Chemoenzymatic Total Synthesis of (+)-10-Keto-Oxycodone from Phenethyl Acetate †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
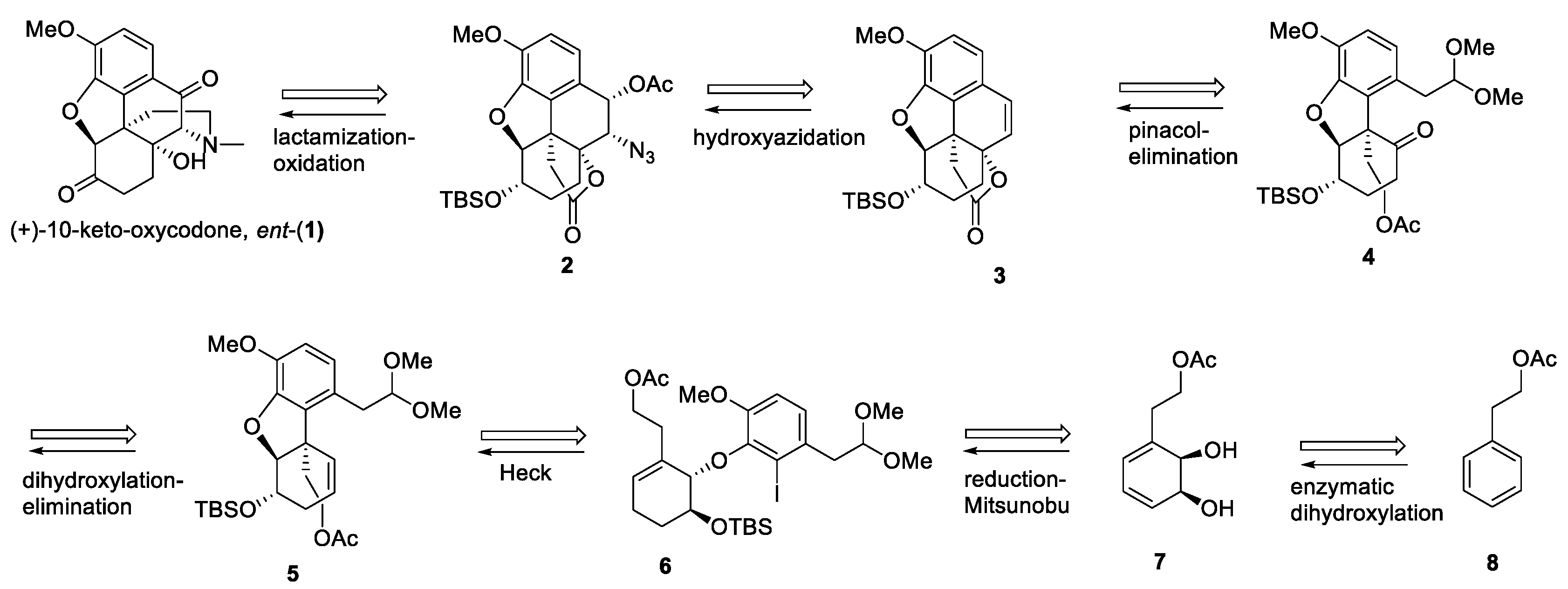
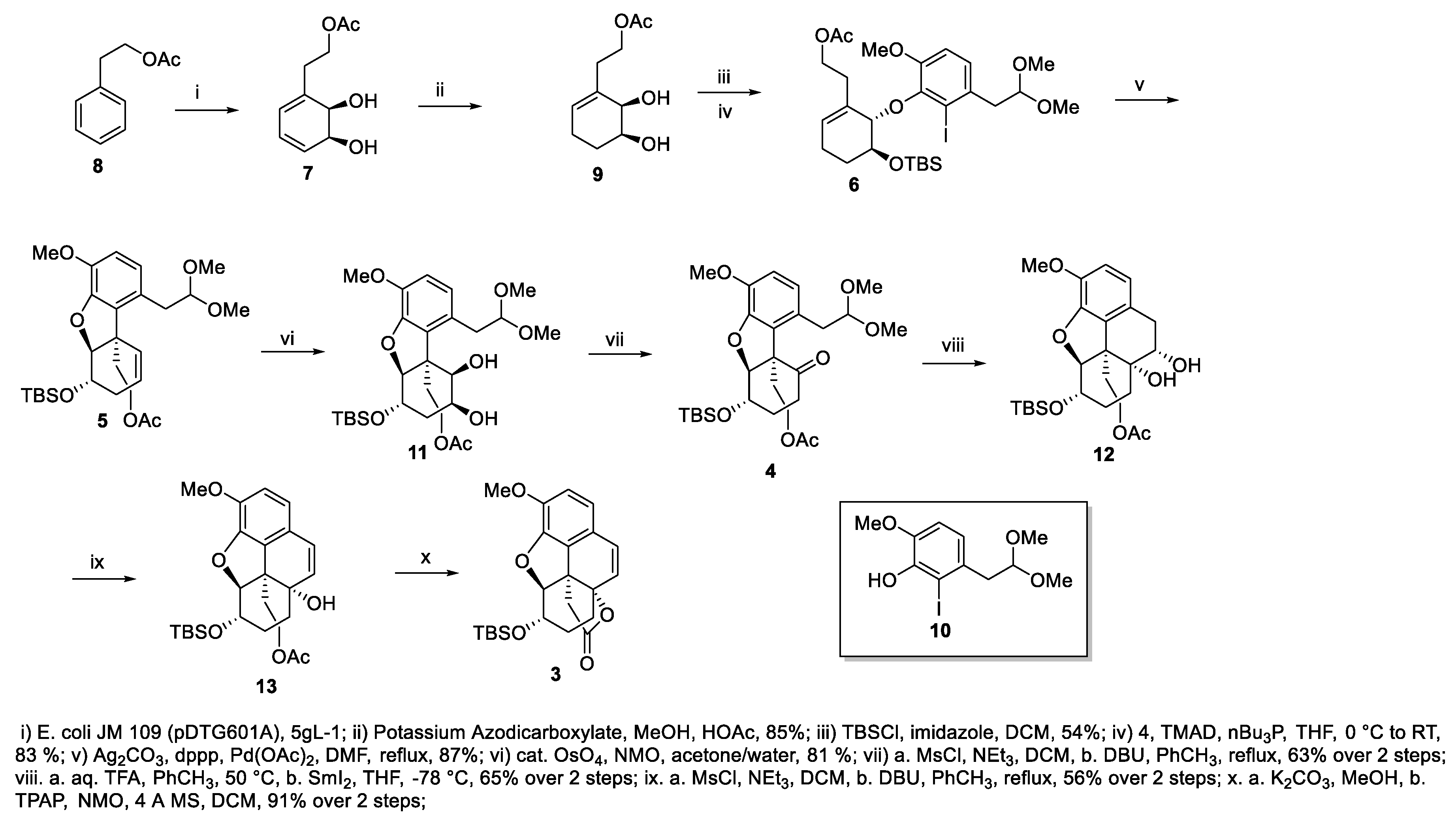
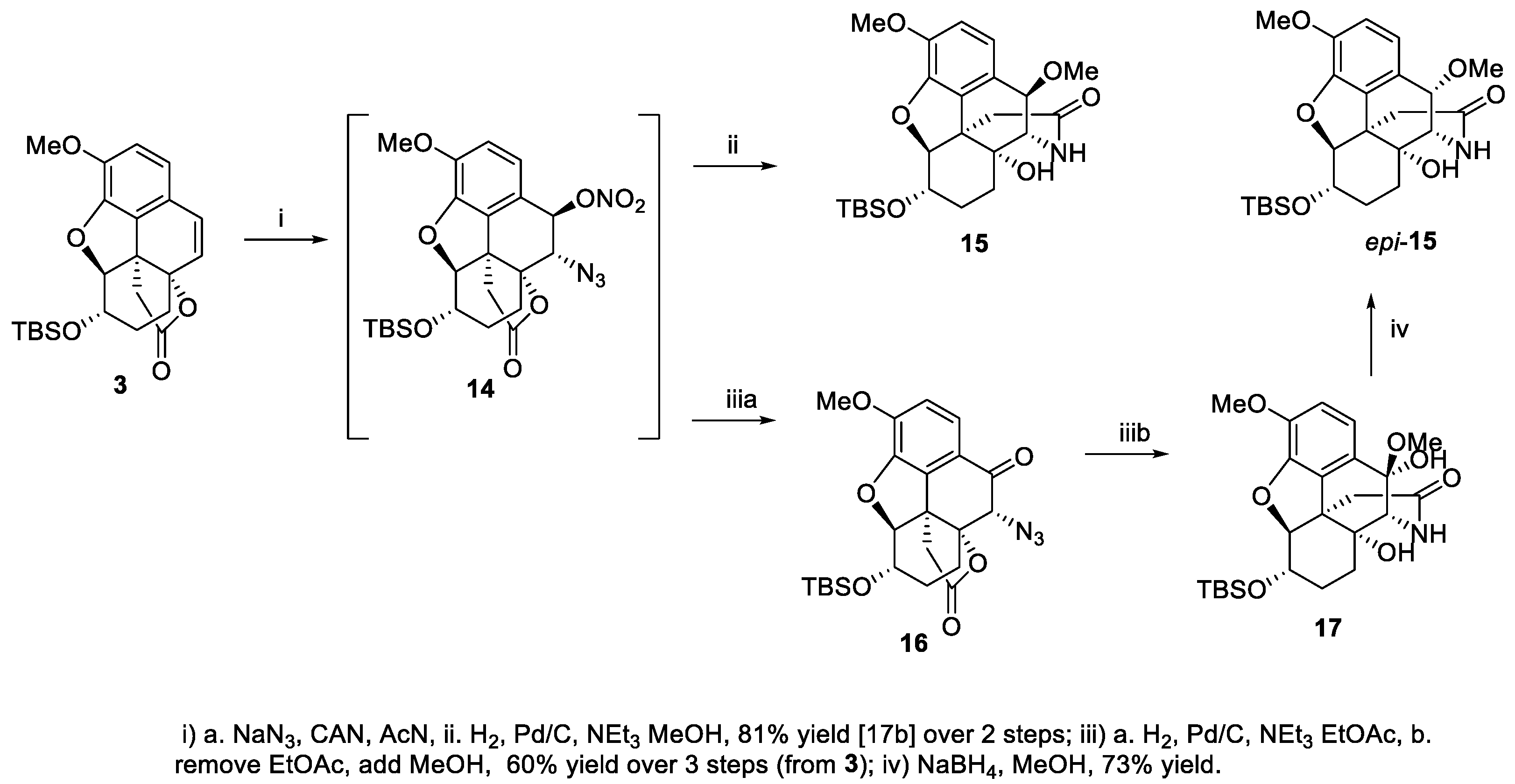
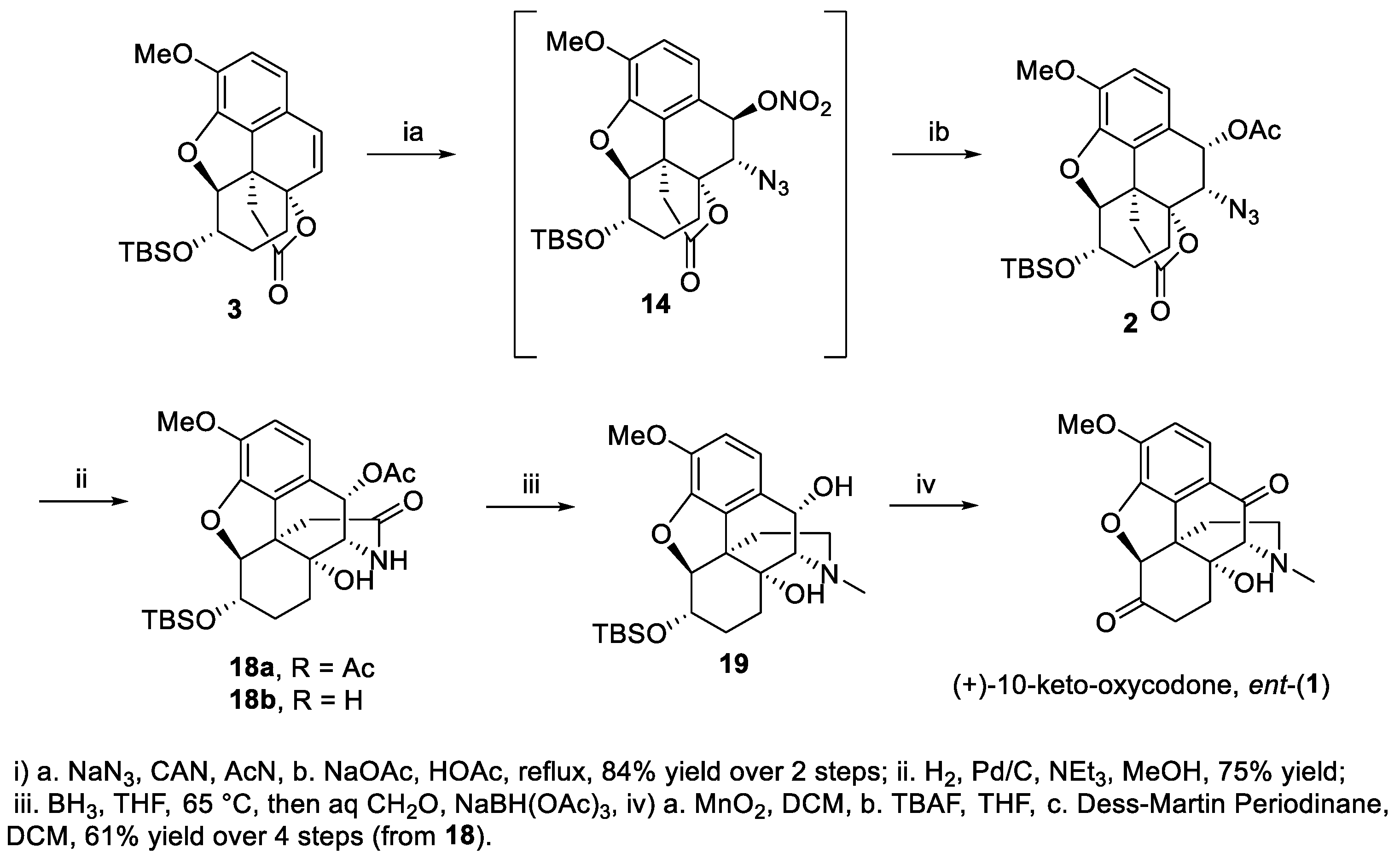
2. Results and Discussion
3. Experimental Section
3.1. General Methods
3.2. (4bR,5S,6S,8aR,9S)-9-azido-6-((tert-butyldimethylsilyl)oxy)-4,5-epoxy3-methoxy-10-oxo-5,6,7,8,9,10-hexahydro-8a,4b-(epoxyethano)phenanthren-12-one (16)
3.3. (5R,5aR,8S,8aS,13bR,14R)-8-((tert-butyldimethylsilyl)oxy)-5a,14-dihydroxy-10,14-dimethoxy-1,2,5,5a,6,7,8,8a-octahydro-5,13-methanobenzo[2,3]benzofuro[4,3a-c]azepin-3(4H)-one (17)
3.4. (5S,5aR,8S,8aS,13bR,14S)-8-((tert-butyldimethylsilyl)oxy)-5a-hydroxy-10,14-dimethoxy-1,2,5,5a,6,7,8,8a-octahydro-5,13-methanobenzo[2,3]benzofuro[4,3a-c]azepin-3(4H)-one (epi-15)
3.5. (4bR,5S,6S,8aR,9S,10R)-9-azido-6-((tert-butyldimethylsilyl)oxy)-4,5-epoxy3-methoxy-12-oxo-5,6,7,8,9,10-hexahydro-8a,4b-(epoxyethano)phenanthren-10-yl nitrate (14)
3.6. (4bR,5S,6S,8aR,9S,10S)-9-azido-6-((tert-butyldimethylsilyl)oxy)4,5-epoxy-3-methoxy-12-oxo-5,6,7,8,9,10-hexahydro-8a,4b-(epoxyethano)phenanthren-10-yl acetate (2)
3.7. (4S,4aR,7S,7aS,12bR,13S)-7-((tert-butyldimethylsilyl)oxy)-4a-hydroxy-9-methoxy-2-oxo-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-13-yl acetate (18a)
3.8. (4S,4aR,7S,7aS,12bR,13S)-7-((tert-butyldimethylsilyl)oxy)-4a,13-dihydroxy-9-methoxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-2(3H)-one (18b)
3.9. (4S,4aR,7S,7aS,12bR,13S)-7-((tert-butyldimethylsilyl)oxy)-9-methoxy-3-methyl-1,2,3,4,5,6,7,7a-octahydro-4aH-4,12-methanobenzofuro[3,2-e]isoquinoline-4a,13-diol (19)
3.10. ent-10-keto-oxycodone (ent-(1))
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Nagase, H.; Hayakawa, J.; Kawamura, K.; Kawai, K.; Takezawa, Y.; Matsuura, H.; Tajima, C.; Endo, T. Discovery of a structurally novel opioid κ-agonist derived from 4,5-epoxymorphinan. Chem. Pharm. Bull. 1998, 46, 366–369. [Google Scholar] [CrossRef]
- Millan, M.J. Kappa-opioid receptors and analgesia. Trends Pharmacol. Sci. 1990, 11, 70–76. [Google Scholar] [CrossRef]
- Cowan, A.; Gmerek, D.E. In-vivo studies on kappa opioid receptors. Trends Pharmacol. Sci. 1986, 7, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Horikiri, H.; Hirano, N.; Tanaka, Y.; Oishi, J.; Hatakeyama, H.; Kawamura, K.; Nagase, H. Syntheses of 10-oxo, 10-hydroxy, and 10-hydroxy derivatives of a potent κ-opioid receptor agonist, TRK-820. Chem. Pharm. Bull. 2004, 52, 664–669. [Google Scholar] [CrossRef]
- Archer, S.; Seyed-Mozaffari, A.; Ward, S.; Kosterlitz, H.W.; Paterson, S.J.; McKnight, A.T.; Corbett, A.D. 10-Ketonaltrexone and 10-ketooxymorphone. J. Med. Chem. 1985, 28, 974–976. [Google Scholar] [CrossRef]
- Cain, G.A.; Drummond, S., Jr. Sequential benzylic oxidation of naloxone 3-methyl ether. Synth. Commun. 2000, 30, 4513–4521. [Google Scholar] [CrossRef]
- Hudlicky, T. Recent advances in process development for opiate-derived pharmaceutical agents. Can. J. Chem. 2015, 93, 492–501. [Google Scholar] [CrossRef]
- Reed, J.W.; Hudlicky, T. The quest for a practical synthesis of morphine alkaloids and their derivatives by chemoenzymatic methods. Acct. Chem. Res. 2015, 48, 674–687. [Google Scholar] [CrossRef]
- Rapoport, H.; Masamune, S. The stereochemistry of 10-hydroxycodeine derivatives. J. Am. Chem. Soc. 1955, 77, 4330–4335. [Google Scholar] [CrossRef]
- Sun, H.; Johnson, J.E., Jr.; Fenton, R.L.; Dorn, S.M. Preparation of 10-keto morphinans by benzylic oxidation. U.S. Patent 2011071296 (A1), 1 September 2011. [Google Scholar]
- Uyeda, R.T.; Wuonola, M.A.; Read, J.M., Jr. 10-Keto opiates. Tetrahedron Lett. 1989, 30, 5725–5728. [Google Scholar] [CrossRef]
- Freund, M.; Speyer, E. DE 286431, 1914.
- Freund, M.; Speyer, E. DE 296916, 1916.
- Freund, M.; Speyer, E. Transformation of thebaine into hydroxycodeione and its derivatives. J. Prakt. Chem. 1916, 94, 135–178. [Google Scholar] [CrossRef]
- Lutz, R.E.; Small, L. Reduction studies in the morphine series. IX. Hydroxycodeinone. J. Org. Chem. 1939, 4, 220–233. [Google Scholar] [CrossRef]
- Kok, G.B.; Scammells, P.J. Improved synthesis of 14-hydroxy opioid pharmaceuticals and intermediates. RSC Adv. 2012, 2, 11318–11325. [Google Scholar] [CrossRef]
- Millgate, A.G.; Pogson, B.J.; Wilson, I.W.; Kutchan, T.M.; Zenk, M.H.; Gerlach, W.L.; Fist, A.J.; Larkin, P.J. Morphine-pathway block in top1 poppies. Nature 2004, 431, 413–414. [Google Scholar] [CrossRef]
- Fist, A.J.; Byrne, C.J.; Gerlach, W.L. Papaver Somniferum Strain with High Concentration of Thebaine. U.S. Patent 606749, 30 May 2000. [Google Scholar]
- Fist, A.J. Papaver Somniferum Strain with High Concentration of Thebaine and Oripavine. U.S. Patent Application No. 20090227796, 10 September 2009. [Google Scholar]
- Thodey, K.; Galanie, S.; Smolke, C.D. A microbial biomanufacturing platform for natural and semisynthetic opioids. Nat. Chem. Biol. 2014, 10, 837–844. [Google Scholar] [CrossRef]
- Tasmanian Alkaloids Pty Ltd.: Westbury, TAS, Australia. Available online: https://tasalk.com.au/ (accessed on 2 September 2019).
- Gates, M.; Tschudi, G. The synthesis of morphine. J. Am. Chem. Soc. 1952, 74, 1109–1110. [Google Scholar] [CrossRef]
- Rinner, U.; Hudlicky, T. Alkaloid synthesis. Top. Curr. Chem. 2012, 309, 33–66. [Google Scholar]
- Zezula, J.; Hudlicky, T. Recent Progress in the synthesis of morphine alkaloids. Synlett 2005, 388–405. [Google Scholar] [CrossRef]
- Taber, D.F.; Neubert, T.D.; Schlecht, M.F. The enantioselective synthesis of morphine. In Strategies and Tactics in Organic Synthesis; Harmata, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 5, p. 353. [Google Scholar]
- Novak, B.H.; Hudlicky, T.; Reed, J.W.; Mulzer, J.; Trauner, D. Recent progress in the synthesis of morphine alkaloids. Curr. Org. Chem. 2000, 4, 343. [Google Scholar] [CrossRef]
- Hudlicky, T.; Butora, G.; Fearnley, S.; Gum, A.; Stabile, M. A historical perspective of morphine syntheses. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 18, p. 43. [Google Scholar]
- Rice, K. Synthetic opium alkaloids and derivatives. A short total synthesis of (±)-dihydrothebainone, (±)-dihydrocodeinone, and (±)-nordihydrocodeinone as an approach to a practical synthesis of morphine, codeine, and congeners. J. Org. Chem. 1980, 45, 3135–3137. [Google Scholar] [CrossRef]
- Kimishima, A.; Umihara, J.; Mizoguchi, A.; Yokoshima, S.; Fukuyama, T. Synthesis of (−)-oxycodone. Org. Lett. 2014, 16, 6244–6247. [Google Scholar] [CrossRef]
- Endoma-Arias, M.A.A.; Makarova, M.; Dela Paz, H.E.; Hudlicky, T. Chemoenzymatic total synthesis of (+)-oxycodone from phenethyl acetate. Synthesis 2019, 225–232. [Google Scholar] [CrossRef]
- Makarova, M.; Endoma-Arias, M.A.A.; Dela Paz, H.E.; Simionescu, R.; Hudlicky, T. Chemoenzymatic total synthesis of ent-oxycodone: Second, third and fourth generation strategies. J. Am. Chem. Soc. 2019, 141, 10883–10904. [Google Scholar] [CrossRef]
- Zylstra, G.J.; Gibson, D.T. Toluene degradation by Pseudomonas putida F1. Nucleotide sequence of the todC1C2BADE genes and their expression in Escherichia coli. J. Biol. Chem. 1989, 264, 14940–14946. [Google Scholar]
- Hudlicky, T. Benefits of unconventional methods in the total synthesis of natural products. ACS Omega 2018, 3, 17326–17340. [Google Scholar] [CrossRef]
- Taher, E.S.; Banwell, M.G.; Buckler, J.N.; Yan, Q.; Lan, P. The exploitation of enzymatically derived cis-1,2-dihydrocatechols and related compounds in the synthesis of biologically active natural products. Chem. Rec. 2018, 18, 239–264. [Google Scholar] [CrossRef]
- Banwell, M.G.; Bolte, B.; Buckler, J.N.; Chang, E.L.; Lan, P.; Taher, E.S.; White, L.V.; Willis, A.C. Chemoenzymatic Pathways for the Synthesis of Biologically Active Natural Products; Royal Society of New South Wales: Bowral, NSW, Australia, 2016; Parts 1&2; pp. 34–50. [Google Scholar]
- Lewis, S.E. Applications of biocatalytic arene ipso, ortho cis-dihydroxylation in synthesis. Chem. Commun. 2014, 50, 2821–2830. [Google Scholar] [CrossRef]
- Rinner, U. Comprehensive Chirality; Carreira, E.M., Yamamoto, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 2, pp. 240–267. [Google Scholar]
- Bon, D.J.-Y.D.; Lee, B.; Banwell, M.G.; Cade, I.A. Biocatalytically-derived cis-1,2-dihydrocatechols—enantiopure and versatile building blocks for chemical synthesis. Chim. Oggi 2012, 30, 22–27. [Google Scholar] [CrossRef]
- Hudlicky, T.; Reed, J.W. Celebrating 20 Years of Synlett—Special account on the merits of biocatalysis and the impact of arene cis-dihydrodiols on eantioselective synthesis. Synlett 2009, 685–703. [Google Scholar] [CrossRef]
- Hudlicky, T.; Reed, J.W. Applications of biotransformations and biocatalysis to complexity generation in organic synthesis. Chem. Soc. Rev. 2009, 38, 3117–3132. [Google Scholar] [CrossRef]
- Boyd, D.R.; Bugg, T.D.H. Arene cis-dihydrodiol formation: from biology to application. Org. Biomol. Chem. 2006, 4, 181–192. [Google Scholar] [CrossRef]
- Johnson, R.A. Organic Reactions; Overman, L.E., Ed.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2004; Volume 63, pp. 117–264. [Google Scholar]
- Banwell, M.G.; Edwards, A.J.; Harfoot, G.J.; Jolliffe, K.A.; McLeod, M.D.; McRae, K.J.; Stewart, S.G.; Vogtle, M. Chemoenzymatic methods for the enantioselective preparation of sesquiterpenoid natural products from aromatic precursors. Pure Appl. Chem. 2003, 75, 223–230. [Google Scholar] [CrossRef]
- Boyd, D.R.; Sharma, N.D. Enzymatic and chemoenzymatic synthesis of arene trans-dihydrodiols. J. Mol. Catal. B Enzym. 2002, 19, 31–42. [Google Scholar] [CrossRef]
- Boyd, D.R.; Sharma, N.D.; Allen, C.C.R. Aromatic dioxygenases: Molecular biocatalysis and applications. Curr. Opin. Biotechnol. 2001, 12, 564–573. [Google Scholar] [CrossRef]
- Hudlicky, T.; Gonzalez, D.; Gibson, D.T. Enzymatic dihydroxylation of aromatics in enantioselective synthesis: Expanding asymmetric methodology. Aldrichim. Acta 1999, 32, 35–62. [Google Scholar]
- Boyd, D.R.; Sheldrake, G.N. The dioxygenase-catalysed formation of vicinal cis-diols. Nat. Prod. Rep. 1998, 15, 309–324. [Google Scholar] [CrossRef]
- Reed, J.W.; Hudlicky, T. Advances in Asymmetric Synthesis; Hassner, A., Ed.; JAI Press: Greenwich, CT, USA, 1995; Volume 1, pp. 271–312. [Google Scholar]
- Brown, S.M.; Hudlicky, T. Organic Synthesis: Theory and Applications; Hudlicky, T., Ed.; JAI Press: Greenwich, CT, USA, 1993; Volume 2, pp. 113–176. [Google Scholar]
- Carless, H.A.J. The use of cyclohexa-3,5-diene-1,2-diols in enantiospecific synthesis. Tetrahedron Asymmetry 1992, 3, 795–826. [Google Scholar] [CrossRef]
- Widdowson, D.A.; Ribbons, D.W.; Thomas, D.D. The use of substituted cyclohexadiene diols as versatile chiral synthons. Janssen Chim. Acta 1990, 8, 3–9. [Google Scholar]
- Hudlicky, T.; Endoma, M.A.A.; Butora, G. Nucleophilic substitution of protected β-bromoethyl cyclohexadiene-cis-diol as an alternative to direct microbial oxidation of β-functionalized phenethyl substrates. J. Chem. Soc. Perkin Trans. 1 1996, 17, 2187–2192. [Google Scholar] [CrossRef]
- Endoma, M.A.; Bui, V.P.; Hansen, J.; Hudlicky, T. Medium-scale preparation of useful metabolites of aromatic compounds via whole-cell fermentation with recombinant organisms. Org. Proc. Res. Dev. 2002, 6, 525–532. [Google Scholar] [CrossRef]
- Endoma-Arias, M.A.A.; Hudlicky, J.R.; Simionescu, R.; Hudlicky, T. Chemoenzymatic formal total synthesis of ent-codeine and other morphinans via nitrone cycloadditions and/or radical cyclizations. Comparison of strategies for control of C-9/C-14 stereogenic centers. Adv. Synth. Cat. 2014, 356, 333–339. [Google Scholar] [CrossRef]
- Hudlicky, T.; Endoma-Arias, M.A.A. Chemoenzymatic total synthesis of (+)-galanthamine and (+)-narwedine from phenethyl acetate. Chem. Eur. J. 2016, 22, 14540–14543. [Google Scholar]
- Koizumi, H.; Yokoshima, S.; Fukuyama, T. Total synthesis of (−)-morphine. Chem. Asian J. 2010, 5, 2191–2198. [Google Scholar] [CrossRef]
- Wiemann, J.; Deckelmann, A.M.; Csuk, R. A remarkably simple and convergent partial synthesis of pomolic acid. Tetrahedron Lett. 2016, 57, 3952–3953. [Google Scholar] [CrossRef]
- Oxidation of the diol 12 to afford a keto alcohol followed by NaBH4 reduction resulted in the formation of an epimeric (tentatively assigned as trans) diol. Attempts to form the acetonide of this supposed trans isomer failed. The diol 12 was smoothly converted to the cyclic carbonate [30]. It was also readily converted to the corresponding acetonide and benzylidene acetal. Unpublished observations.
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar] [CrossRef]
- Meredith, W.; Nemeth, G.A.; Boucher, R.; Carney, R.; Haas, M.; Sigvardson, K.; Teleha, C.A. Isolation and synthesis of 2-chloro-10-α-hydroxynaltrexone, a new naltrexone degradant. Tetrahedron Lett. 2003, 44, 7381–7384. [Google Scholar] [CrossRef]
Sample Availability: Samples are not available from the authors. |





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Endoma-Arias, M.A.; Dela Paz, H.; Hudlicky, T. Chemoenzymatic Total Synthesis of (+)-10-Keto-Oxycodone from Phenethyl Acetate. Molecules 2019, 24, 3477. https://doi.org/10.3390/molecules24193477
Endoma-Arias MA, Dela Paz H, Hudlicky T. Chemoenzymatic Total Synthesis of (+)-10-Keto-Oxycodone from Phenethyl Acetate. Molecules. 2019; 24(19):3477. https://doi.org/10.3390/molecules24193477
Chicago/Turabian StyleEndoma-Arias, Mary Ann, Helen Dela Paz, and Tomas Hudlicky. 2019. "Chemoenzymatic Total Synthesis of (+)-10-Keto-Oxycodone from Phenethyl Acetate" Molecules 24, no. 19: 3477. https://doi.org/10.3390/molecules24193477
APA StyleEndoma-Arias, M. A., Dela Paz, H., & Hudlicky, T. (2019). Chemoenzymatic Total Synthesis of (+)-10-Keto-Oxycodone from Phenethyl Acetate. Molecules, 24(19), 3477. https://doi.org/10.3390/molecules24193477