4,5-Diaryl 3(2H)Furanones: Anti-Inflammatory Activity and Influence on Cancer Growth

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
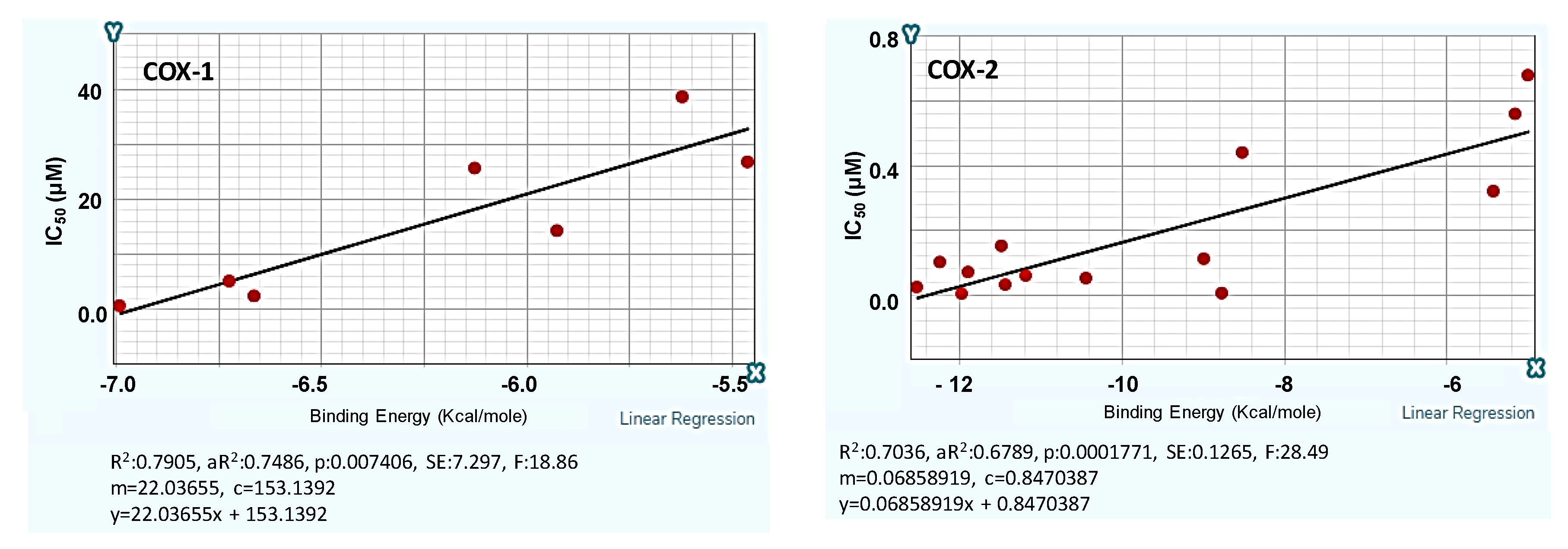
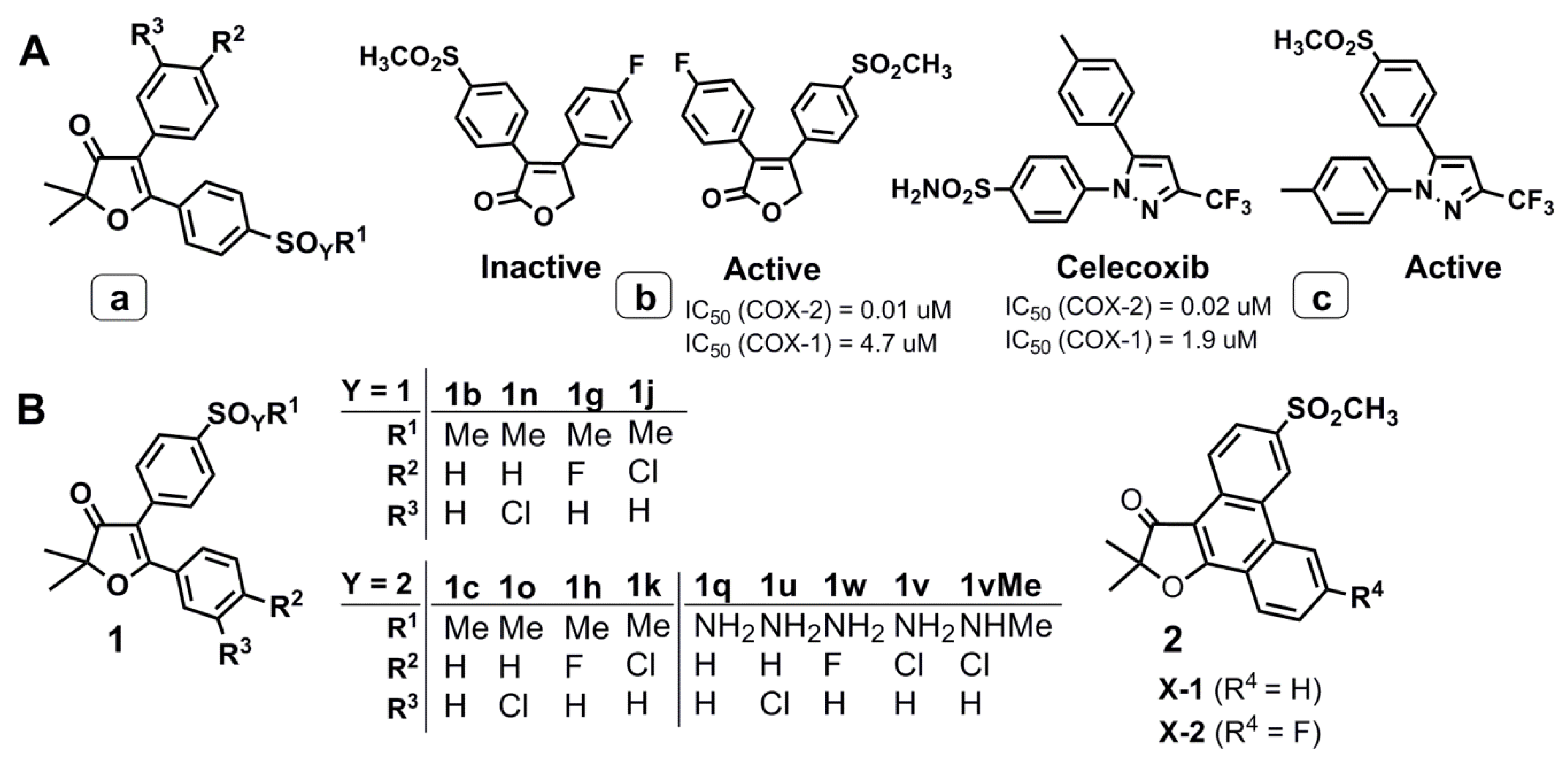
2.1. Design and Computer-Aided Prediction of COX-1/2 Inhibitory Activity
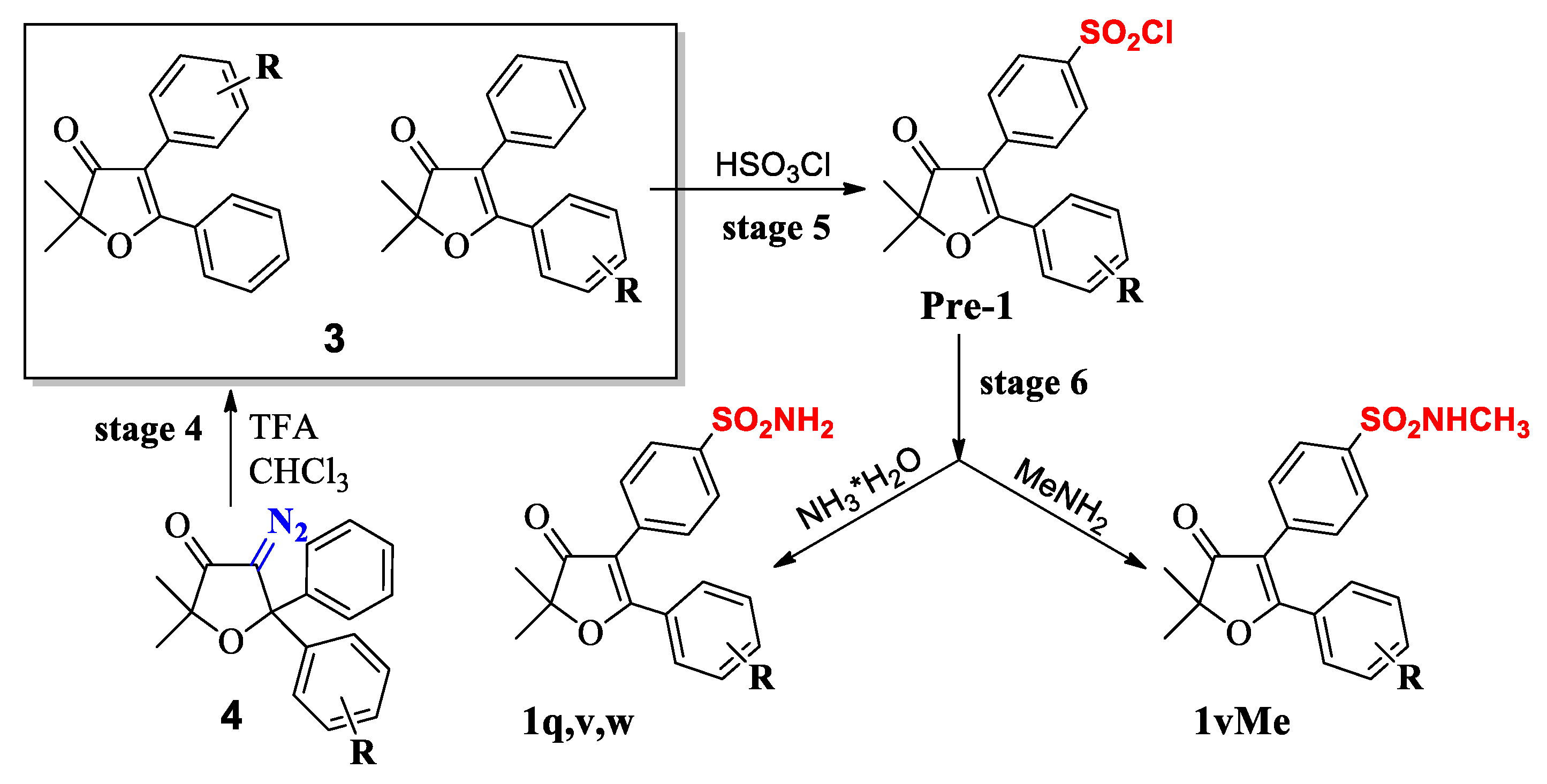
2.2. Chemistry
Synthesis
2.3. Biological Evaluation
2.3.1. In Vitro Evaluation of COX-1/2 Inhibitory Action
2.3.2. In Vivo Anti-Inflammatory Activity
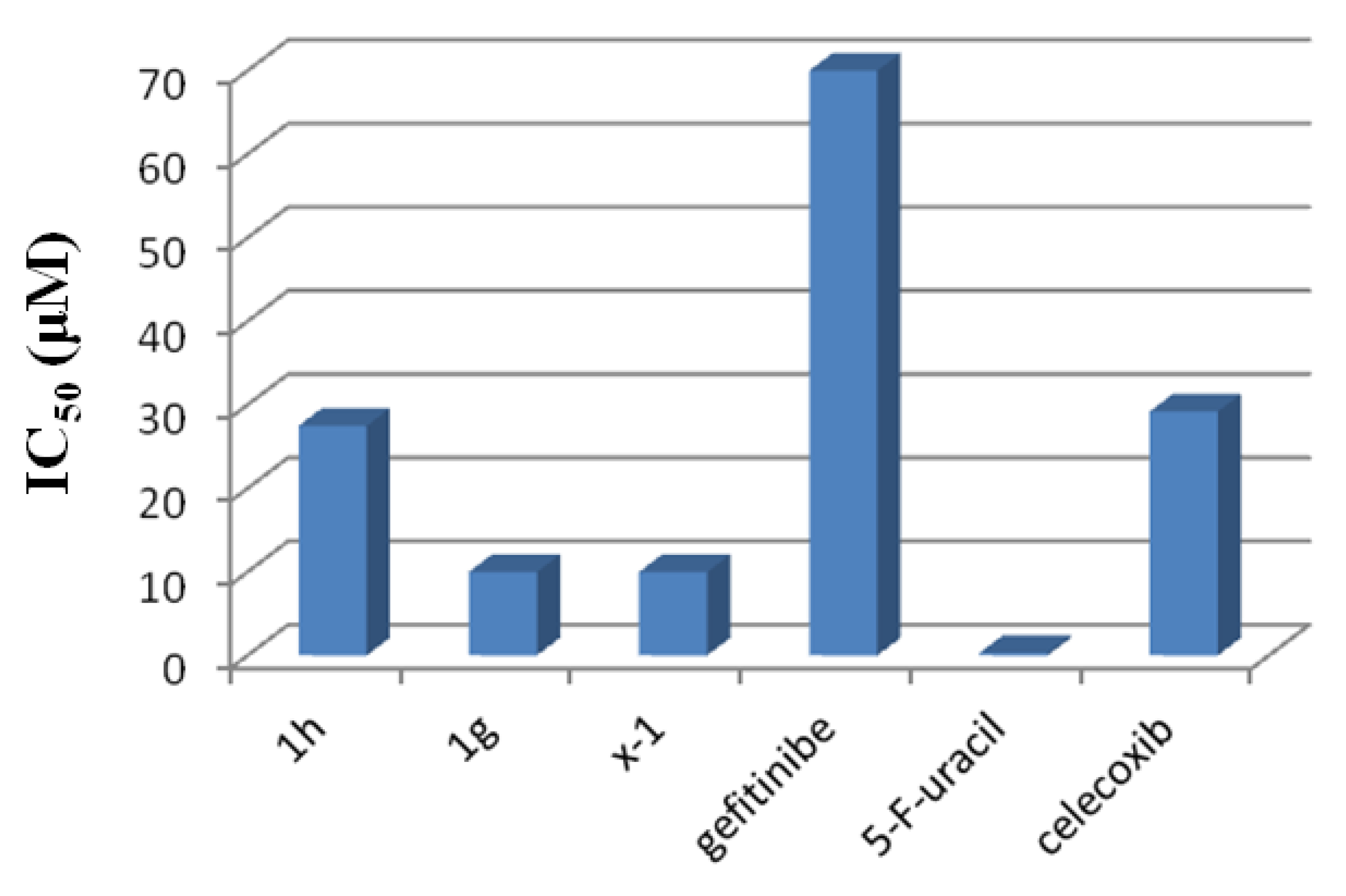
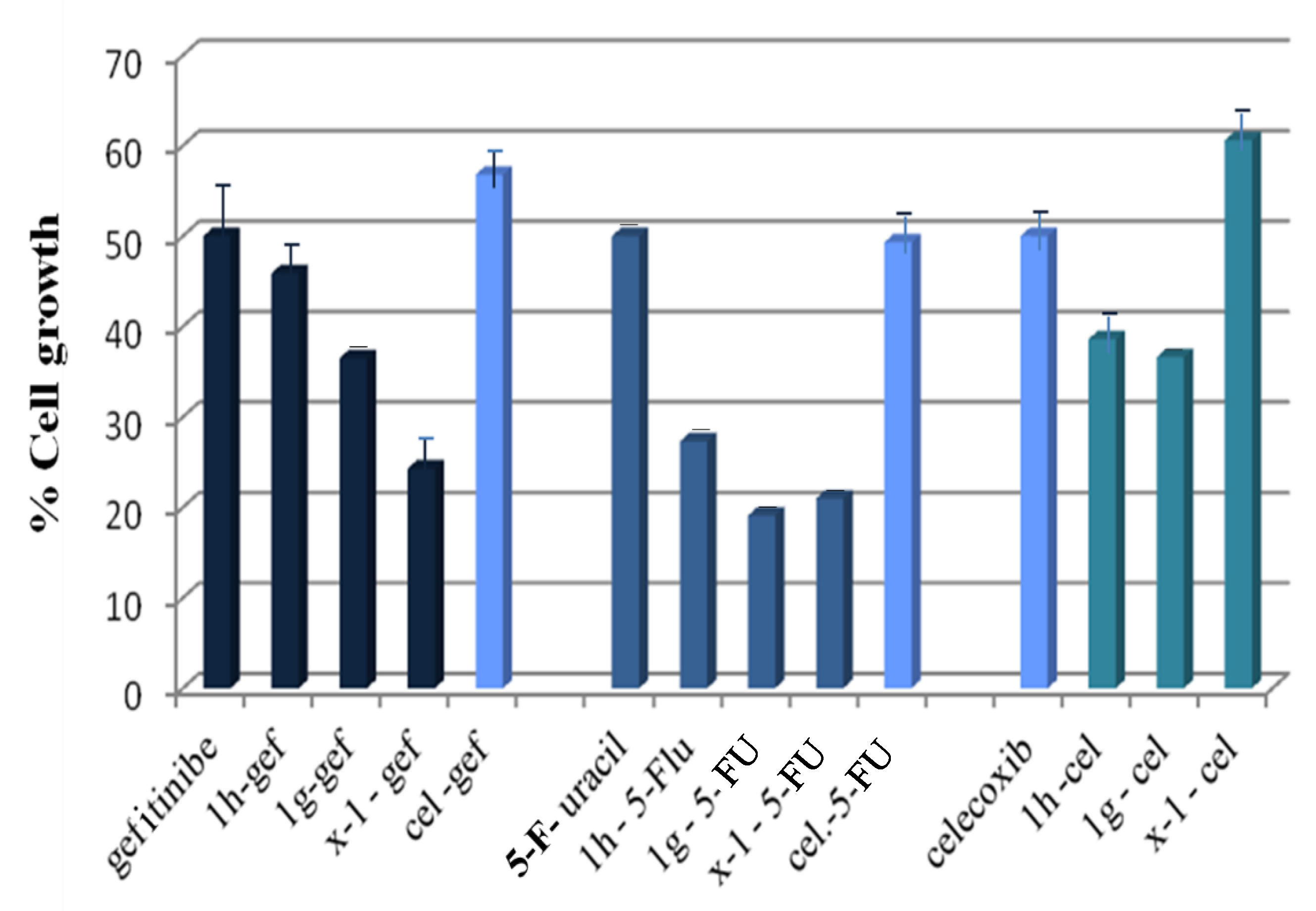
2.3.3. Anticancer Activity against MCF-7 Breast Cancer Cells
3. Materials and Methods
3.1. Computer Simulation Methods
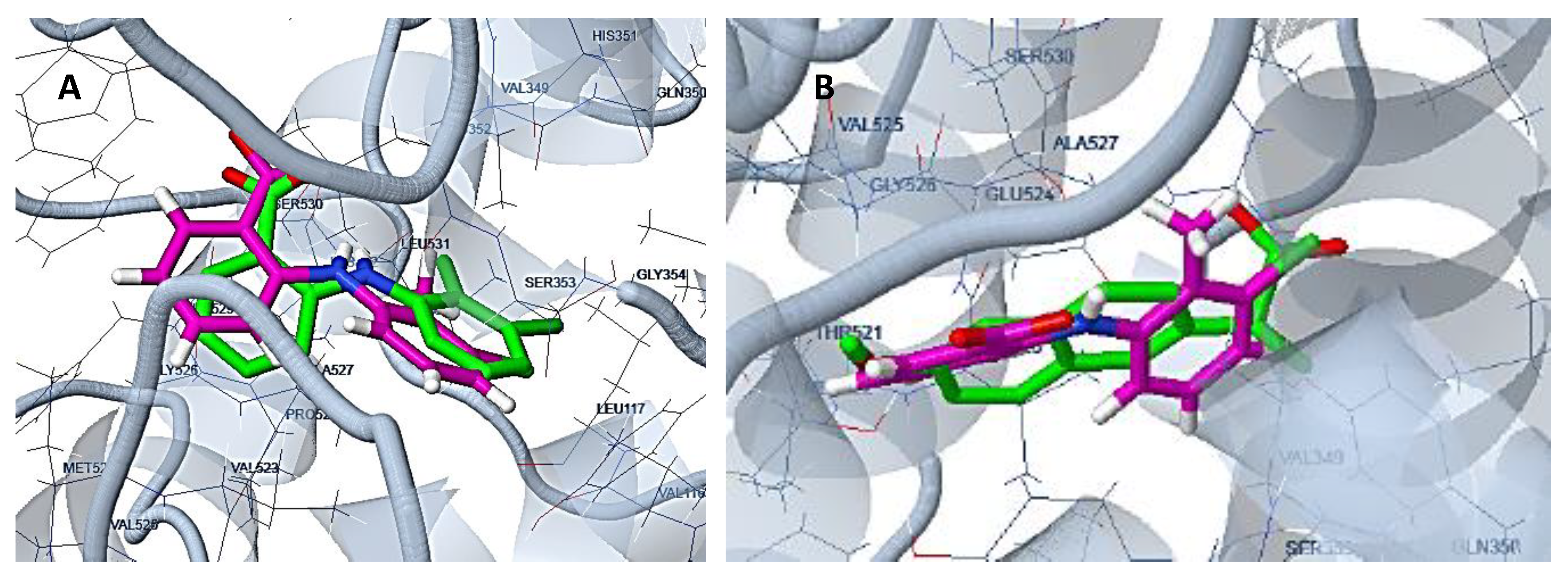
3.1.1. Preparation of Protein Structures and Docking Analysis Based on Ovis aries COX-1, 3KK6 and Mus musculus COX-2, 3LN1
3.1.2. Docking Analysis Based on Ovine COX-1 Structure 2AYL and Human COX-2 Structure 5IKT
3.2. Chemistry
3.2.1. General Method for Preparation of Compounds 1b, 1g, 1j, and 1n
3.2.2. General Method for Preparation of Compounds 1c,1h, 1k, and 1o
3.2.3. General Method for Preparation of Compounds 1q, 1v, and 1w
3.2.4. 5-(4′-Chlorophenyl)-2,2-dimethyl-4-[4′-(methylaminosulfonyl)phenyl]-furan-3(2H)-one (1vMe)
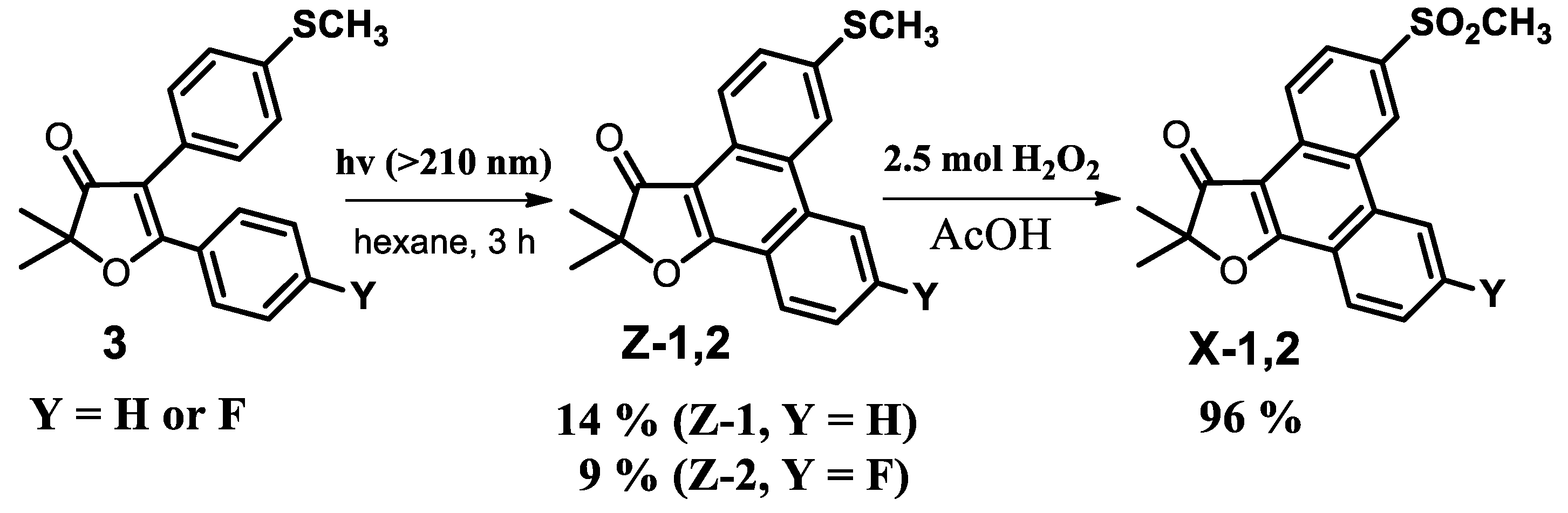
3.2.5. General Method for the Synthesis of Phenanthro[9,10-b]furan-3′(2′H)-ones
2′,2′-dimethyl-3-(methylsulfonyl)-phenanthro[9,10-b]furan-3′(2′H)-one (X-1)
Synthesis of Intermediate 2′,2′-dimethyl-6-(methylthio)-phenanthro[9,10-b]furan-3′(2′H)-one
3-Fluoro-2,2-dimethyl-6-(methylsulfonyl)phenanthro[9,10-b]furan-3(2′H)-one (X-2)
3-Fluoro-2′,2′-dimethyl-6-(methylsulfonyl)-phenanthro[9,10-b]furan-3′(2′H)-one (X-2)
3.3. In Vitro Evaluation of COX Inhibitory Action
3.4. Inhibition of the Carrageenin-Induced Edema
3.5. Cell Cultures
3.6. Growth Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prasit, P.P.; Wang, Z.; Brideau, C.; Chan, C.C.; Charleson, S.E.; Cromlish, W.A.; Ethier, D.; Evans, J.F.; Ford-hutchinson, A.; Gauthier, J.Y.; et al. The discovery of rofecoxib, [MK 966, VIOXX®, 4-(4′-methylsulfonylphenyl)-3-phenyl-2(5H)-furanone—An orally active cyclooxygenase-2 inhibitor]. Bioorg. Med. Chem. Lett. 1999, 9, 1773–1778. [Google Scholar] [CrossRef]
- Penning, T.D.; Talley, J.J.; Bertenshaw, S.R.; Carter, J.S.; Collins, P.W.; Docter, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; et al. Synthesis and Biological Evaluation of the 1.5 Diarylpyrazole Class of Cyclooxygenase-2 Inhibitors: Identification of (4-Methylphenyl)-3-(trifluoromethyl)-1H-pyrazole-1-yl]benzenesulfonamide (SC-58634, Celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef]
- Bresalier, R.S.; Sandler, R.S.; Quan, H.; Bolognese, J.A.; Oxenius, B.; Horgan, K. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N. Engl. J. Med. 2005, 352, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Punganuru, S.R.; Madala, H.R.; Mikelis, C.M.; Dixit, A.; Arutla, V.; Srivenugopal, K.S. Conception, synthesis, and characterization of a rofecoxib-combretastatin hybrid drug with potent cyclooxygenase-2 (COX-2) inhibiting and microtubule disrupting activities in colon cancer cell culture and xenograft models. Oncotarget. 2018, 9, 26109–26129. [Google Scholar] [CrossRef]
- Vizirianakis, I.S.; Chatzopoulou, M.; Bonovolias, I.D.; Nicolaou, I.; Demopoulos, V.J.; Tsiftsoglou, A.S. Toward the development of innovative bifunctional agents to induce differentiation and to promote apoptosis in leukemia: Clinical candidates and perspectives. J. Med. Chem. 2010, 53, 6779–6810. [Google Scholar] [CrossRef] [PubMed]
- Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Ribeiro, M.; Ben-Jacob, E.; Devi, G.R.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bellamkonda, K.; Chandrashekar, N.K.; Osman, J.; Selvanesan, B.C.; Sayeh Savari, S.; Sjölander, A. The eicosanoids leukotriene D4 and prostaglandin E2 promote the tumorigenicity of colon cancer-initiating cells in a xenograft mouse model. BMC Cancer. 2016, 16, 425–439. [Google Scholar] [CrossRef]
- Yao, L.; Liu, F.; Hong, L.; Sun, L.; Liang, S.; Wu, K.; Fan, D. The function and mechanism of COX-2 in angiogenesis of gastric cancer cells. J. Exp. Clin. Cancer Res. 2011, 30, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014, 15, 493–503. [Google Scholar] [CrossRef]
- Karan, D. Inflammasomes: Emerging Central Players in Cancer Immunology and Immunotherapy. Front. Immunol. 2018, 9, 3028. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Al-Raawi, D.; Sabet, S.F.; El-Shinawi, M. Inflammatory breast cancer: New factors contribute to disease etiology: A review. J. Adv. Res. 2014, 5, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Bottazzi, B.; Riboli, E.; Mantovani, A. Aging, inflammation and cancer. Semin. Immunol. 2018, 40, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H. Epithelial-mesenchymal transition: Initiation by cues from chronic inflammatory tumor microenvironment and termination by anti-inflammatory compounds and specialized pro-resolving lipids. Biochem. Pharmacol. 2018, 158, 261–273. [Google Scholar] [CrossRef]
- Fujii, R.; Imanishi, Y.; Shibata, K.; Sakai, N.; Sakamoto, K.; Shigetomi, S.; Habu, N.; Otsuka, K.; Sato, Y.; Watanabe, Y.; et al. Restoration of E-cadherin expression by selective Cox-2 inhibition and the clinical relevance of epithelial-to-mesenchymal transition in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2014, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.-Y.; Wang, W.-L.; Li, C.-F.; Jeng, Y.-M.; Chu, Y.-Y.; Wang, H.-Y.; Tseng, J.T.; Wang, J.-M. IL-18-induced interaction between IMP3 and HuR contributes to COX-2 mRNA stabilization in acute myeloid leukemia. J. Leukoc. Biol. 2016, 99, 131–141. [Google Scholar]
- Pannunzio, A.; Coluccia, M. Cyclooxygenase-1 (COX-1) and COX-1 Inhibitors in Cancer: A Review of Oncology and Medicinal Chemistry Literature. Pharmaceuticals 2018, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Mukae, S.; Murakami, M.; Ishikawa, Y.; Ishii, T.; Ohki, H.; Matsumoto, M.; Komiyama, K. Butyrate Inhibits Oral Cancer Cell Proliferation and Regulates Expression of Secretory Phospholipase A2-X and COX-2. Anticancer Res. 2007, 27, 1493–1502. [Google Scholar] [PubMed]
- Ways, D.K.; Kukoly, C.A.; de Vente, J.; Hooker, J.L.; Bryant, W.O.; Posekany, K.J.; Fletcher, D.J.; Cook, P.P.; Parker, P.J. MCF-7 Breast Cancer Cells Transfected with Protein Kinase C—A Exhibit Altered Expression of Other Protein Kinase C Isoforms and Display a More Aggressive Neoplastic Phenotype. J. Clin. Invest. 1995, 95, 1906–1915. [Google Scholar] [CrossRef]
- Rigas, B.; Goldman, I.; Levine, L. Altered eicosanoid levels in human colon cancer. J. Lab. Clin. Med. 1993, 122, 518–523. [Google Scholar]
- Leahy, K.; Koki, A.; Masferrer, J. Role of cyclooxygenases in angiogenesis. Curr. Med. Chem. 2000, 7, 1163–1170. [Google Scholar] [CrossRef]
- Brunelle, M.; Sartin, E.A.; Wolfe, L.G.; Sirois, J.; Doré, M. Cyclooxygenase-2 expression in normal and neoplastic canine mammary cell lines. Vet Pathol. 2006, 43, 656–666. [Google Scholar] [CrossRef]
- Yusup, G.; Akutsu, Y.; Mutallip, M.; Qin, W.; Hu, X.; Komatsu-Akimoto, A.; Hoshino, I.; Hanari, N.; Mori, M.; Akanuma, N.; et al. A COX-2 inhibitor enhances the antitumor effects of chemotherapy and radiotherapy for esophageal squamous cell carcinoma. Int. J. Oncol. 2014, 44, 1146–1152. [Google Scholar] [CrossRef]
- Samadder, N.J.; Neklason, D.W. Effect of Sulindac and Erlotinib vs Placebo on Duodenal Neoplasia in Familial Adenomatous Polyposis: A Randomized Clinical Trial. JAMA 2016, 315, 1266–1275. [Google Scholar] [CrossRef]
- Shaashua, L.; Shabat-Simon, M.; Haldar, R.; Matzner, P.; Zmora, O.; Shabtai, M.; Sharon, E.; Allweis, T.; Barshack, I.; Hayman, L.; et al. Perioperative COX-2 and β-Adrenergic Blockade Improves Metastatic Biomarkers in Breast Cancer Patients in a Phase-II Randomized Trial. Clin. Cancer Res. 2017, 23, 4651–4661. [Google Scholar] [CrossRef]
- Botti, G.; Fratangelo, F.; Cerrone, M.; Liguori, G.; Cantile, M.; Anniciello, A.M.; Scala, S.; D’Alterio, C.; Trimarco, C.; Ianaro, A.; et al. COX-2 expression positively correlates with PD-L1 expression in human melanoma cells. J. Transl. Med. 2017, 15, 46–58. [Google Scholar] [CrossRef]
- Shin, S.S.; Noh, M.-S.; Byun, Y.J.; Choi, J.K.; Kim, J.Y.; Lim, K.M.; Ha, J.-Y.; Kim, J.K.; Lee, C.H.; Chung, S. 2,2-dimethyl-4,5-diaryl-3(2H)furanone derivatives as selective cyclo-oxygenase-2 inhibitors. Bioorg. Med. Chem. Lett. 2001, 11, 165–168. [Google Scholar] [CrossRef]
- Shin, S.S.; Byun, Y.; Lim, K.M.; Choi, J.K.; Lee, K.W.; Moh, J.H.; Kim, J.K.; Jeong, Y.S.; Kim, J.Y.; Choi, Y.H.; et al. In vitro structure-activity relationship and in vivo studies for a novel class of cyclooxygenase-2 inhibitors: 5-aryl-2,2-dialkyl-4-phenyl-3 (2H)furanone derivatives. J. Med. Chem. 2004, 47, 792–804. [Google Scholar] [CrossRef]
- Praveen, P.N.; Knaus, E.E. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): Cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharmaceut. Sci. 2008, 11, 81–110. [Google Scholar]
- Prasit, P.; Riendeau, D. Selective cyclooxygenase-2 inhibitors. Ann. Rep. Med. Chem. 1997, 32, 211–220. [Google Scholar]
- Leval, X.D.; Julemont, F.; Delarge, J.; Sanna, V.; Pirotte, B.; Dogne, J.M. Advances in the field of COX-2 inhibition. Expert. Opin. Ther. Patents 2002, 12, 969–989. [Google Scholar]
- Chakraborti, A.K.; Garg, S.K.; Kumar, R.; Motiwala, H.F.; Pradeep, S. Progress in COX-2 Inhibitors: A Journey So Far. Curr. Med. Chem. 2010, 17, 1563–1593. [Google Scholar] [CrossRef]
- Rimon, G.; Sidhu, R.S.; Lauver, D.A.; Lee, J.Y.; Sharma, N.P.; Yuan, C.; Frieler, R.A.; Trievel, R.C.; Lucchesi, B.R.; Smith, W.L. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc. Natl. Acad. Sci. USA 2010, 107, 28–33. [Google Scholar] [CrossRef]
- Wang, J.L.; Limburg, D.; Graneto, M.J.; Springer, J.; Hamper, R.J.; Liao, S.; Pawlitz, J.L.; Kurumbail, R.J.; Maziasz, T.; Talley, J.T.; et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 2010, 20, 7159–7163. [Google Scholar] [CrossRef]
- Shin, S.S.; Noh, M.-S.; Byun, Y.J.; Choi, J.K.; Kim, J.K.; Lim, K.M.; Kim, J.Y.; Choi, Y.H.; Ha, J.-Y.; Lee, K.-W.; et al. 4,5-Diaryl-3(2H)furanone Derivatives as Cyclooxygenase-2 Inhibitors. U.S. Patent 6492,416, 2 December 2002. [Google Scholar]
- Semenok, D. Selective Inhibitors of Cyclooxygenase and Method for Their Production” (2017). Russ. Patent RU 2631317 C1, 21 September 2017. [Google Scholar]
- Medvedev, J.; Semenok, D.; Nikolaev, V.; Rodina, L. Method of Producing 2,2-dialkyl-4,5-diarylfuran-3(2H)-ones” (2015). Russ. Patent RU 2563876 C1, 21 September 2015. [Google Scholar]
- Regitz, M.; Maas, G. Diazo Compounds. Properties and Synthesis; Academic Press: New York, NY, USA, 1986. [Google Scholar]
- Medvedev, J.J.; Semenok, D.V.; Azarova, X.V.; Rodina, L.L.; Nikolaev, V.A. A New Powerful Approach to Multi-Substituted 3(2H)-Furanones via Brønsted Acid-Catalyzed Reactions of 4-Diazodihydrofuran-3-ones”. Synthesis 2016, 48, 4525–4532. [Google Scholar]
- Rodina, L.L.; Medvedev, J.J.; Galkina, O.S.; Nikolaev, V.A. Thermolysis of 4-Diazotetrahydrofuran-3-ones: Total Change of Reaction Course Compared to Photolysis. Eur. J. Org. Chem. 2014, 2993–3000. [Google Scholar] [CrossRef]
- Rodina, L.; Medvedev, Y.Y.; Moroz, P.N.; Nikolaev, V.A. Thermolysis and acid-catalyzed decomposition of 4-diazotetrahydrofuran-3-ones. A new efficient synthesis of tetrasubstituted dihydrofuran-3-ones. Rus. J. Org. Chem. 2012, 48, 602–604. [Google Scholar] [CrossRef]
- Semenok, D.V. Synthesis and Some Reactions of Sulfur-Containing 5,5-diaryl-4-diazodihydrofuran-3(2H)-ones-3(2H)-ones; Skolkovo Institute of Science and Technology: Moskva, Russia, 2014. [Google Scholar] [CrossRef]
- Nikolaev, V.A.; Galkina, O.S.; Sieler, J.; Rodina, L.L. Surprising secondary photochemical reactions observed on conventional photolysis of diazotetrahydrofuranones. Tetrahedron Lett. 2010, 51, 2713–2716. [Google Scholar] [CrossRef]
- Moon, D.; Kim, M.; Heo, M.; Lee, J.; Choi, Y.; Kim, G. Gefitinib induces apoptosis and decreases telomerase activity in MDA-MB-231 human breast cancer cells. Arch. Pharmacal Res. 2009, 32, 1351–1360. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, B.; Liu, J.; Liu, J.; Li, C.; Dong, W.; Fang, S.; Li, M.; Song, B.; Tang, B.; et al. Mechanisms of Gefitinib-mediated reversal of tamoxifen resistance in MCF-7 breast cancer cells by inducing ERα re-expression. Sci. Rep. 2015, 5, 7835–7842. [Google Scholar] [CrossRef]
- Yu, L.; Chen, M.; Li, Z.; Wen, J.; Fu, J.; Guo, D.; Jiang, Y.; Wu, S.; Cho, C.-H.; Liu, S. Celecoxib Antagonizes the Cytotoxicity of Cisplatin in Human Esophageal Squamous Cell Carcinoma Cells by Reducing Intracellular Cisplatin Accumulation. Mol. Pharm. 2011, 79, 608–617. [Google Scholar] [CrossRef]
- Liu, X.-H.; Rose, D.R. Differential Expression and Regulation of Cyclooxygenase-1 and -2 in Two Human Breast Cancer Cell Lines’. Cancer Res. 1996, 56, 5125–5127. [Google Scholar]
- Mc Fadden, D.W.; Riggs, D.R.; Jackson, B.J.; Cunningham, C. Additive effects of Cox-1 and Cox-2 inhibition on breast cancer in vitro. Int. J. Oncol. 2006, 29, 1019–1023. [Google Scholar]
- Farrell, N.; Kiley, D.M.; Schmidt, W.; Hacker, M.P. Chemical properties and antitumor activity of complexes of platinum containing substituted sulfoxides [PtCl (R′R″SO) (diamine)]NO3. Chirality and leaving-group ability of sulfoxide affecting biological activity. Inorg. Chem. 1990, 29, 397–403. [Google Scholar] [CrossRef]
- Suiko, M.; Maekawa, K. Synthesis and Antitumor Activity of 2-Alkanesulfinyl (or Alkanesulfonyl)-7-methyl-5H-1,3,4-thiadiazolo[3,2-a]pyrimidin-5-ones. Agric. Biol. Chem. 1977, 41, 2047–2053. [Google Scholar]
- Tang, W.; Eisenbrand, G. Salvia miltiorrhiza Bge. Chin. Drugs Plant Origin 1992, 891–902. [Google Scholar]
- Liu, X.-Q.; Guo, Y.-Q.; Gao, W.-Y.; Zhang, T.-J.; Yan, L.-L. Two new phenanthrofurans from Pleione bulbocodioides. J. Asian Nat. Prod. Res. 2008, 10, 453–457. [Google Scholar] [CrossRef]
- Prostaglandin G/H Synthase 1. Available online: http://www.uniprot.org/uniprot/P23219 (accessed on 1 May 2019).
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminf. 2009, 1, 15–31. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comp. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Solis, F.J.; Wets, R.J.B. Minimization by Random Search Techniques. MATH OPER RES. 1981, 6, 19–30. [Google Scholar] [CrossRef]
- Tsolaki, E.; Eleftheriou, P.; Kartsev, V.; Geronikaki, A.; Saxena, A.K. Application of docking analysis in the prediction and biological evaluation of the lipoxygenase inhibitory action of thiazolyl derivatives of mycophenolic acid. Molecules 2018, 23, 1621. [Google Scholar] [CrossRef]
- Ziakas, G.N.; Rekka, E.A.; Gavalas, A.M.; Eleftheriou, P.T.; Kourounakis, P.N. New analogues of butylated hydroxytoluene asanti-inflammatory and antioxidant agents. Bioorg. Med. Chem. 2006, 14, 5616–5624. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Geronikaki, A.; Eleftheriou, P.T.; Hadjipavlou-Litina, D.I.; Filimonov, D.I.; Poroikov, V.V. Computer-aided discovery of potential anti-inflammatory thiazolidinones with dual 5-LOX/COX inhibition. J. Med. Chem. 2008, 51, 1601–1609. [Google Scholar]
- Garcia, R.; Franklin, R.A.; McCubrey, J.A. Cell Death of MCF-7 Human Breast Cancer Cells Induced by EGFR Activation in the Absence of Other Growth Factors. Cell Cycle 2006, 5, 1840–1846. [Google Scholar] [Green Version]
- Mcclelland, R.A.; Barrow, D.; Madden, T.-A.; Dutkowski, C.M.; Ppamment, J.; Knowlden, J.M.; Gee, J.M.W.; Nicholson, R.I. Enhanced Epidermal Growth Factor Receptor Signaling in MCF7 Breast Cancer Cells after Long-Term Culture in the Presence of the Pure Antiestrogen ICI 182,780 (Faslodex)*. Endocrinology 2001, 142, 2776–2788. [Google Scholar] [CrossRef] [Green Version]
- Suojanen, J.; Sorsa, T.; Salo, T. Tranexamic acid can inhibit tonguesquamous cell carcinoma invasion in vitro. Oral Dis. 2009, 15, 170–175. [Google Scholar] [CrossRef]
- Zhao, L.; Au, J.L.-S.; Wientjes, M.G. Comparison of methods for evaluating drug-drug interaction. Front. Biosci. 2010, 2, 241–249. [Google Scholar] [PubMed]
Sample Availability: Not available. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COX-1 | COX-2 | ||||||
|---|---|---|---|---|---|---|---|
| Structure | Code | R | Binding Energy (Kcal/mole) | IC50 (μΜ) | Binding Energy (Kcal/mole) | IC50 (μΜ) | S * |
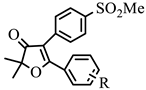 | 1c | H | −5.78 | 25.8 | −6.67 | 0.39 | 66.2 |
| 1o | m–Cl | −6.18 | 17.0 | −10.21 | 0.15 | 113.3 | |
| 1h | p–F | −5.27 | 37.0 | −7.17 | 0.36 | 102.8 | |
| 1k | p–Cl | −4.96 | 43.8 | −7.32 | 0.35 | 125.1 | |
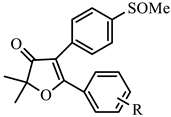 | 1b | H | −5.68 | 27.0 | −6.38 | 0.41 | 65.9 |
| 1n | m–Cl | −5.97 | 21.6 | −7.60 | 0.33 | 65.5 | |
| 1g | p–F | −5.94 | 22.2 | −7.61 | 0.33 | 67.3 | |
| 1j | p–Cl | −6.44 | 11.2 | −6.93 | 0.37 | 30.3 | |
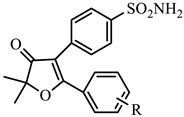 | 1q | H | −6.48 | 10.3 | −9.13 | 0.22 | 46.8 |
| 1v | p–Cl | −6.19 | 16.7 | −9.99 | 0.16 | 1–4.4 | |
| 1w | p–F | −5.83 | 24.7 | −9.61 | 0.19 | 130.0 | |
| Celecoxib | 14(exp) | 0.04(exp) | |||||
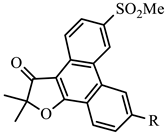 | x-1 | H | −4.92 | 44.7 | −7.28 | 0.28 | 159.6 |
| x-2 | F | −5.23 | 37.9 | −8.86 | 0.24 | 157.9 | |
| R | 1,4-diol (6) | Dihydro furan-3-one (5) | 4-diazodi-hydrofuran-3-one (4) | 4,5-diaryl-3(2H) furanones (2) | Sulfoxides 1b,n,g,j | Sulfones 1c,o,h,k |
|---|---|---|---|---|---|---|
| m-Cl | 57 (65) a | 90 | 83-87 | 83% (94; 5.2:1) c | 92% | 93% |
| m-F | 25 (33) a | 31 | 52 (71) b | 66% (75; 7.3:1) c | - | - |
| p-Cl | 23 (28) a | 91 | 70 | 65% (86; 3.2:1) c | 89% | 98% |
| p-F | 52 (64) a | 84 | 86 | 77% (99; 3.5:1) c | 94% | 80% |
| H | 86–93 | 93 | 71 | 75% (99; 3.2:1) c | 84% | 96% |
| R | 1,4-diol (6) | dihydrofuran-3-one (5) | 4-diazodihyd-rofuran-3-one (4) | 4,5-diaryl-3(2H) furanones (3) | Sulfonyl Chloride | Sulfonyl amide (1q,1u, 1w,1v) |
|---|---|---|---|---|---|---|
| m-Cl | 95 | 98 | 85 | 67 (95;2.45:1) a | 45 b | 79 |
| p-Cl | 96 | 98 | 87 | 55 (99; 1.25:1) a | 50 b | 71 |
| p-F | 98 | 96 | 86 | 47 (99; 1:1.12) a | 62 b | 72 |
| H | 90 | 95 | 69 | 99 (one product) | 50 | 74 |
| R | Sulfones (SO2Me) (1c,1o,1h,1k) | Sulfoxides (SOMe) (1b,1n,1g,1j) | Sulfonamides (SO2NH2) (1q,1u,1w,1v) | Phenanthrenes |
|---|---|---|---|---|
| m-Cl | 33 | 33 | 27 | - |
| p-Cl | 9 | 8 | 29 | - |
| p-F | 23 | 27 | 36 | 3.1 (X-2) |
| H | 43 | 37 | 23 | 6.0 (X-1) |
| Compound | % Inhibition (COX-1) | % Inhibition (COX-2) |
|---|---|---|
| 1c (H) | 0 | 0 |
| 1h (p–F) | 32 | 0 |
| 1k (p–Cl) | 80 | 6 |
| 1j (p–Cl) | 0 | 0 |
| 1g (p–F) | 73 | 0 |
| Experimental IC50, μΜ | Docking Assisted Predicted Values | |||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | 2 Ovis aries COX-1, 3KK6 | 2 Mus musculus COX-2, 3LN1 | 3 Ovine COX-1 4O1Z | 3 Human COX-2 5IKT | ||||
| COX-1 | COX-2 | Ε Kcal/mol | IC50 (μΜ) | Ε Kcal/mol | IC50 (μΜ) | COX-1 | COX-2 | |
| 1k (p–Cl) | 29 | > 50 | −4.96 | 43.8 | −7.32 | 0.345 | −5.69 | +5.91 |
| 1o (m–Cl) | 22 | 71 | −6.18 | 17.0 | −10.21 | 0.147 | −6.27 | −5.49 |
| 1g (p–F) | 2.8 | > 50 | −5.94 | 22.2 | −7.61 | 0.325 | −6.33 | −3.50 |
| 1v (p–Cl) | 28 | 20 | −6.19 | 16.7 | −9.99 | 0.162 | −7.01 | −4.36 |
| 1w (p–F) | 70 | >50 | −5.83 | 24.7 | −9.61 | 0.188 | −4.85 | −2.92 |
| x-1 | −6.09 | −5.87 | ||||||
| Naproxen | 40 | 50 | −5.66 | |||||
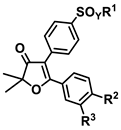
| Class | Compound | Dose, µmol/g | CPE, % * |
|---|---|---|---|
| SO2CH3 | 1c (H) | 0.1 | 50.2 |
| 1o (m–Cl) | 0.1 | 50.7 | |
| 1h (p–F) | 0.1 | 48.8 | |
| 1k (p–Cl) | 0.1 | 50.6 | |
| SOCH3 | 1b (H) | 0.1 | 47 |
| 1n (m–Cl) | 0.1 | 51 | |
| 1g (p–F) | 0.1 | 49 | |
| 1j (p–Cl) | 0.1 | 54 | |
| SO2NH2 | 1q (H) | 0.1 | 38 |
| 1v (p–Cl) | 0.1 | 44.3 | |
| 1w (p–F) | 0.1 | 43.7 | |
| Phen | x-1 (H) | 0.1 | 45 |
| x-2 (p–F) | 0.1 | 40 | |
| Reference | E-1 (H) | 0.1 | 57.7 |
| E-3 (m–Cl) | 0.1 | 52.1 |
| Compound | MCF-7 IC50 Values (µM) * | HSC-3 % Cell ** Growth | HSC-3 IC50 Values (µM) * |
|---|---|---|---|
| 1-h | 24 | 100 ± 10 | 90 |
| 1-g | 10 | 36 ± 4 | 7.5 |
| x-1 | 10 | 91 ± 10 | 60 |
| Gefitinib | 70 | ||
| 5-Fluorouracil | 0.03 | ||
| Celecoxib | 29.186 |
| Compound * Concentrations (μM) | % Cell Growth | CI | Combination Effect |
|---|---|---|---|
| 27.5 μM 1h + 70 μM gef | 45.9 | >1 | antagonism |
| 10 μM 1g + 70 μM gef | 36.5 | 0.780 ± 0.200 < 1 | synergism |
| 10 μM x-1 + 70 μM gef | 24.3 | 0.260 ± 0.033 < 1 | synergism |
| 29 μM cel + 70 μM gef | 56.8 | >1 | antagonism |
| 21.3 μM 1h + 0.3 μM 5-fu | 27.3 | 0.533 ± 0.015 <1 | synergism |
| 2.9 μM 1g + 0.3 μM 5-fu | 19.1 | 0.058 ± 0.010 <1 | synergism |
| 3.8 μM x-1 + 0.3 μM 5-fu | 21.0 | >1 | antagonism |
| 29 μM cel + 00.3 μM 5-fu | 49.3 | >1 | antagonism |
| 21.3 μM 1h + 29 μM cel | 38.6 | >1 | antagonism |
| 2.9 μM 1g + 29 μM cel | 36.6 | 0.750 ± 0.025 < 1 | synergism |
| 3.8 μM x-1 + 29 μM cel | 60.6 | >1 | antagonism |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semenok, D.; Medvedev, J.; Giassafaki, L.-P.; Lavdas, I.; Vizirianakis, I.S.; Eleftheriou, P.; Gavalas, A.; Petrou, A.; Geronikaki, A. 4,5-Diaryl 3(2H)Furanones: Anti-Inflammatory Activity and Influence on Cancer Growth. Molecules 2019, 24, 1751. https://doi.org/10.3390/molecules24091751
Semenok D, Medvedev J, Giassafaki L-P, Lavdas I, Vizirianakis IS, Eleftheriou P, Gavalas A, Petrou A, Geronikaki A. 4,5-Diaryl 3(2H)Furanones: Anti-Inflammatory Activity and Influence on Cancer Growth. Molecules. 2019; 24(9):1751. https://doi.org/10.3390/molecules24091751
Chicago/Turabian StyleSemenok, Dmitrii, Jury Medvedev, Lefki-P. Giassafaki, Iason Lavdas, Ioannis S. Vizirianakis, Phaedra Eleftheriou, Antonis Gavalas, Anthi Petrou, and Athina Geronikaki. 2019. "4,5-Diaryl 3(2H)Furanones: Anti-Inflammatory Activity and Influence on Cancer Growth" Molecules 24, no. 9: 1751. https://doi.org/10.3390/molecules24091751