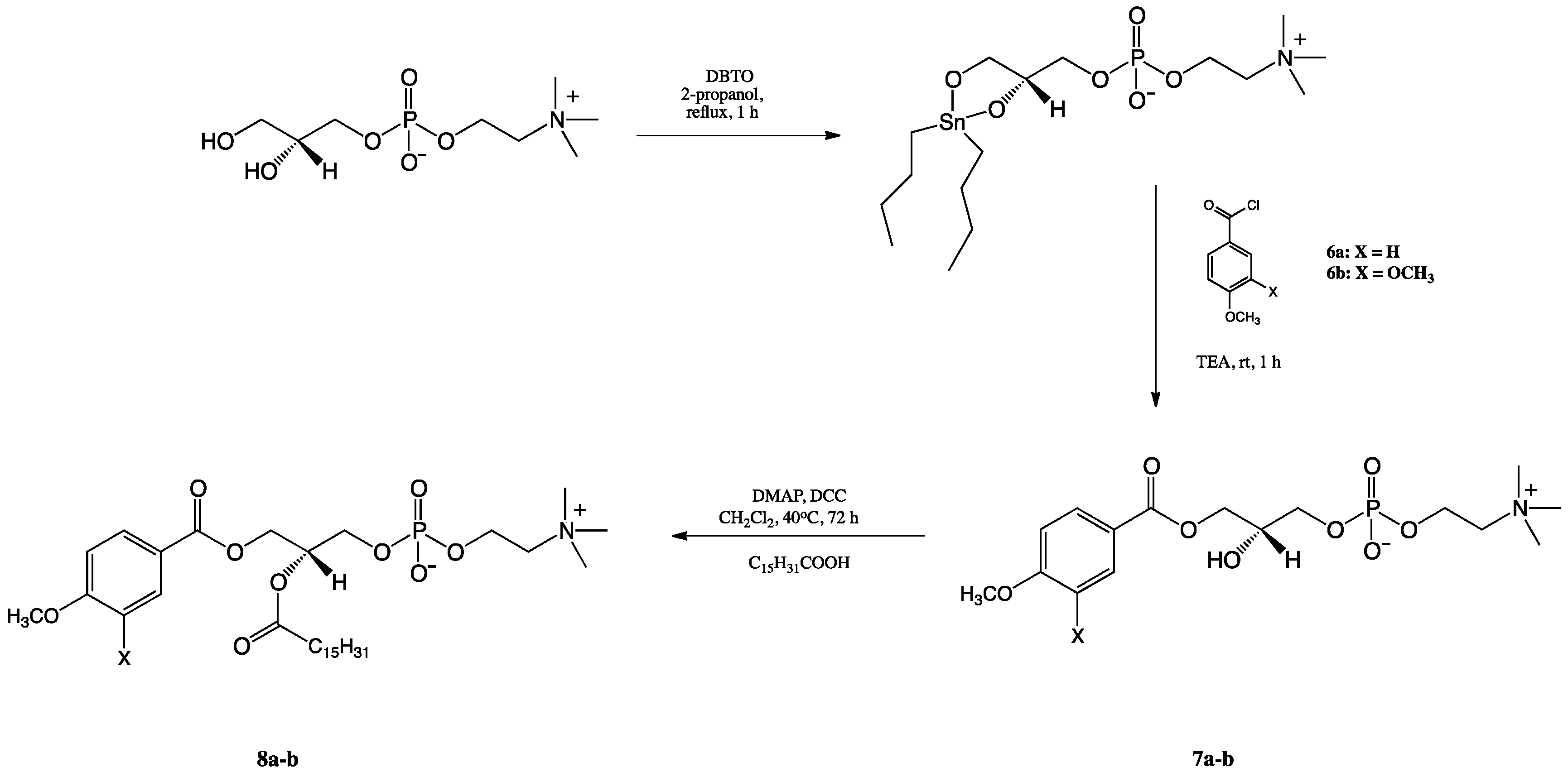
2.1. Synthesis of Structured Phospholipids with Anisic and Veratric Acids
2.2. Cytotoxic Activity In Vitro Against Selected Cancer Cell Lines
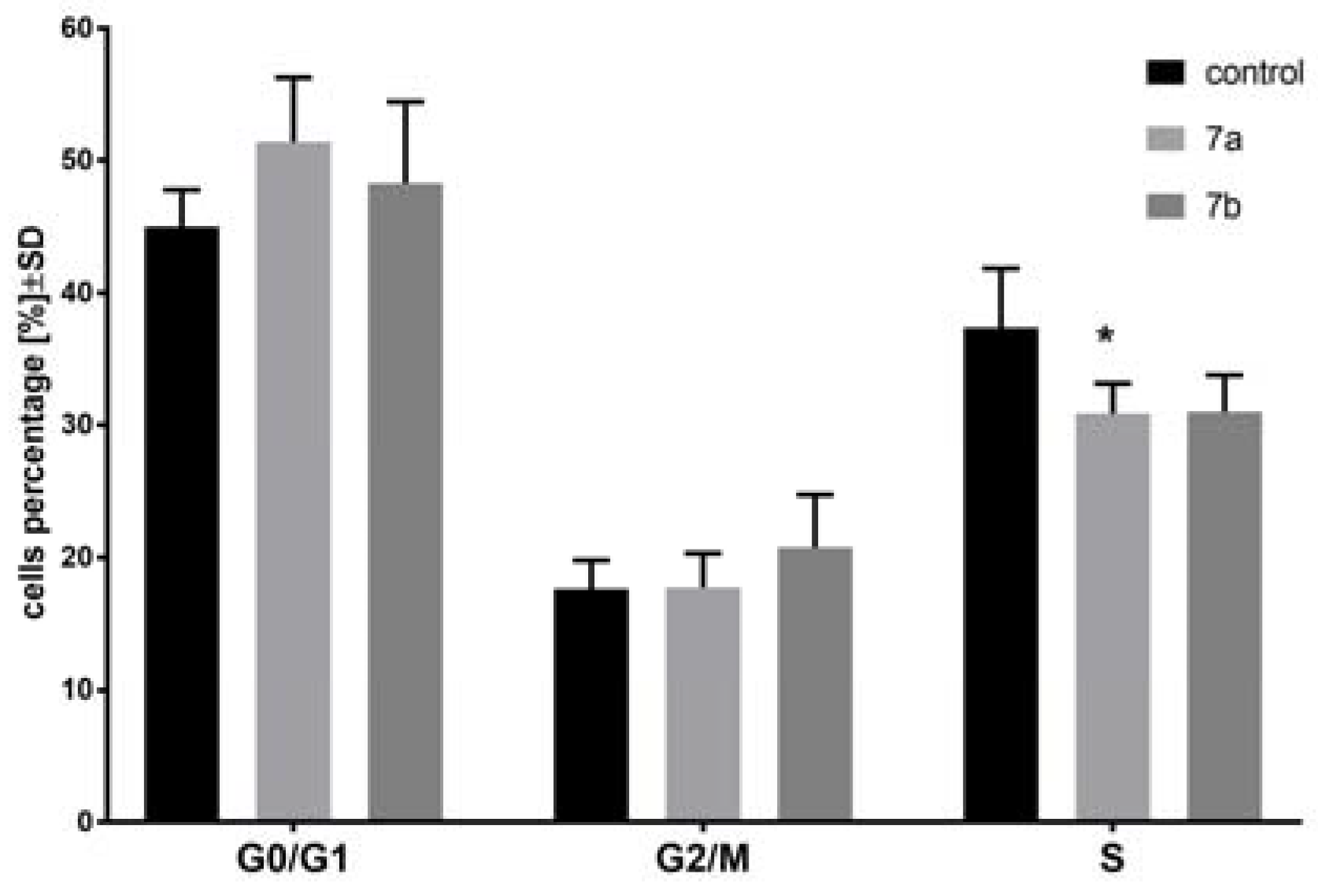
2.3. The Effect of 1-Phenoyl-2-hydroxy-sn-glycero-3-phosphocholines on the Cell Cycle of the MV4-11 Cells
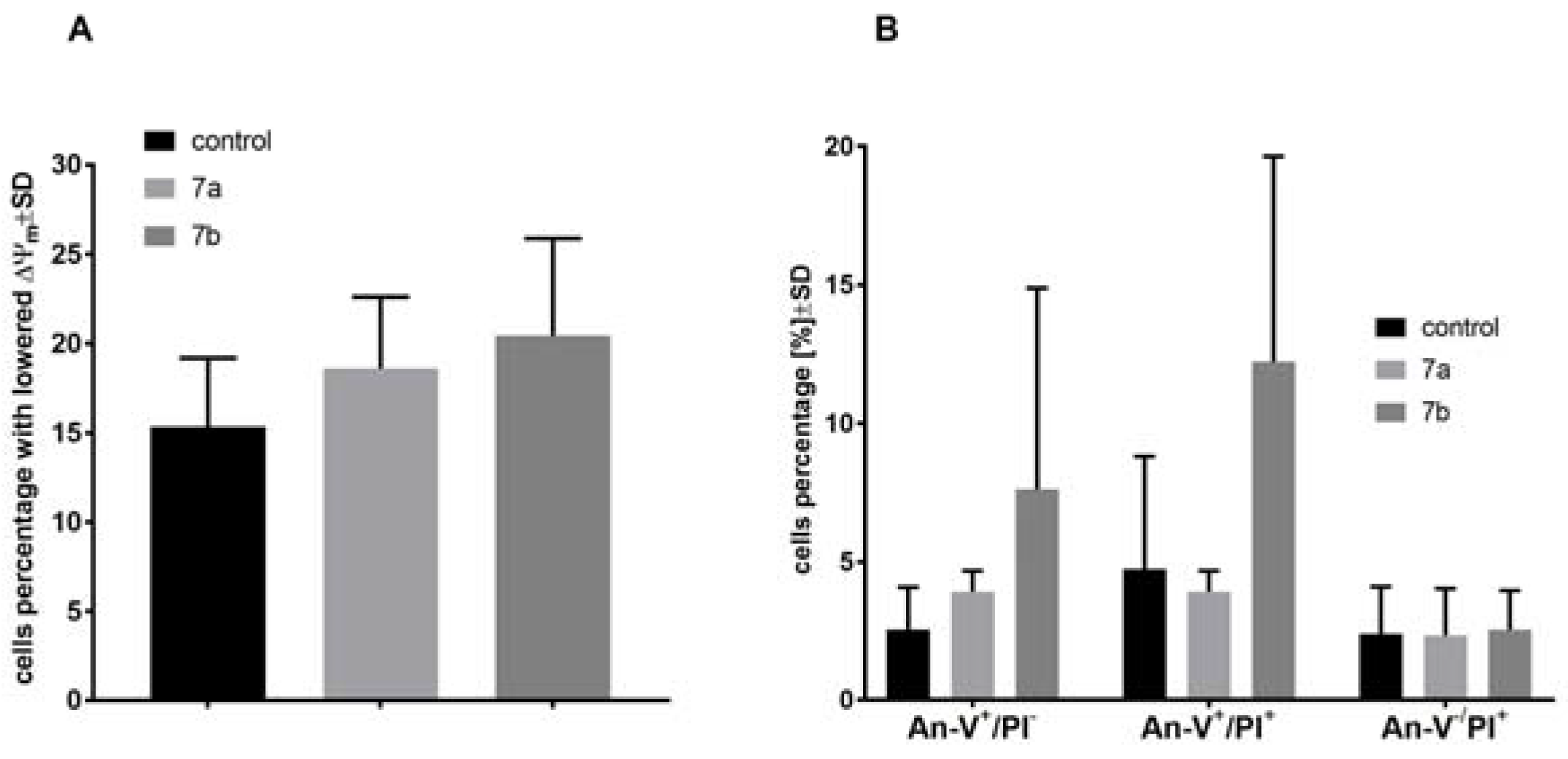
2.4. The Effect of 1-Phenoyl-2-hydroxy-sn-glycero-3-phosphocholines on the Mitochondrial Membrane Potential, Cell Death, and Caspase-3/7 Activity of the MV4-11 Cells
3. Materials and Methods
3.1. Materials
3.2. Methods of Analysis
3.3. Methods of Synthesis
3.3.1. Synthesis of 1,2-Diphenoyl-sn-glycero-3′-phosphatidylcholines
3.3.2. Synthesis of 1-palmitoyl-2-phenoyl-sn-glycero-3′-phosphatidylcholines
3.3.3. Synthesis of 1-Phenoyl-2-hydroxy-sn-glycero-3′-phosphatidylcholines
3.3.4. Synthesis of 1-Phenoyl-2-palmitoyl-sn-glycero-3-phosphatidylcholines
3.4. Biological Studies
3.4.1. Cell Line
3.4.2. Cytotoxicity Assay In Vitro
3.4.3. MTT Assay
3.4.4. SRB Assay
3.4.5. Cell Cycle Analysis
3.4.6. Apoptosis Determination by Annexin V Staining
3.4.7. Mitochondrial Membrane Potential Determination
3.4.8. Caspase-3/7 Activity Determination
3.4.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Scalbert, A.; Morand, C.; Manach, C.; Rémésy, C. Absorption and metabolism of polyphenols in the gut and impact on health. Biomed. Pharmacother. 2002, 56, 276–282. [Google Scholar] [CrossRef]
- Del Rio, D.; Costa, L.G.; Lean, M.E.J.; Crozier, A. Polyphenols and health: What compounds are involved? Nutr. Metab. Cardiovasc. Dis. 2010, 20, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Natella, F.; Nardini, M.; Di Felice, M.; Scaccini, C. Benzoic and cinnamic acid derivatives as antioxidants: Structure- activity relation. J. Agric. Food Chem. 1999, 47, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Hadjipavlou-Litina, D.; Litinas, K.; Geromichalos, G. Novel cinnamic acid derivatives as antioxidant and anticancer agents: Design, synthesis and modeling studies. Molecules 2014, 19, 9655–9674. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-H.; Ou-Yang, J.-P. Pharmacological actions of sodium ferulate in cardiovascular system. Cardiovasc. Drug Rev. 2005, 23, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Sung, S.H.; Jang, Y.P.; Markelonis, G.J.; Oh, T.H.; Kim, Y.C. E-p-Methoxycinnamic acid protects cultured neuronal cells against neurotoxicity induced by glutamate. Br. J. Pharmacol. 2002, 135, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Adisakwattana, S.; Roengsamran, S.; Hsu, W.H.; Yibchok-Anun, S. Mechanisms of antihyperglycemic effect of p-methoxycinnamic acid in normal and streptozotocin-induced diabetic rats. Life Sci. 2005, 78, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Espinoza, M.-C.; Villeneuve, P. Phenolic acids enzymatic lipophilization. J. Agric. Food Chem. 2005, 53, 2779–2787. [Google Scholar] [CrossRef] [PubMed]
- Guyot, B.; Bosquette, B.; Pina, M.; Graille, J. Esterification of phenolic acids from green coffee with an immobilized lipase from Candida antarctica in solvent-free medium. Biotechnol. Lett. 1997, 19, 529–532. [Google Scholar] [CrossRef]
- Buisman, G.J.H.; Van Helteren, C.T.W.; Kramer, G.F.H.; Veldsink, J.W.; Derksen, J.T.P.; Cuperus, F.P. Enzymatic esterifications of functionalized phenols for the synthesis of lipophilic antioxidants. Biotechnol. Lett. 1998, 20, 131–136. [Google Scholar] [CrossRef]
- Choo, W.S.; Birch, E.J. Radical scavenging activity of lipophilized products from lipase-catalyzed transesterification of triolein with cinnamic and ferulic acids. Lipids 2009, 44, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Karam, R.; Karboune, S.; St-Louis, R.; Kermasha, S. Lipase-catalyzed acidolysis of fish liver oil with dihydroxyphenylacetic acid in organic solvent media. Process Biochem. 2009, 44, 1193–1199. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Feddern, V.; Glasius, M.; Guo, Z.; Xu, X. Improved enzymatic production of phenolated acylglycerols through alkyl phenolate intermediates. Biotechnol. Lett. 2011, 33, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Mu, Y.; Chen, H.; Xiu, Z.; Yang, T. Enzymatic synthesis of feruloylated lysophospholipid in a selected organic solvent medium. Food Chem. 2013, 141, 3317–3322. [Google Scholar] [CrossRef] [PubMed]
- Balakrishna, M.; Kaki, S.S.; Karuna, M.S.L.; Sarada, S.; Kumar, C.G.; Prasad, R.B.N. Synthesis and in vitro antioxidant and antimicrobial studies of novel structured phosphatidylcholines with phenolic acids. Food Chem. 2017, 221, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Gliszczyńska, A.; Niezgoda, N.; Gładkowski, W.; Czarnecka, M.; Świtalska, M.; Wietrzyk, J. Synthesis and biological evaluation of novel phosphatidylcholine analogues containing monoterpene acids as potent antiproliferative agents. PLoS ONE 2016, 11, e0157278. [Google Scholar] [CrossRef] [PubMed]
- Gliszczyńska, A.; Niezgoda, N.; Gladkowski, W.; Świtalska, M.; Wietrzyk, J. Isoprenoid-phospholipid conjugates as potential therapeutic agents: Synthesis, characterization and antiproliferative studies. PLoS ONE 2017, 12, e0172238. [Google Scholar] [CrossRef] [PubMed]
- Ohira, H.; Torii, N.; Aida, T.M.; Watanabe, M.; Smith, R.L. Rapid separation of shikimic acid from Chinese star anise (Illicium verum Hook. f.) with hot water extraction. Sep. Purif. Technol. 2009, 69, 102–108. [Google Scholar] [CrossRef]
- Gadgoli, C.; Mishra, S.H. Antihepatotoxic activity of p-methoxy benzoic acid from Capparis spinosa. J. Ethnopharmacol. 1999, 66, 187–192. [Google Scholar] [CrossRef]
- Tao, L.; Wang, S.; Zhao, Y.; Sheng, X.; Wang, A.; Zheng, S.; Lu, Y. Phenolcarboxylic acids from medicinal herbs exert anticancer effects through disruption of COX-2 activity. Phytomedicine 2014, 21, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.W.; Jung, E.; Kim, S.; Lee, K.E.; Youm, J.K.; Park, D. Antagonist effects of veratric acid against UVB-induced cell damages. Molecules 2013, 18, 5405–5419. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.J.; Wu, X.W.; Wang, R.F.; Peng, Y.S.; Yang, X.; Liu, J.X. Absorption properties and mechanism of trolline and veratric acid and their implication to an evaluation of the effective components of the flowers of Trollius chinensis. Chin. J. Nat. Med. 2014, 12, 700–704. [Google Scholar] [CrossRef]
- Saravanakumar, M.; Raja, B. Veratric acid, a phenolic acid attenuates blood pressure and oxidative stress in L-NAME induced hypertensive rats. Eur. J. Pharmacol. 2011, 671, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Raja, B.; Saravanakumar, M.; Sathya, G. Veratric acid ameliorates hyperlipidemia and oxidative stress in Wistar rats fed an atherogenic diet. Mol. Cell. Biochem. 2012, 366, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Sivagami, G.; Karthikkumar, V.; Balasubramanian, T.; Nalini, N. The modulatory influence of p-methoxycinnamic acid, an active rice bran phenolic acid, against 1,2-dimethylhydrazine-induced lipid peroxidation, antioxidant status and aberrant crypt foci in rat colon carcinogenesis. Chem. Biol. Interact. 2012, 196, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Hudson, E.A.; Dinh, P.A.; Kokubun, T.; Simmonds, M.S.J.; Gescher, A. Characterization of potentially chemopreventive phenols in extracts of brown rice that inhibit the growth of human breast and colon cancer cells. Cancer Epidemiol. Biomarkers Prev. 2000, 9, 1163–1170. [Google Scholar] [PubMed]
- Gupta, C.M.; Radhakrishnan, R.; Khorana, H.G. Glycerophospholipid synthesis: Improved general method and new analogs containing photoactivable groups. Proc. Natl. Acad. Sci. USA 1977, 74, 4315–4319. [Google Scholar] [CrossRef] [PubMed]
- Singh, A. An efficient synthesis of phosphatidylcholines. J. Lipid Res. 1990, 31, 1522–1525. [Google Scholar] [PubMed]
- Ichihara, K.; Iwasaki, H.; Ueda, K.; Takizawa, R.; Naito, H.; Tomosugi, M. Synthesis of phosphatidylcholine: An improved method without using the cadmium chloride complex of sn-glycero-3-phosphocholine. Chem. Phys. Lipids 2005, 137, 94–99. [Google Scholar] [CrossRef] [PubMed]
- D’Arrigo, P.; Fasoli, E.; Pedrocchi-Fantoni, G.; Rossi, C.; Saraceno, C.; Tessaro, D.; Servi, S. A practical selective synthesis of mixed short/long chains glycerophosphocholines. Chem. Phys. Lipids 2007, 147, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Mattson, F.H.; Volpenhein, R.A. Synthesis and properties of glycerides. J. Lipid Res. 1962, 3, 281–296. [Google Scholar]
- Nevozhay, D. Cheburator Software for Automatically Calculating Drug Inhibitory Concentrations from in Vitro Screening Assays. PLoS ONE 2014, 9, e106186. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anti- cancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1a-b, 3a-b, 5a-b, 7a-b, and 8a-b are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Acyl Residue | Cell Lines IC50 [μm] | |||||||
|---|---|---|---|---|---|---|---|---|---|
| sn-1 | sn-2 | MV4-11 | A-549 | MCF-7 | LoVo | LoVo/DX | HepG2 | BALB/3T3 | |
| Palmitic acid | - | - | 161 ± 78.9 | 56.4 ± 1.6 | 122.6 ± 63.4 | 42.1 ± 4.6 | 86.3 ± 21.3 | 275.1 ± 85.2 | 60.4 ± 4 |
| DPPC | PA | PA | 178.7 ± 30.3 | 229.7 ± 31.3 | 398.2 ± 1 93.9 | 274.2 ± 17.1 | n.a. | n.a. | 48.8 ± 2.1 |
| 1-PA-LPC | PA | - | 64 ± 0.9 | 62.3 ± 6.5 | 73 ± 4.6 | 52.4 ± 4.3 | 58 ± 3.7 | 282.7 ± 12.5 | 176.2 ± 11.3 |
| 1a (ANISA) | - | - | 264.6 ± 49.2 | n.a. | 483 ± 60.1 | 312.5 ± 29.6 | n.a. | n.a. | n.a. |
| 3a | ANISA | ANISA | 404.3 ± 163.7 ′′ | n.a. | n.a. | 319.2 ± 24.2 ′′ | n.a. | n.a. | n.a. |
| 5a | PA | ANISA | 59.6 ± 6.3 *^′′ | 56.1 ± 1.3 ′′ | 73.1 ± 8.4 *′′ | 52 ± 2.6 *^′′ | 57.7 ± 0.1 ′′ | 289.7 ± 8.2 ′′ | 121 ± 25.9 ′′ |
| 7a | ANISA | - | 21.1 ± 4.9 *^#& | 198.4 ± 14.1 #& | 48.4 ± 2.4 *#& | 43.5 ± 3.2 *^#& | 70.1 ± 2.9 #& | 82.8 ± 9.6 #& | 52.9 ± 8.4 #& |
| 8a | ANISA | PA | 79.5 ± 6.4 *^#′′ | 59.2 ± 0.2 ′′ | 79.4 ± 8.1 *′′ | 59.3 ± 4.6 *^′′ | 59.1 ± 0.9 ′′ | 275.9 ± 12′′ | 201.7 ± 22.4 #′′ |
| 1b (VA) | - | - | 366.3 ± 70.8 | n.a. | 578.6 ± 28.9 | 316.4 ± 30 | n.a. | n.a. | n.a. |
| 3b | VA | VA | 463.7 ± 38.4 ′′ | n.a. | 610.5 ± 5.7 ′′ | 285.5 ± 43.6 ′′ | 120.4 ± 49.9 | 51.4 ± 5.8 ′′ | 354.6 ± 60.2 ′′ |
| 5b | PA | VA | 156.7 ± 66.1 *^′′ | 68.8 ± 1 | 118.6 ± 24.5 *^′′ | 58.8 ± 9.5 *^′′ | 59.9 ± 0.9 ′′ | 274.4 ± 21.7 ^′′ | 251.5 ± 4.3 ^′′ |
| 7b | VA | - | 9.5 ± 2.1 *^#& | 65.4 ± 5 | 20.7 ± 4.3 *^#& | 16.7 ± 1.9 *^#& | 47.8 ± 3.9 #& | 53.1 ± 7.2 #& | 33.2 ± 3.4 ^#& |
| 8b | VA | PA | 164.1 ± 67.1 *^′′ | 82.4 ± 16 | 86.2 ± 8.8 *^′′ | 63.1 ± 6.5 *^′′ | 65.6 ± 0.7 #′′ | 289.6 ± 7.5 ^′′ | 253.1 ± 11.8 ^′′ |
| Cisplatin | 1.3 ± 0.47 | 8.6 ± 0.7 | 8.1 ± 0.03 | 2.56 ± 0.35 | 3.17 ± 0.2 | 2.38 ± 0.64 | 4.2 ± 1.1 | ||
| Doxorubicin | - | - | 0.117 ± 0.012 | 6.53 ± 0.93 | - | - | |||
| Compounds | Acyl Residue | ||
|---|---|---|---|
| sn-1 | sn-2 | RI | |
| 1a (ANISA) | - | - | - |
| 3a | ANISA | ANISA | - |
| 5a | PA | ANISA | 1.11 |
| 7a | ANISA | - | 1.61 |
| 8a | ANISA | PA | 0.99 |
| 1b (VA) | - | - | - |
| 3b | VA | VA | 0.42 |
| 5b | PA | VA | 1.02 |
| 7b | VA | - | 2.86 |
| 8b | VA | PA | 1.04 |
| DOX | - | - | 55.81 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarnecka, M.; Świtalska, M.; Wietrzyk, J.; Maciejewska, G.; Gliszczyńska, A. Synthesis, Characterization, and In Vitro Cancer Cell Growth Inhibition Evaluation of Novel Phosphatidylcholines with Anisic and Veratric Acids. Molecules 2018, 23, 2022. https://doi.org/10.3390/molecules23082022
Czarnecka M, Świtalska M, Wietrzyk J, Maciejewska G, Gliszczyńska A. Synthesis, Characterization, and In Vitro Cancer Cell Growth Inhibition Evaluation of Novel Phosphatidylcholines with Anisic and Veratric Acids. Molecules. 2018; 23(8):2022. https://doi.org/10.3390/molecules23082022
Chicago/Turabian StyleCzarnecka, Marta, Marta Świtalska, Joanna Wietrzyk, Gabriela Maciejewska, and Anna Gliszczyńska. 2018. "Synthesis, Characterization, and In Vitro Cancer Cell Growth Inhibition Evaluation of Novel Phosphatidylcholines with Anisic and Veratric Acids" Molecules 23, no. 8: 2022. https://doi.org/10.3390/molecules23082022
APA StyleCzarnecka, M., Świtalska, M., Wietrzyk, J., Maciejewska, G., & Gliszczyńska, A. (2018). Synthesis, Characterization, and In Vitro Cancer Cell Growth Inhibition Evaluation of Novel Phosphatidylcholines with Anisic and Veratric Acids. Molecules, 23(8), 2022. https://doi.org/10.3390/molecules23082022