
Synthesis of Glycosidic (β-1′′→6, 3′ and 4′) Site Isomers of Neomycin B and Their Effect on RNA and DNA Triplex Stability

Department of Chemistry, University of Turku, 20014 Turku, Finland
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(3), 580; https://doi.org/10.3390/molecules24030580
Submission received: 4 January 2019
/
Revised: 3 February 2019
/
Accepted: 4 February 2019
/
Published: 6 February 2019
(This article belongs to the Special Issue Molecular Tools for Nucleic Acid Manipulation for Biological Intervention)
Abstract
:Glycosidic (β-1′′→6, 3′ and 4′) site isomers of neomycin B (i.e., neobiosamine (β-1′′→6, 3′ and 4′) neamines) have been synthesized in a straightforward manner. Peracetylated neomycin azide was used as a common starting material to obtain neobiosamine glycosyl donor and 6, 3′,4′-tri-O-acetyl neamine azide that after simple protecting group manipulation was converted to three different glycosyl acceptors (i.e., 5,6,4′-, 5,3′,4′- and 5,6,3′-tri-O-acetyl neamine azide). Glycosylation between the neobiosamine glycosyl donor and the neamine-derived acceptors gave the protected pseudo-tetrasaccharides, which were converted, via global deprotection (deacetylation and reduction of the azide groups), to the desired site isomers of neomycin. The effect of these aminoglycosides on the RNA and DNA triplex stability was studied by UV-melting profile analysis.
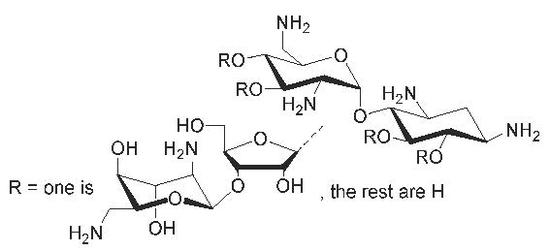
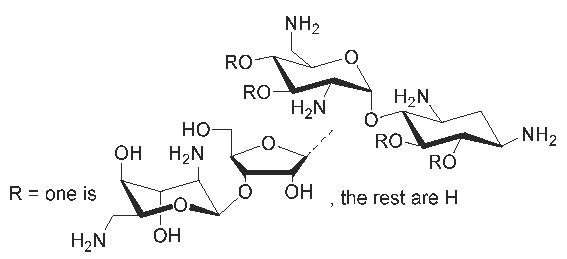
1. Introduction
Among the small molecular ligands that target nucleic acids, aminoglycosides (AGs) deserve special attention [1]. Their binding to a variety of nucleic acid targets has been extensively studied. Binding to the ribosomal decoding site is the basis of AGs’ bactericidal effect [2,3,4,5]. The continuous increase of antibiotic resistant infectious diseases maintains the interest around this RNA target, and a significant effort has been paid to optimization and modification of existing AG-based lead compounds to provide new potential antibacterial drugs [6,7]. Relatively high binding affinities have also been reported for other structurally resembling binding sites in ribozymes [7,8] and important regions of HIV RNAs (Trans Activation Response element (TAR), Reverse Response Element (RRE) and Dimerization Initiation Site (DIS)) [9,10,11,12]). In these RNA targets, the binding sites are bulges and internal loops, which, in contrast to canonical double helices, are able to form appropriate hydrogen bonds and electrostatic interactions with AGs. AGs can also act as groove binders that stabilize DNA- and RNA-triple helices and their hybrids [13,14,15,16,17]. Among the AGs studied for the triplex recognition, neomycin B has shown to be the most effective groove binder. The binding occurs to the Watson–Hoogsteen groove, in which favorable contacts with rings II and IV (cf. Scheme 1), together with the appropriate shape complementarity, may take place [14,17]. In each case, however, electrostatic interactions dominate AGs’ affinity to nucleic acids, which makes the binding promiscuous and may disturb detailed characterization of the binding motifs. Conformational adaption of the nucleic acid targets (in particular RNA) disturbs modelling and discovery of specific ligands even further [1]. Chemically synthesized structural analogs of the known AG-ligands are hence important tools that may be used for the exclusion analysis of the binding requirements.
In the present study, a straightforward synthesis of glycosidic (β-1″→6, 3′ and 4′) site isomers of neomycin B is described (Note: C6–, C3′– and C4′–substituted neamines are not neomycin class AGs, i.e., 4,5-disubstituted deoxystreptamines, and the correct names for these compounds are neobiosamine(β-C1″→C6, 3′ and 4′)neamines). The key steps of the synthesis (Scheme 1) were (1) acid-catalyzed thiolysis of neomycin azide [18], which gave useful intermediates for both glycosyl donor and the acceptors, and (2) selective diacetylation of neamine azide, which after simple protecting group manipulation afforded the glycosyl acceptors. Glycosylation between the neobiosamine donor and triacetylated neamine acceptors resulted in the azide masked pseudo-tetrasaccharides, which, via global deprotection, were converted to the desired site isomers of neomycin B (Scheme 2). In order to evaluate how the changed glycosidic connection (β-1″→3′,4′ or 6) between the neamine and neobiosamine cores affects the groove binding, the stability of RNA and DNA triple helix models in the presence of these structural analogs (and neomycin B) was studied by UV-melting profile analysis.
2. Results and Discussion
2.1. Synthesis of Glycosyl Donor 6 and Acceptors 14–16
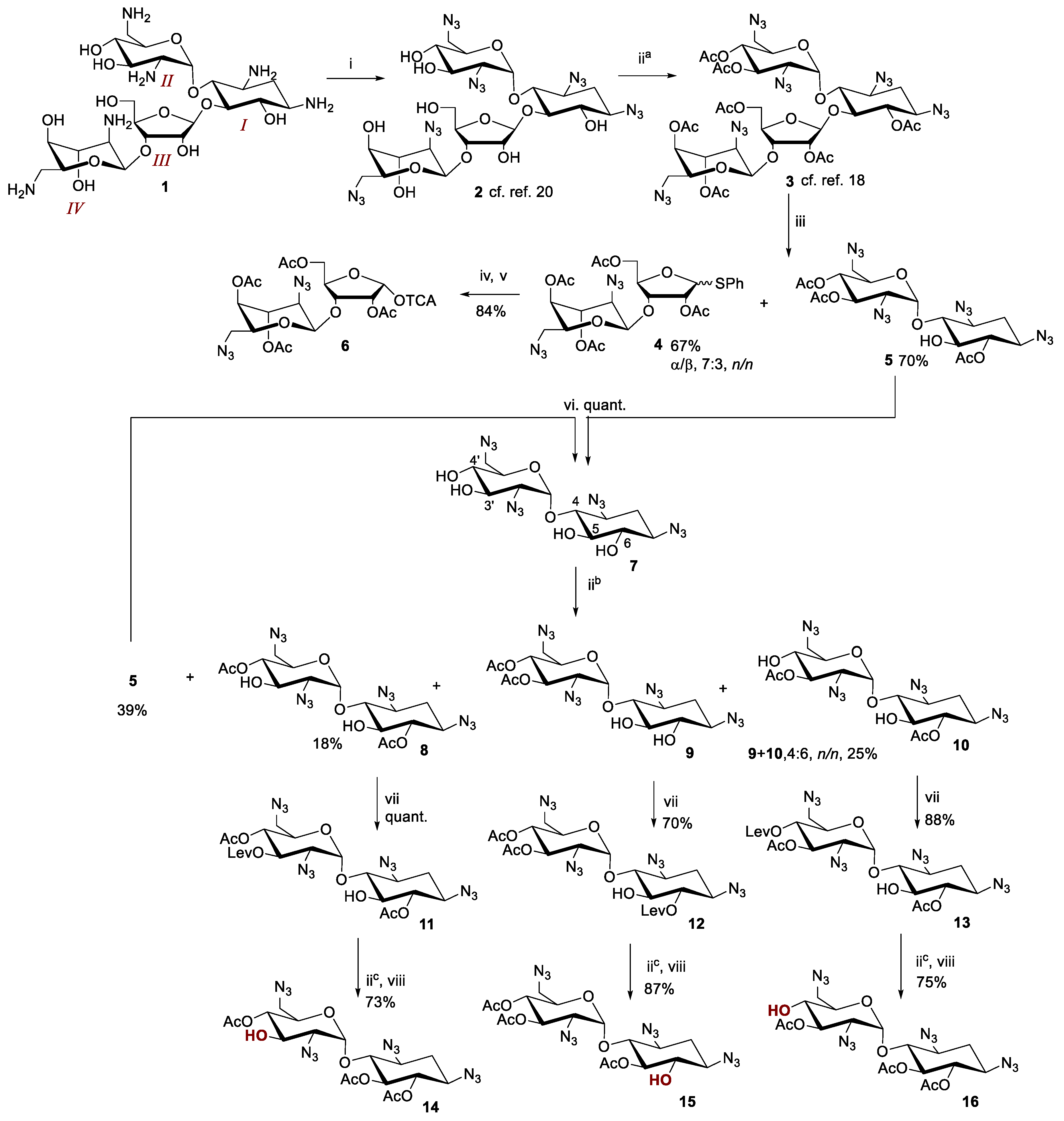
Acid-catalyzed degradation of neomycin B azide (3) has been studied in detail by Wu et al. [18]. Various conditions by using different Lewis acids (SnCl4, TMSOTf and BF3·OEt2) and thiols (propylenedithiol, EtSH and TolSH) have been evaluated for the selective cleavage. According to this data, 3 was cleaved with thiophenol in the presence of BF3·OEt2, to yield an anomeric mixture of phenylthio 2,5,3′4′-tetra-O-acetyl 2′,6′-diazidoneobiosamine (4) and 6,3′,4′-tri-O-acetyl 1,3,2′,6′-tetraazido neamine (5) in high yields (67% and 70%, respectively, iii in Scheme 1).
The anomeric mixture of phenylthio 2,5,3′4′-tetra-O-acetyl 2′,6′-diazidoneobiosamine (4) may be used as such as a glycosyl donor, but the mixture of the donor makes the control and monitoring of the reaction (cf. Scheme 2) complex. Therefore, the phenylthioglycoside (4) was hydrolyzed and the resulted hemiacetal converted to pure α-anomer of tricloroacetimidate 6. Overall, this leaving group conversion (from 4 to 6) could be carried out in 86% yield (iv and v/Scheme 1).
The 5-OH group of neamine (7) is less nucleophilic than the 6-, 3′- and 4′-OH groups. For this reason, selective triacetylation of N-protected neamines (e.g., 5 from 7) may be readily carried out as described previously in several studies [19,20]. On using a smaller excess of acetic anhydride, diacetylated neamines (8–10) may also be obtained, albeit the yield and ratio of the products (8, 9, 10, 5 and monoacetylated neamines) are more sensitive to reaction conditions (temperature, reaction time, and the addition rate and excess of acetic anhydride). Overnight reaction in pyridine in the presence of a catalytic amount of 4-(N,N-dimethylamino)pyridine (DMAP) and 2.5 equiv. of acetic anhydride (slowly added to the reaction mixture) resulted in the desired diacetylated neamines 8, 9 and 10 in 18%, 10% and 15% yield, respectively (9 and 10 isolated as a mixture (4:6, n/n)) (iib/Scheme 1). The triacetylated neamine 5 was obtained as the major product (39% yield), but it could be readily converted back to the starting material (7) and the reaction repeated. It may be worth mentioning that a smaller excess of acetic anhydride did not markedly improve the yields of 8–10. Moreover, the increased ratio of monoacetylated neamines disturbed the chromatographic isolation of the diacetylated products. Thus, the slight ‘overacetylation’ was beneficial. The modest reactivity of the 5-OH group of the diacetylated neamines (8, 9 and 10) was next utilized for the selective levulinoylation of the 3′-, 6- and 4′-OH groups to obtain 11–13 (70% – quant., vii/Scheme 1). The reaction was carried out in pyridine using 1.5 equiv. of levulinic anhydride in the presence of a catalytic amount of DMAP. The remained free 5-OH group of 11–13 was finally acetylated using an excess of acetic anhydride (5 equiv. iic/Scheme 1) and subsequent removal of the levulinoyl group with hydrazine hydrate (viii/Scheme 1) gave the desired glycosyl acceptors 14–16 in 73–87% yield.
2.2. Glycosylation and Global Deprotection
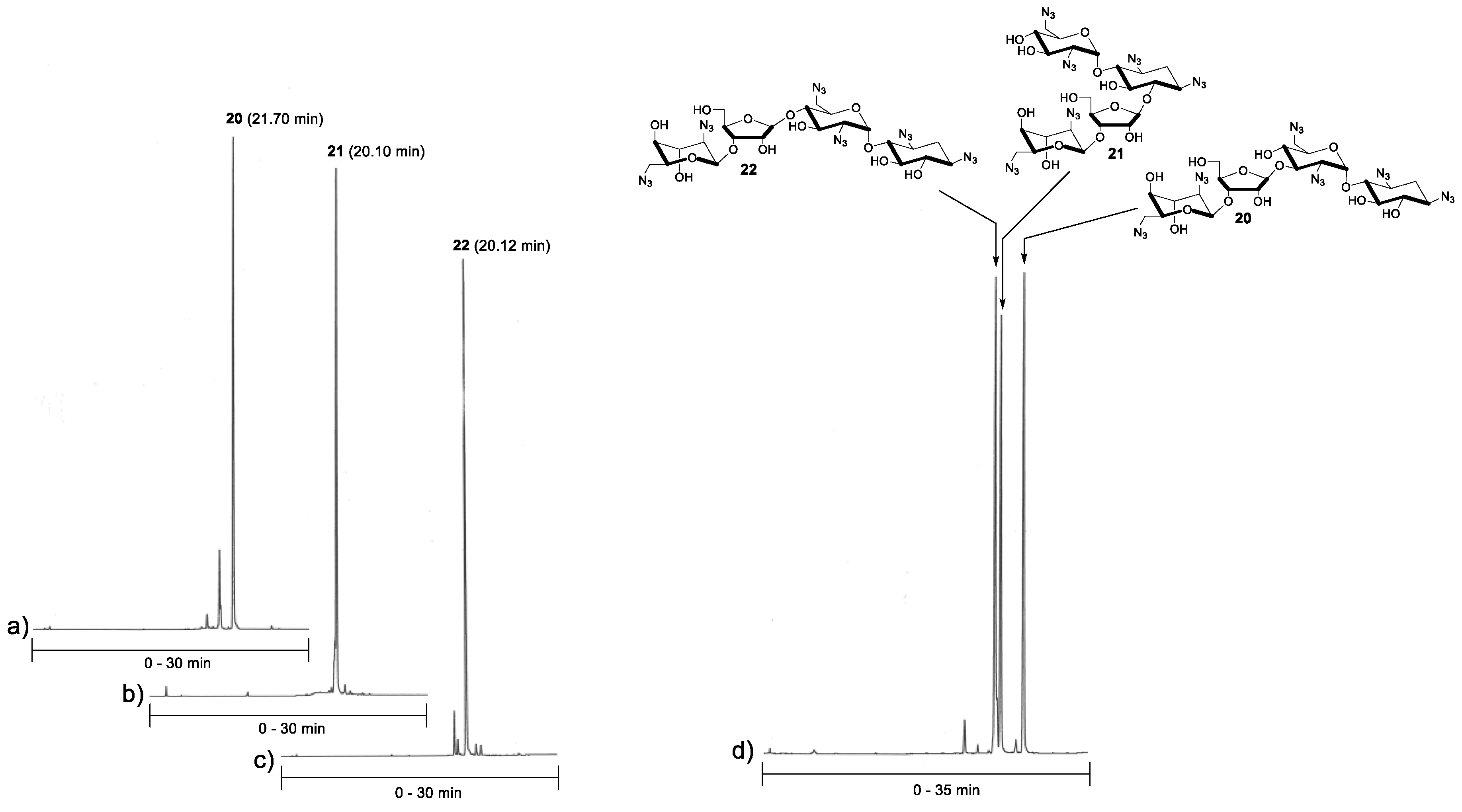
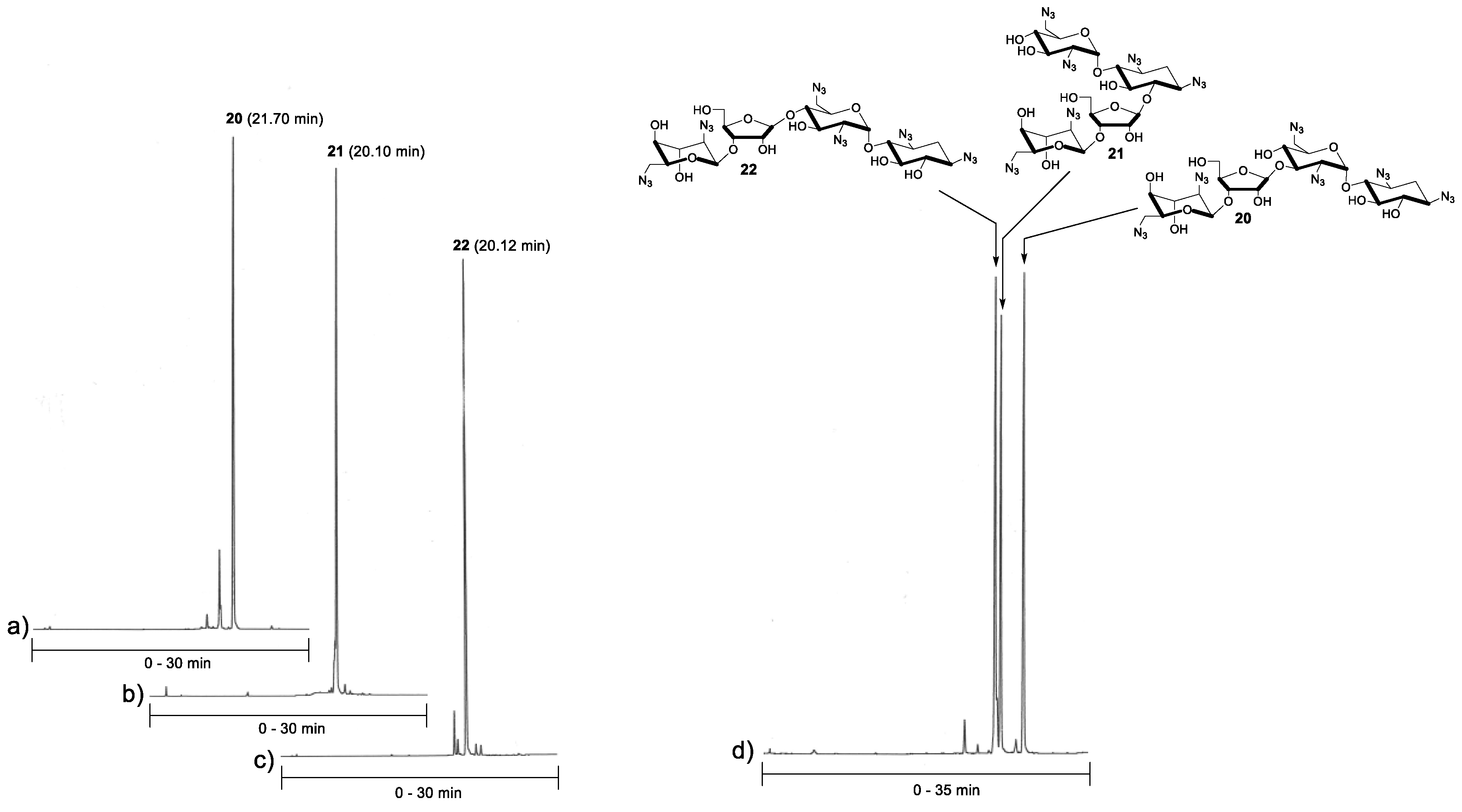
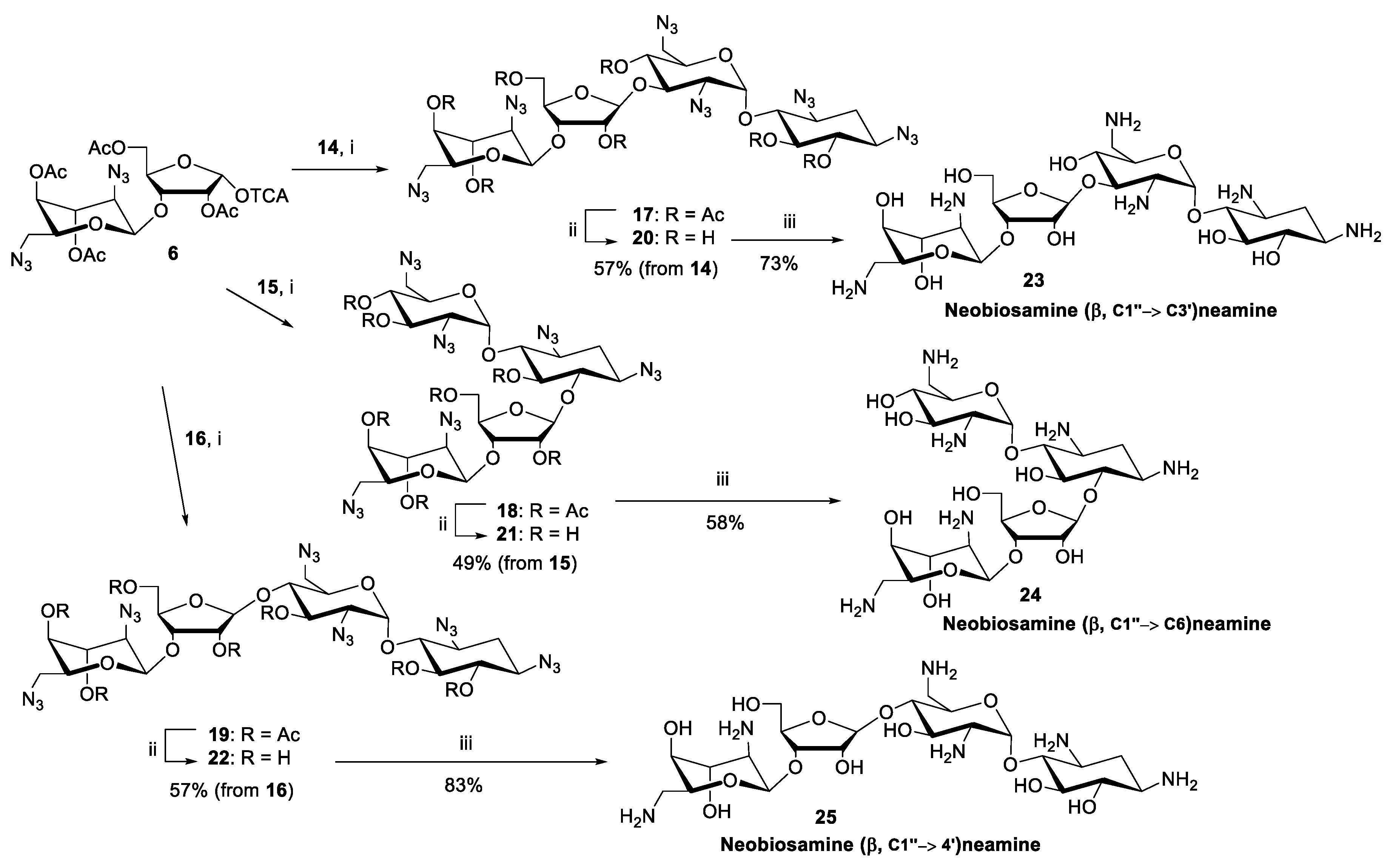
A standard procedure using TMSOTf as a catalyst was applied for the glycosylation between acceptors 14–16 and donor 6 (1.5 equiv.). Each reaction was performed at −20 °C (for 2 h) in dry dichloromethane under nitrogen. For the purification reasons (the fully protected pseudo-tetrasaccharides 17–19 remained contaminated by traces of donor 6, despite a laborious chromatographic purification), the glycosylation was followed by subsequent deacetylation (0.1 mol L−1 NaOMe in MeOH) and the neomycin azides 20–22 could be isolated in acceptable yields (49–69%). The purity of the products (20–22) was confirmed by reversed phase-high performance liquid chromatography (RP HPLC) analysis (Figure 1). Finally, the azide masks were removed by Staudinger reaction using trimethylphosphine and aqueous ammonia, followed by elution through an ion exchange resin, to give the glycosidic (β-C1″→C6, C3′ and C4′) site isomers of neomycin B (23–25) in 58–83% yields.
2.3. The Effect of 23–25 on DNA and RNA Triplex Stability
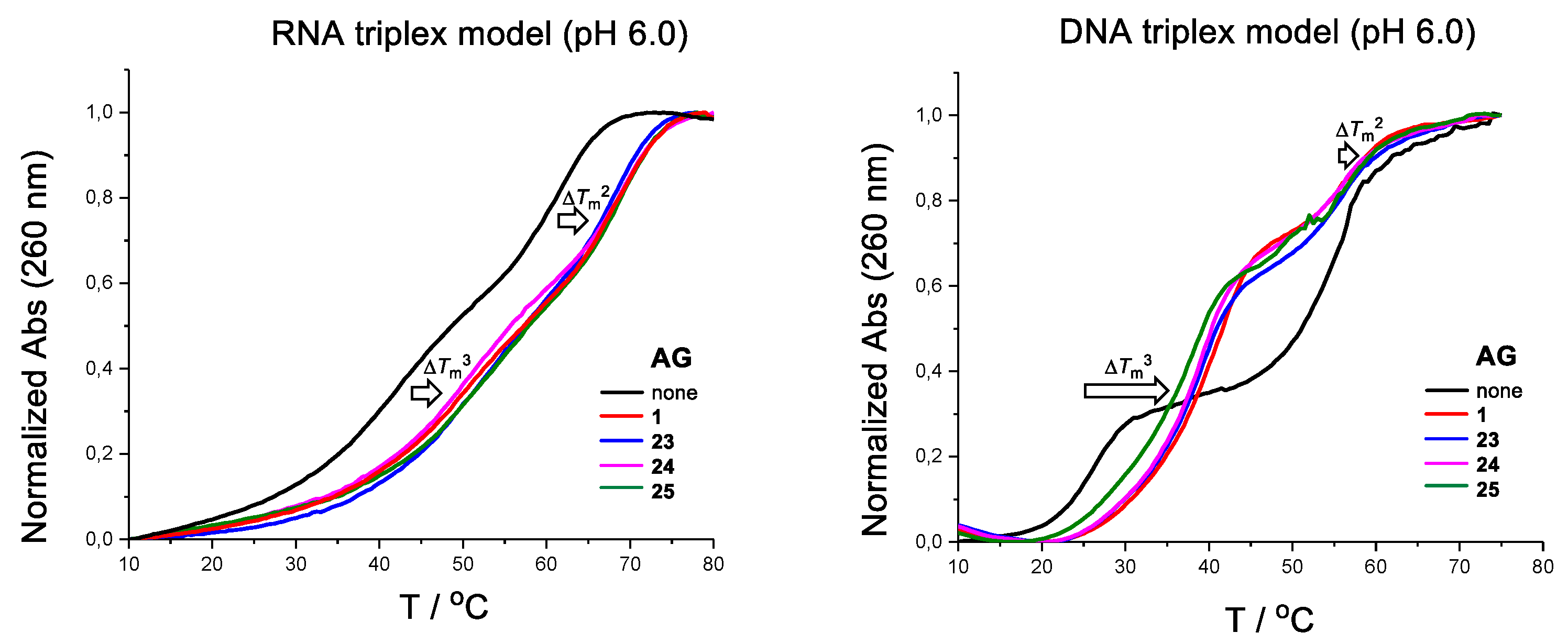
The effect of the glycosidic site isomers of neomycin B (23–25) and neomycin (1) on the stability of simple DNA and RNA triple helices was studied by UV-melting profile experiments (Table 1). The DNA triple helix is consisted of a purine rich region of c-Myc promoter 1 [21], used as a model in several previous studies [22,23,24,25,26,27]. The formation of this triple helix, consisting of CH+•G-C-triplets, requires slightly acidic pH. The intramolecular RNA triplex is also a known model (in fact, this model may exist as a mixture with its dimer [28]). The measurements were carried out using 2 µmol L−1 of the oligonucleotides in a mixture of 10 mmol L−1 sodium cacodylate and 0.1 mol L−1 NaCl at pH 6.0 (both RNA and DNA model) and at pH 7.0 (RNA model only) in the presence of 5 and 10 eq. of the AGs (1 and 23–25). The temperature was changed at a rate of 0.2 °C min−1. The Tm3- and Tm2-values (triplex and duplex melting points) were extracted from the first and second inflection points of the biphasic melting curves at λ = 260 nm, respectively (Figure 2). Consistent with the previous findings (affinity of neomycin follows the trend: DNA duplex < DNA triplex < RNA duplex ~ RNA triplex [17]), AGs stabilized both the triplex and the duplex on the RNA model at pH 6.0, whereas basically only the triplex on the DNA model (ΔTm3 vs. ΔTm2 = +11.1−18.6 °C vs. +0.7−2.3 °C). A slight selectivity of the stabilization on the RNA triplex vs. duplex may be observed at pH 7.0 (ΔTm3 vs. ΔTm2 = +6.9−7.8 °C vs. +1.1−1.9 °C). Most importantly, AGs’ effect on the triplex (both on RNA and DNA model) did not show a marked difference when AGs are compared to each other. Only a slight variation was observed. For example, on the DNA model, the stability of the triplex increased by ΔTm = +18.6 °C, +17.4 °C, +15.8 °C and +13.9 °C in the presence of 10 equiv. of neomycin B (1), the C1″→C4′ (25), C1″ →C3′ (23) and C1″→C6 (24) site isomers, respectively. On the RNA triplex, the effect with the C1″→C3′ (23) and C1″→C6 (24) site isomers was, in fact, slightly higher compared to that with neomycin B (1) (ΔTm3 = +11.3 and +10.6 °C vs. +9.3 °C). Probably, the carbohydrate core and spatial orientation of the amino groups on it do not play a marked enough role to result in discrimination between the affinities of the glycosidic site isomers (1, 23–25). This may be an unexpected observation as, e.g., previous studies with kanamycin class aminoglycosides (kanamycin = a 4,6-disubstituted deoxystreptamine consisted of neamine and α-d-glucos-3-amine at C6) show a modest or no effect on the poly U-A-U DNA triplex [14]. The effect of 24 (a 4,6-disubstituted deoxystreptamine) on the DNA triplex was the smallest (ΔTm3 = +13.9 °C) of the isomers (23–25), but still comparable to neomycin B (1, ΔTm3 = +18.6 °C), the most effective aminoglycosidic DNA triplex groove binder to date. The biosamine moiety, or the l-neosamine (Ring IV in Scheme 1), hence plays a marked role in the groove binding despite the glycosidic site on the neamine core.
3. Materials and Methods
General Remarks
Pyridine, methanol and dichloromethane were dried over 3Å molecular sieves. Nuclear magnetic resonance (NMR) spectra were recorded using a 500 MHz instrument. The chemical shifts for 1H and 13C-NMR resonances are given in parts of million from the residual signal of the deuterated solvents (CD3OD and CD3Cl3). Mass spectra were recorded using electrospray ionization (ESI-TOF). The NMR spectral data for all new compounds are showed in the Supplementary Materials.
2,5,3′,4′-Tetra-O-acetyl-2′,6′-diazido-1-phenylthio-α/β-neobiosamine (4). Peracetylated neomycin azide (3, 5.8 g, 5.6 mmol, synthesized according to the literature [18,20]) and PhSH (0.64 mL, 6.2 mmol) were dissolved in dry dichloromethane (50 mL). The mixture was cooled down to 0 °C, and BF3·Et2O (2.1 mL, 17 mmol) was slowly added under nitrogen. The mixture was stirred first at 0 °C for 20 min, and allowed then to warm up to room temperature. After overnight reaction, triethylamine (1.5 mL) was added, the mixture was diluted with dichloromethane (150 mL), and washed with water (20 mL). The organic phase was dried over Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel chromatography (30-40% EtOAc in petroleum ether) to give an anomeric mixture of 4 (2.3 g, 67%, α/β = 7:3, v/v, pure fractions used for the NMR characterization) and 1,3,2′,6′-tetra azido-6,3′,4′-tri-O-acetyl neamine (5, 2.2 g, 70%). 4: β-anomer: 1H (500 MHz, CDCl3): δ 7.52–7.50 (m, 2H), 7.34–7.28 (m, 3H), 5.74 (d, 1H, J = 4.7 Hz), 5.48 (t, 1H, J = 4.2 Hz), 5.06 (t, 1H, J = 2.7 Hz), 5.03 (d, 1H, J = 1.5 Hz), 4.73 (b, 1H), 4.50–4.47 (m, 3H), 4.26 (m, 1H), 4.14 (m, 1H), 3.64 (dd, 1H, J = 13.1 and 8.6 Hz), 3.42 (b, 1H), 3.22 (dd, 1H, J = 13.1 and 3.7 Hz), 2.20, 2.20, 2.18 and 2.09 (4 × s, 3H each); 13C (125 MHz, CDCl3): δ 170.7, 170.2, 169.9, 168.5, 134.5, 131.5, 129.1, 127.5, 98.5, 89.1, 78.4, 75.8, 73.9, 72.6, 68.8, 65.8, 63.0, 56.4, 50.7, 20.8, 20.8, 20.7, 20.6; HRMS (ESI-TOF) calculated for C25H30N6KO11S [M + K]+: 661.1330, observed: 661.1332. α-anomer: 1H (500 MHz, CDCl3): δ 7.54–7.52 (m, 2H), 7.38–7.33 (m, 3H), 5.43 (d, 1H, J = 2.9 Hz), 5.26 (dd, 1H, J = 5.1 and 3.0 Hz), 5.05 (dd, 1H, J = 2.9 and 2.8 Hz), 4.92 (d, 1H, J = 1.8 Hz), 4.73 (t, 1H, J = 2.0 Hz), 4.51 (dd, 1H, J = 12.1 and 2.6 Hz), 4.45 (dd, 1H, J = 7.3 and 5.2 Hz), 4.31 (m, 1H), 4.23 (dd, 1H, J = 12.1 and 5.1 Hz), 4.11 (ddd, 1H, J = 8.1, 4.4 and 1.8 Hz), 3.61 (dd, 1H, J = 13.0 and 8.2 Hz), 3.34 (dd, 1H, J = 2.1 and 2.0 Hz), 3.29 (dd, 1H, J = 13.0 and 4.4 Hz), 2.19, 2.18, 2.13 and 2.10 (4 × s, each 3H); 13C (125 MHz, CDCl3): δ 170.7, 170.3, 169.8, 168.6, 132.9, 132.2, 129.1, 128.3, 98.9, 88.7, 80.1, 76.5, 75.0, 73.5, 68.7, 65.7, 63.2, 56.4, 50.6, 20.9, 20.8, 20.7, 20.7; HRMS (ESI-TOF) calculated for C25H30N6NaO11S [M + Na]+: 645.1591, observed: 645.1563.
2,5,3′,4′-Tetra-O-acetyl-2′,6′-diazido-neobiosamine trichloroacetimidate (6). N-bromosuccinimide (NBS) (0.77 g, 4.3 mmol) and trifluoroacetic acid (0.30 mL) were added to a two-phase mixture of 4 (1.4 g, 2.2 mmol, as an anomeric mixture) in dichloromethane (15 mL), acetonitrile (7.5 mL) and H2O (15 mL) at 0 °C. The mixture was allowed to warm up to ambient temperature, stirred for 1 h, poured to saturated NaHCO3 and extracted with dichloromethane. The organic layers were combined, dried over Na2SO4, filtered and evaporated to dryness. The residue was purified by silica column chromatography (5% MeOH in DCM) to give an anomeric mixture of the neobiosamine hemiacetal (1.1 g, 98%) as a white foam. 1H-NMR (500 MHz, CDCl3) δ: 5.52 (dd, 0.25H, J = 10.3 and 4.1 Hz), 5.41 (d, 1H, J = 2.5 Hz), 5.23 (d, 1H, J = 4.5 Hz), 5.08 (dd, 0.25 H, J = 3.2 and 3.1 Hz), 5.05–5.02 (m, 1.25H), 5,00 (d, 0.25H, J = 2.7 Hz), 4.99 (d, 1H, J = 1.8 Hz), 4.75 (t, 0.25H, J = 2,3 Hz), 4.72 (dd, 1H, J = 1.9 and 1.8 Hz), 4.67 (dd, 1H, J = 7.5 and 4.5 Hz), 4.53 (m, 0.25H), 4.50 (dd, 1H, J = 11.6 and 2.3 Hz), 4.46 (t, 0.25H, J = 5.2 Hz), 4.38 (dd, 0.25H, J = 12.2 and 2.8 Hz), 4.31 (m, 1H), 4.28–4.21 (m, 1.25H), 4.15 (ddd, 1H, J = 8.3, 4.1 and 1.8 Hz), 4.11 (ddd, 0.25H, J = 8.5, 4.0 and 2.0 Hz), 3.66–3.60 (m, 1.25H), 3.46 (t, 0.25H, J = 2.4 Hz), 3.42 (d, 0.25H, J = 4.1 Hz), 3.39 (d, 1H, J = 3.0 Hz), 3.36 (t, 1H, J = 1.9 Hz), 3.29 (dd, 0.25H, J = 13.1 and 4.1 Hz), 3.24 (dd, 1H, J = 13.1 and 4.1 Hz), 2.20, 2.18, 2.17, 2.13, 2.11 and 2.11 (each s, 15H); 13C-NMR (125 MHz, CDCl3) δ: 171.0, 170.3, 169.9, 168.6, 100.2, 99.1, 98.6, 95.8, 79.3, 79.1, 76.4, 76.2, 74.4, 73.8, 73.5, 71.9, 68.8, 68.7, 65.8, 65.8, 64.5, 63.4, 50.8, 20.9, 20.8, 20.7, 20.7; MS (ESI-tOF): calculated for C19H26N6NaO12 [M + Na]+: 553.1506, observed 553.1488. The hemiacetal (1.0 g, 1.9 mmol) was dissolved in a mixture of dichloromethane (1.0 mL) and trichloroacetonitrile (1.9 mL, 19 mmol). The mixture was cooled down to 0 °C and 1.8-diazabicyclo(5.4.0)undec-7-ene (DBU) (56 μL, 0.38 mmol) was added. The mixture was stirred for 1 h at 0 °C and eluted as such (without a work up) through a silica gel column (1% Et3N, 50% EtOAc in petroleum ether) to give 1.2 g (91%, 84% overall yield from 4) of the trichloroacetimidate product (6) as a white foam. 1H-NMR (500 MHz, CDCl3): δ 8.36 (s, 1H), 6.36 (s, 1H), 5.47 (d, 1H, J = 4.6 Hz), 5.06–5.04 (m, 2H) 4.73 (dd, 1H, J = 1.9 and 1.8 Hz), 4.68 (dd, 1H, J = 8.1 and 4.5 Hz), 4.53 (dd, 1H, J = 12.1 and 2.8 Hz), 4.45 (m, 1H), 4.22 (dd, 1H, J = 12.2 and 6.4 Hz), 4.19 (ddd, 1H, J = 8.6, 3.7 and 1,8 Hz), 3.63 (dd, 1H, J = 13.1 and 8,6 Hz), 3.35 (dd, 1H, J = 2.1 and 1.9 Hz), 3.21 (dd, 1H, J = 13.1 and 3.7 Hz), 2.20, 2.19, 2.18, 2.11 (each s, each 3H); 13C-NMR (125 MHz, CDCl3): δ 170.8, 170.1, 169.8, 168.6, 160.3, 102.7, 98.4, 90.6, 80.2, 75.3, 73.9, 73.0, 68.7, 65.8, 64.0, 56.3, 50.7, 20.8, 20.8, 20.7, 20.6; MS (ESI-TOF): calculated for C21H26Cl3N7NaO12 [M + Na]+: 696.0603, observed 696.0631.
Synthesis of diacetylated neamine azides (8, 9 and 10). Acetic anhydride (2.3 mL, 24 mmol) was slowly added to a mixture of neamine azide (7, 4.0 g, 9.3 mmol) in pyridine (20 mL). The mixture was stirred overnight at ambient temperature, concentrated, poured to saturated NaHCO3 and extracted with ethyl acetate. The organic layers were combined, washed with brine, dried over Na2SO3, filtered and evaporated to dryness. The residue was purified by silica gel chromatography (40% EtOAc in petroleum ether) to give three fractions of products. The first eluting compound was 6,3′,4′-tri-O-acetyl-1,3,2′,6′-tetra-azido-neamine (5, 2.0 g, 39%, the main product). The second fraction was a mixture of 3′,4′-di-O-acetyl- (9) and 6, 3′-di-O-acetyl- (10) 1,3,2′,6′-tetra-azidoneamines (9:10, 4:6, n/n, 1.3 g, 25%) and the third fraction was pure 6,4′-di-O-acetyl-1,3,2′,6′-tetra-azidoneamine (8, 0.87 g, 18%). Each product was obtained as a white foam. Compound 8: 1H (500 MHz, CDCl3): δ 5.32 (d, 1H, J = 3.4 Hz), 4.96 (t, 1H, J = 10.0 Hz), 4.91 (t, 1H, J = 9.7 Hz), 4.24 (ddd, 1H, J = 10.0, 5.0 and 2.8Hz), 4.12 (dd, 1H, J = 9.0 and 8.6 Hz), 3.86 (d, 1H, J = 3.3 Hz), 3.67 (ddd, 1H, J = 12.2, 9.5 and 2.9 Hz), 3.60 (dd, 1H, J = 10.1 and 3.7 Hz), 3.54 (ddd, 1H, J = 12.5, 10.0 and 4.6 Hz), 3.46–3.41 (m, 2H), 3.38–3.33 (m, 2H), 3.03 (b, 1H), 2.40 (ddd, 1H, J = 13.4, 4.6 and 4.5 Hz), 2.20 (s, 3H), 2.18 (s, 3H), 1.63 (ddd, 1H, J = 12.6 Hz, each); 13C (125 MHz, CDCl3): δ 171.4, 170.5, 98.9, 83.7, 74.9, 74.4, 72.3, 71.7, 69.5, 64.2, 58.6, 58.0, 51.0, 32.0, 20.9. A mixture of 9 + 10: 1H (500 MHz, CDCl3): δ 5.51 (dd, 0.4H, J = 10.3 and 9.4 Hz), 5.36 (d, 0.4H, J = 5.0 Hz), 5.34 (d, 0.6H, J = 3.7 Hz), 5.30 (dd, 0.6H, J = 10.1 and 9.7 Hz), 5.07 (dd, 0.4H, J = 9.9 and 9.6 Hz), 4.94 (dd, 0.6H, J = 9.8 and 9.7 Hz), 4.37 (ddd, 0.4H, J = 10.2, 4.9 and 2.7 Hz), 4.17 (ddd, 0.6H, J = 10.1, 4.3 and 2.8 Hz), 3.77 (d, 0.4H, J = 2.7 Hz), 3.72–3.62 (m, 3.4H), 3.59–3.52 (m, 1.6H), 3.47–3.31 (m, 3.2H), 2.98 (b, 0.4H), 2.78 (d, 0.6H, J = 5.6 Hz), 2.43–2.36 (m, 1H), 2.22 (s, 1.8H), 2.20 (s, 1.8 H), 2.12 (s, 1.2 H), 2.08 (s, 1.2 H), 1.66–1.55 (m, 1H); 13C (125 MHz, CDCl3): δ 172.1, 170.5, 170.2, 169.8, 99.0, 98.6, 83.7, 83.2, 75.9, 75.4, 74.9, 74.8, 74.4, 71.9, 71.3, 69.9, 69.5, 69.3, 61.72, 61.71, 59.7, 58.6, 58.4, 57.9, 53.5, 50.9, 50.8, 32.0, 31.9, 20.9, 20.8, 20.7, 20.6.
6,4′-Di-O-acetyl-3′-O-levulinyl-1,3,2′,6′-tetra-azidoneamine (11). Freshly prepared levulinic anhydride (63 mg, 0.29 mmol) was added to a mixture of 8 (0.10 g, 0.20 mmol) in pyridine (2.0 mL) and a catalytic amount of 4-(N,N-dimethylamino)pyridine was added. The mixture was stirred overnight at ambient temperature, poured to saturate NaHCO3, and the product was extracted with dichloromethane. The organic layer was separated, dried over Na2SO4 and evaporated to dryness. The residue was purified by silica gel chromatography (5% MeOH in DCM) to give 120 mg (quant.) of the product (11) as a white foam. 1H-NMR (500 MHz, CDCl3): δ 5.52 (dd, 1H, J = 10.1 and 9.9 Hz), 5.43 (d, 1H, J = 3.6 Hz), 5.07 (t, 1H, J = 9.8 Hz), 4.94 (dd, 1H, J = 10.0 & 9.9 Hz), 4.34 (ddd, 1H, J = 7.9, 5.0 and 2.7 Hz), 3.88 (b, 1H), 3.69 (t, 1H, J = 9.5 Hz), 3.61 (dd, 1H, J = 10.6 and 3.6 Hz), 3.54 (ddd, 1H, J = 12.4, 10.2 and 4.4 Hz), 3.47 (dd, 1H, J = 9.8 and 9.2 Hz), 3.43–3.35 (m, 2H), 3.32 (dd, 1H, J = 13.5 and 5.2 Hz), 2.82 (m, 1H), 2.71 (m, 1H), 2.62 (m, 1H), 2.54 (m, 1H), 2.41 (ddd, 1H, J = 13.4, 4.5 and 4.5 Hz), 2.19 (s, 3H), 2.17 (s, 3H), 2.13 (s, 3H), 1.63 (ddd, 1H, J = 12.6 Hz. each); 13C (125 MHz, CDCl3): δ 206.0, 171.9, 170.5, 170.0, 98.8, 83.2, 75.1, 74.4, 71.0, 69.6, 68.9, 61.8, 58.3, 57.9, 50.9, 37.6, 31.9, 29.7, 27.9, 20.8, 20.7; HRMS (ESI-TOF) calculated for C21H28N12NaO10 [M + Na]: 631.1949, observed: 631.1924.
3′,4′-Di-O-acetyl-6-O-levulinyl-1,3,2′,6′-tetra-azidoneamine (12) and 6,3′-Di-O-acetyl-4′-levulinyl-1,3,2′,6′-tetra-azidoneamine (13). The mixture of 9 and 10 (1:2, n/n, 0.38 g, 0.74 mmol) was treated as described for 11 (from 8) above. 0.11 g (70%) of 12 and 0.27g (88%) of 13 were obtained as white foams. 12: 1H-NMR (500 MHz, CDCl3): δ 5.57 (d, 1H, J = 3.7 Hz), 5.48 (dd, 1H, J = 11.1 and 9.3 Hz), 5.04 (dd, 1H, J = 10.1 and 9.4 Hz), 4.94 (t, 1H, J = 9.9 Hz), 4.39 (ddd, 1H, J = 10.1, 5.0 and 2.8 Hz), 3.87 (d, 1H, J = 3.1 Hz), 3.76 (ddd, 1H, J = 9.4, 9.4 and 3.0 Hz), 3.53–3.42 (m, 2H), (t, 1H, J = 7.0 Hz), 3.50 (dd, 1H, J = 10.7 and 3.7 Hz), 3.39 (dd, 1H, J = 13.5 and 2.9 Hz), 3.31 (dd, 1H, J = 13.4 and 5.1 Hz), 2.93 (m, 1H), 2.81 (m, 1H), 2.66 (m, 1H), 2.57 (m, 1H), 2.40 (ddd, 1H, J = 13.3, 4.5 and 4.5 Hz), 2.21 (s, 3H), 2.10 (s, 3H), 2.06 (s, 3H), 1.64 (ddd, 1H, J = 12.5 Hz, each); 13C (125 MHz, CDCl3): δ 207.8, 172.3, 170.2, 169.7, 98.4, 81.4, 75.8, 74.7, 70.6, 69.4, 69.3, 61.2, 58.5, 57.7, 50.8, 38.4, 31.9, 29.8, 28.1, 20.7, 20.6; HRMS (ESI-TOF) calculated for C21H28N12NaO10 [M + Na]: 631.1949, observed: 631.1970. 13: 1H-NMR (500 MHz, CDCl3): δ 5.52 (dd, 1H, J = 10.3 and 9.6 Hz), 5.38 (d, 1H, J = 3.6 Hz), 5.07 (t, 1H, J = 9.8 Hz), 4.94 (t, 1H, J = 9.8 Hz), 4.32 (ddd, 1H, J = 10.2, 4.9 and 2.8 Hz), 3.69 (m, 1H), 3.64 (dd, 1H, J = 10.6 and 3.7 Hz), 3.55 (ddd, 1H, J = 12.6, 10.1 and 4.6 Hz), 3.48–3.36 (m, 3H), 2.76 (m, 2H), 2.51 (m, 2H), 2.41 (ddd, 1H, J = 13.5, 4.6 and 4.5 Hz), 2.18 (s, 6H), 2.14 (s, 3H), 1.63 (ddd, 1H, J = 12.5 Hz, each); 13C (125 MHz, CDCl3): δ 206.2, 171.6, 170.3, 98.8, 83.5, 75.0, 74.4, 70.7, 69.6, 69.2, 61.8, 58.3, 57.9, 50.7, 37.7, 31.9, 29.6, 27.8, 20.7, 20.7; HRMS (ESI-TOF) calculated for C21H28N12NaO10 [M + Na]: 631.1949, observed: 631.1961.
5,6,4′-Tri-O-acetyl-1,3,2′,6′-tetra-azidoneamine (14). Acetic anhydride (85 μL, 0.90 mmol) was added to a mixture of 11 (0.11 g, 0.18 mmol) and a catalytic amount of 4-(N,N-dimethylamino)pyridine in pyridine (2.0 mL). The mixture was stirred at ambient temperature for 3 h, poured to saturated NaHCO3, and the product was extracted with dichloromethane. The organic layers were combined, dried over Na2SO4, and evaporated to dryness. The residue was dissolved in a mixture dichloromethane (6.0 mL) and methanol (1.0 mL) and hydrazine acetate (34 mg, 0.36 mmol) was added. The mixture was stirred at ambient temperature for 3 h, poured to 10% aqueous KH2PO4, and the product was extracted with dichloromethane. The organic layers were combined, washed with brine, dried over Na2SO4, and evaporated to dryness. The residue was purified by silica gel chromatography (2% MeOH in DCM) to give 73 mg (73%) of the product 14 as a white foam. 1H-NMR (500 MHz, CDCl3): δ 5.15 (dd, 1H,m J = 9.8 Hz, both), 5.14 (d, 1H, J = 3.3 Hz), 4.95 (t, 1H, J = 10.0 Hz), 4.90 (dd, 1H, J = 9.7 and 9.6 Hz), 4.32 (m, 1H), 4.07 (t, 1H, J = 9.7 Hz), 3.69–3.64 (m, 2H), 3.51 (m, 1H), 3.39–3.24 (m, 4H), 2.44 (ddd, 1H, J = 13.3, 4.4 and 4.4 Hz), 2.15 (s, 3H), 2.10 (s, 3H), 2.09 (s, 3H), 1.64 (ddd, 1H, J = 12.8 Hz, each); 13C (125 MHz, CDCl3): δ 171.0, 169.9, 169.8, 98.9, 78.6, 74.1, 73.4, 72.1, 69.7, 69.6, 62.9, 58.7, 57.7, 51.0, 31.8, 20.9, 20.7, 20.5; HRMS (ESI-TOF) calculated for C18H24N12NaO9 [M + Na]: 575.1687, observed: 575.1680.
5,3′,4′-Tri-O-acetyl-1,3,2′,6′-tetra-azidoneamine (15). Compound 15 was synthesized from 12 as described for 14 above. 12 (0.13 g, 0.22 mmol) gave 98 mg (87%) of the product (15) as a white foam. 1H-NMR (500 MHz, CDCl3): δ 5.46 (dd, 1H, J = 10.7 and 9.3 Hz), 5.26 (d, 1H, J = 3.8 Hz), 5.09–5.04 (m, 2H), 4.48 (ddd, 1H, J = 10.2, 4.8 and 2.8 Hz), 3.63–3.53 (m, 3H), 3.48 (m, 1H), 3.42–3.30 (m, 3H), 2.94 (d, 1H, J = 6.3 Hz), 2.41 (ddd, 1H, J = 13.3, 4.4 and 4.4 Hz), 2.20 (s, 3H), 2.10 (s, 3H), 2.08 (s, 3H), 1.55 (ddd, 1H, J = 12.3 Hz, each); 13C (125 MHz, CDCl3): δ 171.2, 170.0, 169.8, 98.7, 78.7, 76.2, 75.6, 69.8, 69.5, 69.3, 60.7, 60.3, 58.6, 50.7, 31.6, 20.9, 20.6, 20.6; HRMS (ESI-TOF) calculated for C18H24N12NaO9 [M + Na]: 575.1687, observed: 575.1668.
5,6,3′-Tri-O-acetyl-1,3,2′,6′-tetra-azidoneamine (16). Compound 16 was synthesized from 13 as described for 14 above. 13 (0.28 g, 0.47 mmol) gave 98 mg (75%) of the product (16) as a white foam. 1H-NMR (500 MHz, CDCl3): δ 5.28 (dd, 1H, J = 10.6 and 9.3 Hz), 5.18 (d, 1H, J = 3.8 Hz), 5.15 (t, 1H, J = 9.8 Hz), 4.95 (t, 1H, J = 10.1 Hz), 4.29 (m, 1H), 3.74 (ddd, 1H, J = 14.6, 10.1 and 4.6 Hz), 3.67 (t, 1H, J = 9.8 Hz), 3.67–3.54 (m, 4H), 3.34 (dd, 1H, J = 10.7 and 3.8 Hz), 3.09 (d, 1H, J = 6.8 Hz), 2.47 (ddd, 1H, J = 13.4, 4.6 and 4.6 Hz), 2.19 (s, 3H), 2.10 (s, 6H), 1.64 (ddd, 1H, J = 12.6 Hz, each); 13C (125 MHz, CDCl3): δ 172.0, 169.9, 169.6, 98.9, 78.5, 74.1, 73.6, 73.1, 72.0, 69.9, 60.7, 58.5, 57.6, 50.8, 49.2, 31.9, 21.0, 20.6, 20.5; HRMS (ESI-TOF) calculated for C18H24N12NaO9 [M + Na]: 575.1687, observed: 575.1676.
1,3,2′,6′,2′′′,6′′′ Hexa-azido-neobiosamine-(β-1′′→3′) neamine (20). To a mixture of 14 (70 mg, 0.13 mmol) and 6 (0.13 g, 0.19 mmol) in dry DCM (2.0 mL), trimethylsilyl trifluoromethanesulfonate (14 µL, 0.075 mmol) was added at −20 °C under N2. The resulting mixture was stirred for 2 h, then quenched with Et3N (100 µL) and purified by silica gel chromatography (10% EtOAc/DCM) to obtain peracetylated product (17, contaminated by traces from 6). The residue 17 was then dissolved in dry MeOH (2 mL) and sodium methoxide solution (2 mL of 0.2 mol L−1 solution in MeOH) was added. After 1 h, the reaction solution was neutralized with a strong cation-exchange resin (Dowex H+ 50W), filtered and evaporated to dryness. The residue was purified by silica gel chromatography (10% MeOH/DCM) to obtain 56 mg (57%) of the product 20 as a white foam. 1H-NMR (500 MHz, CD3OH): δ 5.64 (d, 1H, J = 3.7 Hz), 5.13 (s, 1H), 5.21 (d, 1H, J = 1.8 Hz), 4.64 (dd, 1H, J = 7.7 and 4.3 Hz), 4.27 (d, 1H, J = 4.3 Hz), 4.23-4.17 (m, 2H), 4.02 (ddd, 1H J = 8.3, 4.7 and 1.9 Hz), 3.94 (t, 1H, J = 3.4 Hz), 3.85-3.78 (m, 2H), 3.74 (dd, 1H, J = 12.1 and 3.7 Hz), 3.68-3.64 (m, 2H), 3.54-3.34 (m, 10H), 3.26 (t, 1H, J = 9.4 Hz), 2.26 (ddd, 1H, J = 12.9, 4.4 and 4.4 Hz), 1.42 (ddd, 1H, J = 12.3 Hz, each); 13C (125 MHz, CD3OH): δ 110.0, 99.7, 99.3, 83.3, 81.3, 81.2, 78.0, 77.9, 76.3, 75.8, 74.0, 73.1, 71.2, 71.1, 69.7, 64.8, 61.9, 61.9, 61.9, 60.8, 52.8, 52.5, 33.2; HRMS (ESI-TOF) calculated for C23H34N18NaO13 [M + Na]: 793.2450, observed: 793.2427.
1,3,2′,6′,2′′′,6′′′ Hexa-azido-neobiosamine-(β-1′′→6) neamine (21). Compound 21 was synthesized from 15 (94 mg, 0.17 mmol) as described for 20 (from 14) above. 64 mg (49%) of 21 was obtained as a white foam. 1H-NMR (500 MHz, CD3OH): δ 5.56 (d, 1H, J = 3.8 Hz), 5.46 (d, 1H, J = 1.0 Hz), 5.10 (d, 1H, J = 1.8 Hz), 4.46 (dd, 1H, J = 7.3 and 4.5 Hz), 4.24 (dd, 1H, J = 4.5 Hz and 1.0 Hz), 4.20–4.13 (m, 2H), 4.00 (ddd, 1H, J = 8.3, 4.7 and 1.9 Hz), 3.94 (t, 1H, J = 3.4 Hz), 3.87-3.83 (m, 2H), 3.78 (dd, 1H, J = 11.9 and 6.4 Hz), 3.68-3.61 (m, 3H), 3.55–3.61 (m, 7H), 3.40–3.35 (m, 2H), 3.18 (dd, 1H, J = 10.5 and 3.8 Hz), 2.32 (m, 1H), 1.51 (m, 1H); 13C (125 MHz, CD3OH): δ 109.2, 100.2, 99.8, 83.6, 81.8, 81.7, 78.2, 77.6, 75.7, 74.6, 73.4, 72.7, 72.6, 71.3, 69.7, 65.0, 64.4, 61.9, 60.7, 60.3, 52.7, 52.5, 33.2; HRMS (ESI-TOF) calculated for C23H34N18NaO13 [M + Na]: 793.2450, observed: 793.2478.
1,3,2′,6′,2′′′,6′′′ Hexa-azido-neobiosamine-(β-1′′→4′) neamine (22). Compound 22 was synthesized from 16 (70 mg, 0.13 mmol) as described for 20 (from 14) above. 67 mg (69%) of 22 was obtained as a white foam. 1H-NMR (500 MHz, CD3OH): δ 5.63 (d, 1H, J = 3.8 Hz), 5.13 (d, 1H, J = 1.8 Hz), 4.99 (d, 1H, J = 1.1 Hz), 4.57-4.55 (m, 1H), 4.19-4.16 (m, 3H), 4.03 (ddd, 1H, J = 8.5, 4.4 and 1.9 Hz), 3.96-3.91 (m, 2H), 3.82 (dd, 1H, J = 12.1 and 2.5 Hz), 3.72 (dd, 1H, J = 12.1 and 4.1 Hz), 3.69-3.63 (m, 2H), 3.57 (dd, 1H, J = 13.4 and 2.4 Hz), 3.52-3.38 (m, 7H), 3.36 (dd, 1H, J = 13.0 and 4.5 Hz), 3.26 (t, 1H, J = 9.4 Hz), 3.19 (dd, 1H, J = 10.5 and 3.8 Hz), 2.24 (ddd, 1H, J = 12.8, 4.3 and 4.3 Hz), 1.41 (ddd, 1H, J = 12.3 Hz, each); 13C (125 MHz, CD3OH): δ 110.0, 99.7, 99.5, 83.7, 81.3, 81.2, 78.0, 78.0, 76.4, 75.8, 74.6, 71.9, 71.3, 70.9, 69.7, 64.3, 62.1, 61.9, 61.9, 61.0, 52.5, 52.1, 33.3; HRMS (ESI-TOF) calculated for C23H34N18NaO13 [M + Na]: 793.2450, observed: 793.2467.
RP HPLC analysis of 20–23. The purity of the isolated products (20–23) was further confirmed by an RP HPLC analysis. An aliquot of each compound was dissolved in a mixture of acetonitrile and water (1:9, v/v) and injected to an analytical RP (C18, 250 × 4.6 mm, 5 µm) column. Gradient elutions from 10% to 80% acetonitrile in H2O over 30 min. (chromatograms a–c/Figure 1: purified products 20-23) and from 10% to 60% acetonitrile over 35 min. (chromatogram d/Figure 1: a mixture of 20–23, flow rate of 1.0 mL min−1, and detection at λ = 220 nm were used.
Neobiosamine (β-1′′→3′) neamine (23). Trimethylphosphine (1 mol L−1 Me3P in toluene, 0.99 mL, 1.0 mmol) was added to a mixture of 20 (51 mg, 0.066 mmol) in water-dioxane (1:4, v/v, 2.0 mL). The mixture was stirred at ambient temperature for 4 h under nitrogen and then concentrated ammonia (1.0 mL) was added. After overnight reaction the mixture was evaporated to dryness. The crude product was purified by an ion exchange chromatography (Dowex 1 × 2 200 anion exchanger resin, activated by 2 mol L−1 NaOH, eluent carbon dioxide free H2O) to give 30 mg (73%) of the product (23) as a yellowish powder. 1H-NMR (500 MHz, D2O): δ 5.36 (d, 1H, J = 3.9 Hz), 5.24 (d, 1H, J = 0.8 Hz), 4.99 (d, 1H, J = 1.8 Hz), 4.62 (dd, 1H, J = 7.1 and 4.7 Hz), 4.40 (m, 1H), 4.23 (m, 1H), 4.05 (dd, 1H, J = 3.2 Hz, both), 3.97 (m, 1H), 3.90 (dd, 1H, J = 12.5 and 2.8 Hz), 3.86–3.78 (m, 2H), 3.71–3.66 (m, 2H), 3.54 (t, 1H, J = 9.2 Hz), 3.42 (t, 1H, J = 9.5 Hz), 3.32 (t, 1H, J = 9.3 Hz), 3.17 (t, 1H, J = 9.6 Hz), 3.08–3.01 (m, 3H), 2.94–2.84 (m, 4H), 2.75 (m, 1H), 2.00 (ddd, 1H, J = 12.9, 4.1 and 4.1 Hz), 1.23 (ddd, 1H, ddd, J = 12.1 Hz, each); 13C (125 MHz, D2O): δ 108.4, 100.3, 99.1, 86.2, 83.2, 81.5, 77.4, 76.0, 75.6, 75.2, 73.1, 72.2, 70.6, 69.8, 68.5, 60.3, 54.6, 52.6, 50.3, 49.3, 41.5, 41.1, 35.0; HRMS (ESI-TOF) calculated for C23H47N6O13 [M + H]: 615.3196, observed: 615.3221.
Neobiosamine (β-1′′→6) neamine (24). 24 was synthesized as described for 23 above. 61 mg (0.079 mmol) of 21 gave 29 mg (58%) of 24 as a yellowish powder. 1H-NMR (500 MHz, D2O): δ 5.34 (d, 1H, J = 3.9 Hz), 5.25 (s, 1H), 4.98 (d, 1H, J = 1.8 Hz), 4.62 (dd, 1H, J = 7.4 and 4.7 Hz), 4.38 (m, 1H), 4.20 (m, 1H), 4.04 (t, 1H, J = 3.2 Hz), 3.99 (m, 1H), 3.90 (dd, 1H, J = 12.6 and 2.6 Hz), 3.82–3.74 (m, 2H), 3.68–3.57 (m, 3H), 3.35–3.29 (m, 3H), 3.10–3.03 (m, 3H), 2.94 (dd, 1H, J = 13.5 and 4.1 Hz), 2.88–2.75 (m, 4H), 2.03 (ddd, 1H, J = 12.9, 4.1 and 4.1 Hz), 1.26 (ddd, 1H, J = 12.4 Hz, each); 13C (125 MHz, D2O): δ 108.8, 100.4, 98.9, 85.9, 85.8, 81.3, 75.9, 75.4, 75.0, 73.4, 73.0, 72.4, 71.4, 70.5, 68.5, 60.2, 55.2, 52.6, 49.1, 48.9, 41.4, 41.0, 36.1; HRMS (ESI-TOF) calculated for C23H47N6O13 [M + H]: 615.3196, observed: 615.3192.
Neobiosamine (β-1′′→4′) neamine (25). 25 was synthesized as described for 23 above. 51 mg (0.066 mmol) of 22 gave 32 mg (83%) of 25 as a yellowish powder. 1H-NMR (500 MHz, D2O): δ 5.31 (d, 1H, J = 3.9 Hz), 5.09 (s, 1H), 4.97 (d, 1H, J = 1.6 Hz), 4.60 (dd, 1H, J = 7.0 and 4.6 Hz), 4.31 (m, 1H), 4.23 (m, 1H), 4.04 (m, 1H, 3.96-3.88 (m, 2H), 3.85–3.79 (m, 2H), 3.68–3.64 (m, 2H), 3.52 (dd, 1H, J = 9.2 Hz, both), 3.42 (t, 1H, J = 9.4 Hz), 3.30 (t, 1H, J = 9.2 Hz), 3.16 (t, 1H, J = 9.6 Hz), 3.04 -2.98 (m, 3H), 2.90–2.81 (m, 4H), 2.72 (m, 1H), 1.99 (m, 1H), 1.22 (ddd, 1H, J = 12.4 Hz, each); 13C (125 MHz, D2O): δ 108.1, 100.3, 99.2, 86.9, 81.5, 80.5, 77.4, 75.9, 75.7, 75.5, 73.0, 72.1, 71.9, 70.6, 68.5, 60.2, 54.9, 52.7, 50.3, 49.3, 41.2, 41.1, 35.5; HRMS (ESI-TOF) calculated for C23H47N6O13 [M + H]: 615.3196, observed: 615.3202.
UV measurements. Melting curves (absorbance vs. temperature) were measured at 260 nm on a Perkin-Elmer Lambda 35 UV/Vis spectrophotometer equipped with a multiple cell holder and a Peltier temperature controller. An internal thermometer was additionally used to verify the validity of the target temperature. The temperature was changed from 10 to 80 °C at a rate of 0.2 °C min. Tm values were determined as the maximum of the first derivate of the melting curve.
4. Conclusions
A straightforward synthesis of glycosidic (1′′→6, 3′ and 4′) site isomers (23–25) of neomycin B has been described. The synthesis includes (1) acid-catalyzed thiolysis of neomycin azide (3) to obtain useful intermediates (4 and 5) for both the glycosyl donor (6) and the acceptors (14–16), and (2) selective diacetylation of neamine azide (7), which after simple protecting group manipulation (i.e., levulinoylation, acetylation and delevulinoylation) afforded the glycosyl acceptors (14–16). Glycosylation between the neobiosamine donor (6) and triacetylated neamine azide acceptors (14–16) was carried out in standard condition to give the azide masked pseudo tetrasaccharides (21–22) in acceptable yields. The azide masks were removed by Staudinger reaction to obtain the desired glycosidic site isomers of neomycin B (23–25). The potential of these aminoglycosides (23–25) to stabilize DNA and RNA triple helix models was studied by UV-melting profile analysis. In each case a marked stabilization was observed. The variable neobiosamine site (6-O, 3′-O or 4′-O) on the neamine core did not show marked difference in the stability compared to neomycin B. It may be concluded that the carbohydrate core and spatial orientation of the amino groups (i.e., spatial orientation of l and d-neosamines provided by the variable glycoside site between neobioasamine and neamine) on it do not play a marked enough role to result in discrimination between the affinities of the site isomers.
Supplementary Materials
The NMR spectral data for all new compounds are available online.
Author Contributions
L.G. performed the part of the aminoglycoside synthesis and UV-melting profile analysis, V.T. performed part of the UV-melting profile analysis, P.V. (the corresponding author) supervised the project and performed part of the aminoglycoside synthesis.
Funding
The financial support from the Academy of Finland (No. 251539, 256214 and 308931) is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Thomas, J.R.; Hergenrother, P.J. Targeting of RNA with Small Molecules. Chem. Rev. 2008, 108, 1171–1224. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Gorini, L.; Davis, B.D. Misreading of RNA codewords induced by aminoglycoside antibiotics. Mol. Pharmacol. 1965, 1, 93–106. [Google Scholar] [PubMed]
- Moazed, D.; Noller, H.F. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 1987, 327, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Von Ahsen, U.; Noller, H.F. Footprinting the sites of interaction of antibiotics with catalytic group I intron RNA. Science 1993, 260, 1500–1503. [Google Scholar] [CrossRef] [PubMed]
- Francois, B.; Russell, R.J.M.; Murray, J.B.; Aboul-ela, F.; Masquida, B.; Vicens, Q.; Westhof, E. Crystal structures of complexes between aminoglycosides and decoding site oligonucleotides: Role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acid Res. 2005, 33, 5677–5690. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Chen, W.; Juskeviciene, R.; Teo, Y.; Shcherbakov, D.; Vasella, A.; Böttger, E.C.; Crich, D. Influence of 4′-O-glycoside constitution and configuration on ribosomal selectivity of paromomycin. J. Am. Chem. Soc. 2015, 137, 7706–7717. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, W.A.; Priestley, S.; Sears, P.S.; Alper, P.B.; Rosenbohm, A.; Hendrix, M.; Hung, S.-C.; Wong, C.-H. Design and synthesis of new aminoglycoside antibiotics containing neamine as an optimal core structure: Correlation of antibiotic activity with in vitro inhibition of translation. J. Am. Chem. Soc. 1999, 121, 6527–6541. [Google Scholar] [CrossRef]
- Von Ahsen, U.; Davies, J.; Schroeder, R. Antibiotic inhibition of group I ribozyme function. Nature 1991, 353, 368–370. [Google Scholar] [CrossRef]
- Herman, T. Drugs targeting the ribosome. Curr. Opin. Struct. Biol. 2005, 15, 355–366. [Google Scholar] [CrossRef]
- Mei, H.-Y.; Galan, A.A.; Halim, N.S.; Mack, D.P.; Morelan, D.W.; Sanders, K.B.; Truong, H.N.; Czarnik, A.W. Inhibition of an HIV-1 Tat-derived peptide binding to TAR RNA by aminoglycoside antibiotics. Bioorg. Med. Chem. Lett. 1995, 5, 2755–2760. [Google Scholar] [CrossRef]
- Zapp, M.L.; Stern, S.; Green, M.R. Small molecules that selectively block RNA bindingo f HIV-1 rev protein inhibit rev function and viral production. Cell 1993, 74, 969–978. [Google Scholar] [CrossRef]
- Tam, V.K.; Kwong, D.; Tor, Y. Fluorescent HIV-1 dimerization initiation site: Design, properties, and use for ligand discovery. J. Am. Chem. Soc. 2007, 129, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Ennifar, E.; Paillart, J.-C.; Bodlenner, A.; Walter, P.; Weibel, J.-M.; Aubertin, A.-M.; Pale, P.; Dumas, P.; Marquet, R. Targeting the dimerization initiation site of HIV-1 RNA with aminoglycosides: From crystal to cell. Nucleic Acid Res. 2006, 34, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Arya, D.P.; Coffee, R.L., Jr. DNA triple helix stabilization by aminoglycoside antibiotics. Bioorg. Med. Chem. Lett. 2000, 10, 1897–1899. [Google Scholar] [CrossRef]
- Arya, D.P.; Micovic, L.; Charles, I.; Lane Coffee, R., Jr.; Willis, B.; Xue, L. Neomycin Binding to Watson-Hoogsteen (W-H) DNA triplex groove: A model. J. Am. Chem. Soc. 2003, 125, 3733–3744. [Google Scholar] [CrossRef] [PubMed]
- Arya, D.P.; Xue, L.; Tennant, P. Combining the best in triplex recognition: Synthesis and nucleic acid binding of a BQQ-neomycin conjugate. J. Am. Chem. Soc. 2003, 125, 8070–8071. [Google Scholar] [CrossRef] [PubMed]
- Arya, D.P.; Coffee, R.L., Jr.; Charles, I. Neomycin induced hybrid triplex formation. J. Am. Chem. Soc. 2001, 123, 11093–11094. [Google Scholar] [CrossRef]
- Arya, D.P. New approaches towards recognition of nucleic acid triple helices. Acc. Chem. Res. 2011, 44, 134–146. [Google Scholar] [CrossRef]
- Wu, B.; Yang, J.; He, Y.; Swayze, E.E. Reexamination of neomycin B degradation: Efficient preparation of its CD and D rings as protected glycosyl donors. Org. Lett. 2002, 4, 3455–3458. [Google Scholar] [CrossRef]
- Van den Broek, S.A.M.W.; Gruijters, B.W.T.; Rutjes, F.P.J.T.; van Delft, F.L.; Blaauw, R.H. A short and scalable route to orthogonally O-protected 2-deoxystreaptamine. J. Org. Chem. 2007, 72, 3577–3580. [Google Scholar] [CrossRef]
- Alper, P.B.; Hung, S.-C.; Wong, C.-H. Metal catalyzed diazo transfer. Tetrahedron Lett. 1996, 37, 6029–6032. [Google Scholar] [CrossRef]
- Geny, S.; Moreno, P.M.D.; Krzywkowski, T.K.; Gissberg, O.; Andersen, N.K.; Isse, A.J.; El-Madani, A.M.; Lou, C.; Pabon, Y.V.; et al. Next-generation bis-locked nucleic acids with stacking linker and 2′-glycylamino-LNA show enhanced DNA invasion into supercoiled duplexes. Nucleic Acids Res. 2016, 44, 2007–2019. [Google Scholar] [CrossRef]
- Tähtinen, V.; Granqvist, L.; Virta, P. Synthesis of C-5, C-2′ and C-4′-neomycin-conjugated triplex forming oligonucleotides and their affinity to DNA-duplexes. Bioorg. Med. Chem. 2015, 23, 4472–4480. [Google Scholar] [CrossRef] [PubMed]
- Granqvist, L.; Virta, P. 4′-C-[(4-trifluoromethyl-1H-1,2,3-triazol-1-yl)methyl]thymidine as a sensitive 19F NMR sensor for the detection of oligonucleotide secondary structures. J. Org. Chem. 2014, 79, 3529–3536. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Sugiura, M.; Nishimoto, S. Monitoring of duplex and triplex formation by 19F NMR using oligodeoxynucleotides possessing 5-fluorodeoxyuridine unit as 19F signal transmitter. Bioorg. Med. Chem. 2010, 18, 6690–6694. [Google Scholar] [CrossRef] [PubMed]
- Granqvist, L.; Kraszewski, A.; Tähtinen, V.; Virta, P. Synthesis of aminoglycoside-2′-O-methyl oligoribonucleotide fusions. Molecules 2017, 22, 760. [Google Scholar] [CrossRef] [PubMed]
- Bhuma, N.; Tähtinen, V.; Virta, P. Synthesis and Applicability of Base-Discriminating DNA-Triplex-Forming 19F NMR Probes. Eur. J. Org. Chem. 2018, 605–613. [Google Scholar] [CrossRef]
- Granqvist, L.; Virta, P. 2′-O-[(4-CF3-triazol-1-yl)methyl] uridine—A sensitive 19F NMR sensor for the detection of RNA secondary structures. J. Org. Chem. 2015, 80, 7961–7970. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 23–25 are available from the authors. |
Scheme 1.
Reagents and conditions: i) CuSO4, TfN3, dichloromethane, H2O, iia-c) Ac2O (a: 10 equiv., b: 2.5 equiv., c: 5 equiv.), DMAP, pyridine, overnight at r.t. iii) PhSH, BF3 · Et2O, DCM, iv) N-bromosuccinimide, trifluoroacetic acid, H2O, MeCN, dichloromethane, for 1h at 0 °C–r.t., v) CCl3CN, 1.8-diazabicyclo(5.4.0)undec-7-ene, DCM, for 1h at 0 °C, vi) 0.1 mol L−1 NaOMe/MeOH, for 1h at r.t., vii) Lev2O, 4-(N,N-dimethylamino)pyridine, pyridine, overnight at r.t., viii) NH2NH2·AcOH, MeOH, dichloromethane, for 3 h at r.t.
Scheme 1.
Reagents and conditions: i) CuSO4, TfN3, dichloromethane, H2O, iia-c) Ac2O (a: 10 equiv., b: 2.5 equiv., c: 5 equiv.), DMAP, pyridine, overnight at r.t. iii) PhSH, BF3 · Et2O, DCM, iv) N-bromosuccinimide, trifluoroacetic acid, H2O, MeCN, dichloromethane, for 1h at 0 °C–r.t., v) CCl3CN, 1.8-diazabicyclo(5.4.0)undec-7-ene, DCM, for 1h at 0 °C, vi) 0.1 mol L−1 NaOMe/MeOH, for 1h at r.t., vii) Lev2O, 4-(N,N-dimethylamino)pyridine, pyridine, overnight at r.t., viii) NH2NH2·AcOH, MeOH, dichloromethane, for 3 h at r.t.

Figure 1.
RP HPLC profiles of the azide-masked neobiosamine neamines (20–22). Conditions: An analytical RP HPLC (C18, 250 × 4.6 mm, 5 µm) column, detection at λ = 220 nm, gradient elutions from 10% to 80% (a–c) and from 10% to 60% (d) acetonitrile in H2O over 30 min (a–c) and 35 min (d).
Figure 1.
RP HPLC profiles of the azide-masked neobiosamine neamines (20–22). Conditions: An analytical RP HPLC (C18, 250 × 4.6 mm, 5 µm) column, detection at λ = 220 nm, gradient elutions from 10% to 80% (a–c) and from 10% to 60% (d) acetonitrile in H2O over 30 min (a–c) and 35 min (d).
Scheme 2.
Reagents and conditions: i) TMSOTf, DCM, 2 h at −20 °C; ii) 0.1 mol L−1 NaOMe/MeOH, 1 h at r.t.; iii) 1: Me3P, toluene, water, dioxane, 4 h at r.t. under N2, 2: conc. ammonia, overnight at r.t.
Scheme 2.
Reagents and conditions: i) TMSOTf, DCM, 2 h at −20 °C; ii) 0.1 mol L−1 NaOMe/MeOH, 1 h at r.t.; iii) 1: Me3P, toluene, water, dioxane, 4 h at r.t. under N2, 2: conc. ammonia, overnight at r.t.

Figure 2.
Representative melting profiles of the DNA and RNA triplex models in the presence of aminoglycosides (AGs) (2 μmol oligonucleotides in the presence of 5 eq. 1, 23–25, 10 mmol L−1 sodium cacodylate, 0.1 mol L–1 NaCl, pH 6.0.
Figure 2.
Representative melting profiles of the DNA and RNA triplex models in the presence of aminoglycosides (AGs) (2 μmol oligonucleotides in the presence of 5 eq. 1, 23–25, 10 mmol L−1 sodium cacodylate, 0.1 mol L–1 NaCl, pH 6.0.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
UV-melting experiments (Tm/°C).
 | ||
|---|---|---|
| RNA Triplex Model | ||
| AG | Tm3(°C) pH 6.0 | Tm2(°C) pH 6.0 |
| none | 41.9 | 60.1 |
| 1 (5 eq) | 51.2 (+9.3) | 68.0 (+7.9) |
| 23 (5 eq) | 53.2 (+11.3) | 66.7 (+6.6) |
| 24 (5 eq) | 52.5 (+10.6) | 67.7 (+7.6) |
| 25 (5 eq) | 51.4 (+9.5) | 68.0 (+7.9) |
| AG | Tm3(°C) pH 7.0 | Tm2(°C) pH 7.0 |
| none | 22.6 | 60.1 |
| 1 (5 eq) | 29.4 (+6.9) | 62.0 (+1.9) |
| 23 (5 eq) | 30.4 (+7.8) | 61.2 (+1.1) |
| 24 (5 eq) | 30.4 (+7.8) | 61.6 (+1.5) |
| 25 (5 eq) | 29.4 (+6.9) | 61.9 (+1.8) |
| DNA Triplex Model | ||
| AG | Tm3(°C) pH 6.0 | Tm2(°C) pH 6.0 |
| none | 25.5 | 54.6 |
| 1 (5 eq) | 40.1 (+14.6) | 56.3 (+1.7) |
| 23 (5 eq) | 38.1 (+12.6) | 55.5 (+0.9) |
| 24 (5 eq) | 36.6 (+11.1) | 56.3 (+1.7) |
| 25 (5 eq) | 38.6 (+13.1) | 55.3 (+0.7) |
| 1 (10 eq) | 44.1 (+18.6) | 56.3 (+1.7) |
| 23 (10 eq) | 41.3 (+15.8) | 55.9 (+1.3) |
| 24 (10 eq) | 39.4 (+13.9) | 56.9 (+2.3) |
| 25 (10 eq) | 42.9 (+17.4) | 56.5 (+1.9) |
ΔTm-values in parentheses. Error limits for each Tm-value (an average of three temperature ramps) were less than 1 °C. Tm3 and Tm2 correspond to melting values of the triplex and duplex, respectively.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Granqvist, L.; Tähtinen, V.; Virta, P. Synthesis of Glycosidic (β-1′′→6, 3′ and 4′) Site Isomers of Neomycin B and Their Effect on RNA and DNA Triplex Stability. Molecules 2019, 24, 580. https://doi.org/10.3390/molecules24030580
AMA Style
Granqvist L, Tähtinen V, Virta P. Synthesis of Glycosidic (β-1′′→6, 3′ and 4′) Site Isomers of Neomycin B and Their Effect on RNA and DNA Triplex Stability. Molecules. 2019; 24(3):580. https://doi.org/10.3390/molecules24030580
Chicago/Turabian StyleGranqvist, Lotta, Ville Tähtinen, and Pasi Virta. 2019. "Synthesis of Glycosidic (β-1′′→6, 3′ and 4′) Site Isomers of Neomycin B and Their Effect on RNA and DNA Triplex Stability" Molecules 24, no. 3: 580. https://doi.org/10.3390/molecules24030580